
Abstract
Monogenic muscular dystrophies (MDs), such as Duchenne muscular dystrophy (DMD) and limb-girdle muscular dystrophy (LGMD), are characterized by chronic inflammation, progressive fibrosis, and impaired muscle regeneration. Central to these pathological processes are macrophages, which exhibit dynamic polarization states that influence the dystrophic microenvironment. In early disease stages, macrophages support tissue repair and regeneration, but chronic inflammation skews their activity toward pro-fibrotic phenotypes, driving excessive extracellular matrix (ECM) deposition and muscle dysfunction. Macrophages also interact with other immune cells, such as T cells and neutrophils, and non-immune cells, including fibroblasts and satellite cells, to regulate inflammatory and fibrotic responses. These interactions establish a dysregulated immune environment that exacerbates muscle damage and impairs effective regeneration. Preclinical studies using the mdx mouse model of DMD highlight the critical role of macrophages in sustaining inflammation and fibrosis, particularly through transforming growth factor-beta (TGF-β) signaling and fibro-adipogenic progenitor (FAP) activation. Therapeutically, targeting macrophages offers significant potential to mitigate disease progression. Strategies include modulating macrophage polarization toward a pro-regenerative M2 phenotype, inhibiting macrophage recruitment via chemokine signaling, and reprogramming macrophage metabolism to support oxidative phosphorylation and mitochondrial function. Additionally, anti-fibrotic interventions targeting TGF-β signaling or macrophage-FAP crosstalk have shown promise in reducing ECM deposition and preserving muscle architecture. In this review, we curate relevant studies and provide insights into the molecular mechanisms governing macrophage behavior in dystrophic muscle. Herein, we discuss how emerging therapeutic strategies targeting macrophage-mediated pathways can be leveraged to mitigate inflammation and fibrosis, enhance muscle regeneration, and improve clinical outcomes.
Keywords
Macrophage, muscular dystrophy, muscle injury, chronic muscle inflammationIntroduction
Muscular dystrophies (MDs) are a group of genetic disorders characterized by progressive muscle wasting, weakness, and eventual loss of functional mobility [1]. These disorders result from mutations in genes encoding structural, enzymatic, or regulatory proteins essential for muscle integrity and function. While MDs vary in their genetic causes, clinical severity, and patterns of muscle involvement, they commonly feature inflammation and fibrosis, which contribute to disease progression and tissue regeneration.
Among the diverse forms of MDs, dystrophinopathy [due to mutations in the Duchenne muscular dystrophy (DMD) gene encoding dystrophin] and dysferlinopathy (resulting from DYSF gene mutations affecting dysferlin) are two of the most extensively studied and clinically significant subtypes due to their well-characterized genetic basis and significant clinical burden. Despite their genetic origins, these disorders share key pathological features, including persistent inflammation and fibrosis, accompanied by accumulating macrophages highlighting the critical role of immune responses in disease progression.
Macrophages, a heterogeneous and highly plastic population of immune cells, are pivotal players in the immune response to muscle injury. Under physiological conditions, macrophages participate in a tightly regulated process of inflammation and resolution, clearing debris and promoting tissue repair [2, 3]. However, in the context of chronic muscle diseases such as dystrophinopathy and dysferlinopathy, macrophages adopt maladaptive roles that perpetuate inflammation and fibrosis. These cells are capable of polarizing into distinct functional states, pro-inflammatory (M1) and anti-inflammatory (M2), which can either exacerbate tissue damage or contribute to repair and fibrosis, depending on the context (Figure 1). Understanding the factors that drive macrophage behavior in MDs is critical for uncovering new therapeutic strategies.
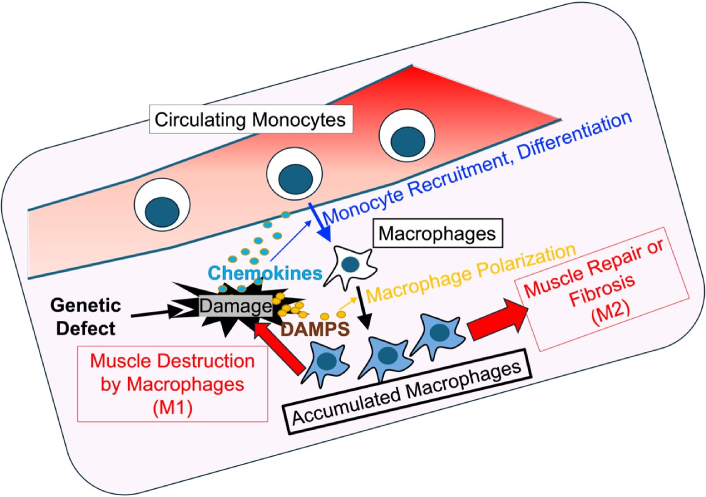
Macrophage dynamics in monogenic MDs. Bone marrow-derived monocytes are recruited to the site of muscle damage caused by genetic defects, where they proliferate and differentiate into macrophages. Macrophages polarize into two subsets: M1 macrophages (pro-inflammatory), which exacerbate tissue damage, and M2 macrophages (anti-inflammatory), which contribute to tissue repair and fibrosis. Muscle damage caused by necrosis and apoptosis initiates this immune response. Chronic cycles of inflammation and repair lead to excessive extracellular matrix deposition, resulting in muscle fibrosis, a hallmark of dystrophic pathology. DAMPs: damage-associated molecular patterns; MDs: muscular dystrophies
The chronic inflammatory environment in MDs creates a positive feedback loop, where persistent macrophage activation leads to the release of cytokines and growth factors that further damage muscle tissue or promote fibrosis. Additionally, the interactions between macrophages and other cellular players, such as T cells, fibroblasts, and myogenic cells, amplify the pathological outcomes. Recent studies have shed light on key signaling pathways and molecular mechanisms governing macrophage activity in these conditions, including NF-κB, transforming growth factor-beta (TGF-β), and JAK/STAT signaling cascades [4–6]. These pathways offer potential targets for therapeutic intervention aimed at reprogramming macrophages to mitigate inflammation and fibrosis.
In this review, we aim to provide a comprehensive overview of the roles and functions of macrophages in monogenic MDs, with a particular focus on their contributions to inflammation and fibrosis. We focus on the molecular mechanisms governing macrophage polarization, their crosstalk with immune and non-immune cells, and the intricate signaling pathways that regulate their functional states within the dystrophic microenvironment. Furthermore, we critically evaluate emerging therapeutic strategies targeting macrophage-mediated pathways, emphasizing their potential to reshape the therapeutic paradigm for MDs.
Overview of common MDs
Duchenne muscular dystrophy
DMD is the most common form of dystrophinopathy, a severe, X-linked recessive genetic disorder characterized by progressive muscle degeneration, chronic inflammation, and fibrosis. DMD is the most prevalent type of muscular dystrophy and results from mutations in the dystrophin gene, which is an essential structural component of muscle fibers [7].
Dystrophin plays a critical role in maintaining the structural integrity of the muscle cell membrane by linking the actin cytoskeleton to the dystrophin-associated glycoprotein complex (DGC), a multiprotein complex crucial for muscle function and membrane stability during contraction [8, 9]. In the absence of functional dystrophin, muscle cells cannot withstand the mechanical stress caused by repeated contractions, leading to cellular damage and leakage of intracellular components. This triggers degeneration and regeneration, where damaged muscle fibers attempt to repair, but ultimately fail, resulting in progressive muscle weakness [10]. The weakness primarily affects proximal muscles, including those of the hips, thighs, and shoulders, and is often noticeable in early childhood as delayed motor milestones, such as difficulty in standing, walking, and running [8].
As the disease advances, muscle weakness worsens, leading to loss of ambulation by early adolescence. Most patients are unable to walk between the age of 12 and 14 [11]. DMD is often accompanied by severe complications, including respiratory failure and cardiomyopathy, which are the primary causes of morbidity and mortality in affected individuals [8, 12]. As muscle fibers continue to deteriorate, they are progressively replaced by fibrosis and adipose tissue, further impairing muscle function and mobility [13]. This replacement of healthy muscle tissue exacerbates the disability and significantly reduces the patient’s quality of life, ultimately leading to premature death, typically due to cardiac and respiratory failure in the late teens or early twenties [13].
The mdx mouse strain serves as a widely used animal model for human DMD. Identified in the early 1980s within a colony of C57Bl/10ScSn mice, the primary mdx mouse model (C57Bl/10ScSn-Dmdmdx) carries a spontaneous nonsense point mutation (C>T) within exon 23 in the dystrophin (Dmd) gene, resulting in a premature stop codon [14, 15]. Consequently, mdx mice exhibit a deficiency of functional dystrophin in their muscles. Similar to human patients, these mice display muscle fiber necrosis and degeneration, elevated levels of serum creatine kinase, and a gradual onset of muscle weakness [14]. The mdx mice closely mirrors the pathology observed in affected humans, making it an invaluable tool for studying disease mechanisms and potential therapeutic interventions [16].
Dysferlinopathy
Dysferlinopathy refers to a group of muscle disorders caused by mutations in the DYSF gene, which encodes the dysferlin protein. These disorders include limb-girdle muscular dystrophy type 2B (LGMD2B) and Miyoshi myopathy (MM) [17]. Dysferlin is 237 kDa membrane protein that plays a crucial role in repairing muscle cell membranes, stabilizing calcium signaling, and supporting the development of the T-tubule system essential for muscle function [18–20]. The absence or dysfunction of dysferlin disrupts these processes, leading to progressive muscle degeneration, impaired calcium signaling, and chronic inflammation [18, 21, 22].
LGMD2B primarily affects the proximal muscles of the shoulders, hips, and limbs, causing weakness that typically manifests in adolescence or early adulthood. In contrast, MM often begins with weakness in the distal muscles, particularly in the calves [17]. While LGMD2B progresses more slowly than DMD, it can still result in severe disability and loss of ambulation in later stages. Unlike DMD, which predominantly affects boys, dysferlinopathy affect both men and women equally [18, 23].
In dysferlin-deficient muscles, the inability to repair sarcolemma damage leads to repeated phases of muscle breakdown and regeneration, exacerbated by inflammatory responses. These mechanisms further contribute to muscle atrophy and functional impairment over time [24]. Although no definitive cure exists, ongoing research into gene therapy, stem cell treatments, and other therapeutic approaches shows promise for managing symptoms and improving quality of life for patients with dysferlinopathy. Dysferlinopathy has been extensively studied using the A/J mouse strain and its backcross variants [25]. The deficiency of dysferlin in A/J mice arises. This condition in A/J mice arises from a homozygous retrotransposon insertion in the dysferlin (Dysf) gene [26]. Muscular dystrophy in these mice typically manifests later in life, around four to five months of age, and is progressive. The muscle pathology is characterized by repeated myofiber degeneration and regeneration, with centrally positioned nuclei [25]. Notably, the proximal muscles in A/J mice are more severely affected by this condition compared to the distal muscles.
Inflammation and fibrosis in muscular dystrophy
While inflammation is a natural response to muscle injury, repeated cycles of damage in muscular dystrophy result in chronic inflammation that impairs repair mechanisms and promotes fibrosis. Upon muscle injury, damage-associated molecular patterns (DAMPs), such as ATP and as high-mobility group box 1 (HMGB1), activate pattern recognition receptors (PRRs) on immune cells, recruiting neutrophils and macrophages. Initially, M1 macrophages dominate and release pro-inflammatory cytokines like TNF-α, IL-1β, and IFN-γ, which aid in debris clearance but exacerbate damage when prolonged [27]. In healthy muscle, M1 macrophages transition to M2 macrophages to promote anti-inflammatory signaling and repair. However, in muscular dystrophy, this transition is impaired, leading to persistent inflammation and inadequate regeneration. This chronic inflammation contributes to fibrosis, characterized by excessive extracellular matrix (ECM) deposition, such as collagen and fibronectin. TGF-β is a central mediator in this process, activating fibroblasts and fibro-adipogenic progenitors (FAPs) to produce ECM components [28]. Dysregulated ECM remodeling, caused by an imbalance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), leads to scar tissue formation that disrupts muscle architecture and reduces satellite cell function essential for regeneration [29]. The interplay between chronic inflammation and fibrosis accelerates muscle degeneration and dysfunction. Common mediators like TGF-β and NF-κB signaling pathways highlight the interconnected nature of these processes, emphasizing the need for targeted therapies to mitigate fibrosis and preserve muscle function [30].
The role and function of intra-muscle macrophages
In skeletal muscle, resident macrophages play a crucial role in maintaining tissue homeostasis and promoting muscle growth and regeneration [31]. These macrophages are part of the broader group of tissue-resident macrophages, which are distinct from infiltrating macrophages that appear during injury or disease [32]. While infiltrating macrophages primarily arise from blood monocytes, tissue-resident macrophages have diverse origins, including both embryonic and adult hematopoiesis [33–35]. Skeletal muscle-resident macrophages are predominantly derived from two sources: embryonic progenitors, including primitive yolk sac macrophages and fetal liver monocytes, and adult bone marrow hematopoietic stem cells (HSCs) [36]. These macrophages are present in interstitial tissues of the skeletal muscle and exhibit distinct molecular and functional characteristics. For example, skeletal muscle macrophages express a set of transcription factors, such as Maf, Mef2c, and Tcf4, which are unique to muscle tissue, underlining their tissue-specific functions [37]. Functionally, skeletal muscle-resident macrophages can be divided into different subsets based on their origin and activation status. One subset, characterized by high MHC-II and low Lyve1 expression (CCR2+MHC-IIhighLyve1low), predominantly arises from adult blood monocytes and plays an active role in antigen presentation. Another subset, which is more phagocytic, arises from both embryonic and adult progenitors and expresses low MHC-II and high Lyve1 (CCR2–MHC-IIlowLyve1high) [38]. These macrophages are involved in tissue maintenance and homeostasis, helping to protect muscle fibers and facilitate their repair after injury [37]. The functions of these intra-muscle macrophages extend beyond simple tissue surveillance. They have been shown to quickly respond to microlesions in muscle tissue by sensing DAMPs and preventing excessive inflammation. Through a process known as “cloaking”, these macrophages seal off the damaged areas, preventing tissue destruction and allowing for tissue healing without inflammation. This function is vital for maintaining the structural integrity of muscle fibers and preventing the loss of myofibers due to microlesions [39]. Moreover, skeletal muscle-resident macrophages also exhibit muscle type-specific features, with respiratory muscle macrophages expressing a higher level of stress response genes compared to those in limb muscles. This suggests that the macrophages’ functions are tailored to the specific demands and stressors of different muscle types [37]. Despite the recognition of their importance in muscle homeostasis, much remains unknown about the detailed interactions between muscle-resident macrophages and other cells in the muscle environment, such as myogenic cells. Future research is needed to explore how these macrophages contribute to muscle development, growth, and repair, as well as their roles in postnatal muscle regeneration. Understanding the full scope of their function could lead to new therapeutic strategies for muscle diseases and injuries.
Influence of macrophages in monogenic MDs
Dystrophinopathy (e.g., DMD) and dysferlinopathy (e.g., LGMD2B) are distinct MDs driven by different genetic mutations. Despite these differences, both conditions involve progressive muscle degeneration, chronic inflammation, and fibrosis, with macrophages playing a central role in mediating these processes. These immune cells contribute to inflammation and fibrosis, exacerbating muscle damage and impairing regeneration. Understanding macrophage-mediated mechanisms provides critical insights into disease progression and therapeutic opportunities.
Macrophage contribution to inflammation in monogenic MDs
In DMD, macrophages infiltrate the damaged muscle tissue in response to continuous muscle degeneration and regeneration. The persistent muscle damage leads to the recruitment of various immune cells, with macrophages being one of the most significant contributors. Initially, pro-inflammatory M1 macrophages dominate the damaged area, releasing a cascade of cytokines, including TNF-α, IL-1β, and IL-6. These cytokines not only amplify the inflammatory response but also directly contribute to the degradation of muscle fibers by inducing oxidative stress and promoting apoptosis of myocytes [40].
M1 macrophages are particularly specialized for free-radical production, driven by the expression of inducible nitric oxide synthase (iNOS). iNOS is induced by TNF-α through the activation of the NF-κB signaling pathway, resulting in the production of nitric oxide (NO) and citrulline [41]. The high levels of NO generated by iNOS disrupt the redox environment within the injured tissue, react with other free radicals, and amplify their reactivity and toxicity. This contributes to the lysis of muscle cell membranes, which has been demonstrated in vitro using mdx muscle-derived macrophages, where iNOS inhibition prevented muscle cell damage [41]. Similarly, mdx mice, a murine model of human DMD, with an iNOS-null mutation exhibited reduced muscle membrane damage during the acute phase of pathology [41]. However, the partial reduction of damage highlights the involvement of iNOS-independent mechanisms, emphasizing the multifactorial role of M1 macrophages in driving muscle pathology [42].
The regulation of M1 macrophage activation in DMD involves multiple interconnected signaling pathways. The activation of M1 macrophages in DMD is regulated through multiple signaling pathways triggered by DAMPs and pro-inflammatory cytokines. DAMPs, such as high-mobility HMGB1 and extracellular ATP released from damaged muscle fibers, bind to PRRs like Toll-like receptor 4 (TLR4) on macrophages [43–45]. This interaction activates downstream signaling cascades, including the MyD88-dependent pathway, which leads to the activation of NF-κB and the production of pro-inflammatory cytokines such as TNF-α and IL-1β [46]. Additionally, ATP binding to purinergic receptors (e.g., P2X7) enhances NLRP3 inflammasome assembly, promoting the secretion of IL-1β and IL-18, further amplifying the inflammatory response [47, 48]. Furthermore, cytokines such as IFN-γ, predominantly secreted by infiltrating T cells, reinforce the M1 phenotype by enhancing STAT1 signaling in macrophages and also suppressing M2 macrophage activation [49–52]. Together, these pathways sustain the pro-inflammatory state of M1 macrophages in DMD, perpetuating oxidative stress and muscle damage.
Additionally, macrophage-secreted chemokines such as CCL2 play a pivotal role in enhancing the recruitment of more immune cells, perpetuating the inflammatory environment and sustaining the dominance of M1 macrophages during early stages of the response [53]. This inflammatory state leads to further damage by recruiting neutrophils and other cytotoxic immune cells into the dystrophic tissue.
In parallel, M2 macrophages, which are involved in anti-inflammatory and reparative processes, are recruited as a counter-regulatory mechanism. These macrophages produce IL-10 and TGF-β to attenuate inflammation and support muscle regeneration [54]. However, in DMD, the dynamic balance between M1 and M2 macrophages is disrupted, resulting in a chronic inflammatory state. This disruption is exacerbated by chronic deterioration and attempted muscle renewal, which prevent the resolution of inflammation, leading to fibrosis and impaired tissue repair. Consequently, prolonged M1 macrophage activity amplifies muscle damage and perpetuates the pathological cycle [54].
This imbalance is exacerbated by the repeated cycles of degeneration and regeneration, which prevent effective resolution of inflammation and hinder proper tissue repair. Consequently, the prolonged presence of M1 macrophages and their cytotoxic activity further aggravates muscle damage, perpetuating the pathological cycle of inflammation and fibrosis.
In dysferlinopathy, defective membrane repair caused by mutations in the dysferlin gene results in chronic muscle damage, creating a sustained trigger for macrophage recruitment and activation [31]. Pro-inflammatory M1 macrophages are predominant during the early stages of inflammation, releasing cytokines such as TNF-α and IL-1β, which amplify the inflammatory response and exacerbate muscle fiber damage [55, 56]. The persistent activation of M1 macrophages leads to the production of reactive oxygen species (ROS), further contributing to oxidative stress and destabilizing the muscle environment [57].
Macrophage activity in dysferlinopathy is significantly influenced by dysregulated signaling pathways, particularly those involving TLRs. MyD88, an adapter protein critical for TLR signaling, plays a central role in the recruitment of macrophages to dysferlin-deficient muscles. When muscles are exposed to DAMPs such as ssRNA, MyD88-dependent signaling through TLR7 and TLR8 is upregulated, driving macrophage activation and invasion. Although MyD88 deletion reduces inflammatory cell recruitment in the presence of DAMPs, its ablation has minimal effect in the absence of external stimuli, suggesting that additional mechanisms contribute to macrophage-driven inflammation in dysferlinopathy [57].
Thrombospondin-1 (TSP1), a TLR4 ligand, further modulates macrophage activity in dysferlin-deficient muscles. TSP1 is expressed at elevated levels in dysferlin-deficient myotubes and activates TLR4/NF-κB signaling in macrophages, enhancing their inflammatory responses. Importantly, this increase in TSP1 expression is directly linked to dysferlin deficiency rather than being secondary to muscle degeneration. Moreover, TSP1 also acts as a chemoattractant for macrophages, and its neutralization significantly reduces monocyte/macrophage recruitment to dysferlin-deficient muscle cultures, underlining its specific role in macrophage-driven inflammation [57–59].
Ultimately, the persistent activation of macrophages and their dysregulated signaling pathways contribute to the chronic inflammatory environment in dysferlinopathy. This prolonged inflammation not only exacerbates muscle degeneration but also impairs effective tissue repair. Thus, macrophage-driven inflammation emerges as a critical driver of disease progression, reinforcing the need for therapeutic interventions targeting these pathways [57–59].
Macrophage role in fibrosis progression in monogenic MDs
Fibrosis is a pathological hallmark of dystrophinopathy and dysferlinopathy, resulting from the complex interaction between immune responses and tissue repair mechanisms, with macrophages playing a pivotal role. These immune cells regulate the local microenvironment, influencing both inflammation and fibrosis through dynamic functional phenotypes.
In DMD, macrophages undergo dynamic polarization, transitioning between the pro-inflammatory M1 phenotype and the anti-inflammatory M2 phenotype. Early in the disease process, M2 macrophages promote tissue repair by aiding in the removal of debris and stimulating muscle regeneration. However, chronic activation of M2 macrophages over time results in excessive secretion TGF-β, a key cytokine for fibroblast activation [60, 61]. TGF-β signaling enhances the synthesis and deposition of ECM components, such as collagen, which progressively replaces functional muscle tissue with fibrotic scar tissue. This pathological remodeling disrupts muscle elasticity and impedes effective force transmission, worsening muscle weakness and loss of function. As the disease progresses, the continuous cycle of muscle injury and ineffective regeneration fosters the accumulation of fibroadipose tissue, further impairing muscle performance and mobility [62], reinforcing the cycle of fibrosis and muscle degeneration in DMD.
Unlike typical muscle injury, where macrophage polarization follows a clear timeline towards tissue repair and regeneration, DMD features an ongoing cycle of muscle damage with insufficient repair. This results in a sustained inflammatory response, where macrophages often fail to transition fully into the regenerative M2 phenotype. Instead, there is a persistent presence of both M1 and M2-like macrophages, with a shift towards a pro-fibrotic M2 phenotype in the later stages of the disease. This shift is marked by the upregulation of arginase-1 (Arg1) and other fibrosis-associated molecules, which promote collagen deposition in the muscle, exacerbating fibrosis and impairing the ability of muscle satellite cells to regenerate myofibers [41, 63].
In dysferlinopathy, a similar inflammatory and fibrotic response is observed, though the underlying mechanisms differ due to the dysferlin deficiency. Dysferlin is essential for proper membrane repair in muscle fibers, and its absence leads to chronic muscle damage and inflammation [64]. Macrophages in dysferlinopathy also exhibit polarized phenotypes, with an initial M2 response attempting repair and regeneration. However, the sustained muscle damage and ineffective repair in dysferlinopathy, similar to DMD, promote the shift of macrophages toward a more fibrotic M2 phenotype. This results in chronic secretion of TGF-β and subsequent collagen deposition, contributing to fibrosis. While the exact contribution of dysferlin to macrophage activation is still under investigation, the fibrotic response in dysferlinopathy involves both macrophages and fibroblasts in a similar manner to DMD.
The immune response in both dystrophinopathy and dysferlinopathy, the profibrotic actions of macrophages are further amplified by secondary damage driven by the immune response. Fibrinogen, a soluble acute phase protein released during stress, plays a critical role in this process. It not only contributes to blood clotting following vascular injury but also extravasates into sites of inflammation, where it can interact with macrophages through receptors such as Mac-1. This interaction induces the production of pro-inflammatory cytokines such as IL-1β, which in turn stimulates TGF-β production. TGF-β then promotes collagen synthesis by fibroblasts, leading to the excessive deposition of ECM proteins, particularly type I and III collagen, which drive fibrosis in the dystrophic muscle [65].
Recent studies in the mdx mouse model of DMD have shown that macrophages play an integral role in sustaining inflammation and promoting fibrosis through their diverse phenotypes. During the early stages of the disease, macrophages contribute to the removal of damaged tissue, but chronic inflammation triggers a shift towards a more fibrotic environment. This ongoing fibrotic remodeling creates a pro-fibrotic niche that impedes muscle regeneration by satellite cells, contributing to the progressive loss of muscle function [4, 41].
Macrophage interactions with other immune cells in monogenic MDs
In both dystrophinopathy and dysferlinopathy, macrophages interact dynamically with other immune cells, creating a complex inflammatory microenvironment that influences disease progression (Figure 2).
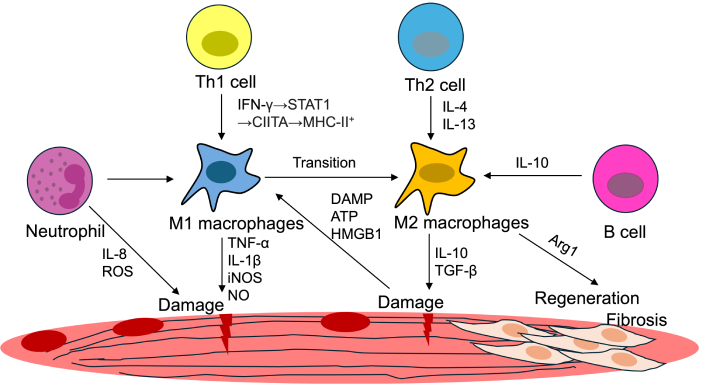
Pathological mechanisms in MDs. Damage-associated molecular patterns (DAMPs), such as ATP and HMGB1, activate immune responses, recruiting M1 and M2 macrophages, neutrophils, and T cells. M1 macrophages contribute to inflammation, while M2 macrophages, influenced by IL-4 and IL-13, are associated with tissue repair and fibrosis. Transitioning macrophage phenotypes and interactions with Th1/Th2 cells, B cells, and pro-regenerative signals (e.g., IL-10, TGF-β) mediate the balance between regeneration and fibrosis. ROS: reactive oxygen species; iNOS: inducible nitric oxide synthase; NO: nitric oxide; HMGB1: high-mobility group box 1; TGF-β: transforming growth factor-beta; Arg1: arginase-1; MDs: muscular dystrophies
T cells, particularly the CD4+ and CD8+ subsets, play an essential role in regulating macrophage activity within inflammatory environments, especially during muscle pathology. Th1 cytokines, such as IFN-γ, which activates the CIITA transcription factor and leads to MHC-II+ upregulation and promote the activation of M1 macrophages, which are pro-inflammatory and can exacerbate muscle damage by increasing the release of inflammatory mediators and cytotoxic molecules [66]. Conversely, Th2 cytokines, such as IL-4 and IL-13, drive a shift toward the M2 macrophage phenotype, characterized by anti-inflammatory properties and the promotion of tissue repair [67, 68]. While M2 macrophages are beneficial in acute tissue injury by aiding repair, under chronic inflammatory conditions, they can contribute to fibrosis by promoting ECM deposition and scar formation. This dynamic interplay between T cells and macrophages underscores the complex immune modulation that influences disease progression in MDs.
Neutrophils, recruited early during muscle damage, release pro-inflammatory mediators such as IL-8 and ROS, which enhance macrophage activation and recruitment, amplifying the inflammatory response. In vitro studies reveal that neutrophils induce muscle cell death via superoxide-dependent mechanisms, while macrophages rely on NO-dependent pathways. When neutrophils and macrophages co-exist with muscle cells in proportions seen in injured muscle, cytotoxicity shifts to a NO-dependent, superoxide-independent mechanism, with their combined presence significantly increasing muscle cell death compared to either cell type alone [69].
In addition to T cells and neutrophils, other immune cell interactions with macrophages play a critical role in the pathogenesis of dystrophinopathy and dysferlinopathy. B cells, for example, contribute to immune modulation not only through antibody production but also via cytokine secretion. IL-10 produced by B cells can promote the polarization of macrophages toward an M2 phenotype, which may be beneficial in acute injury but could exacerbate fibrosis under chronic conditions [70].
Finally, non-immune cells such as fibroblasts and satellite cells also interact with macrophages, influencing muscle regeneration and repair. Macrophages regulate satellite cell activation, which is essential for muscle regeneration, and interact with fibroblasts to mediate ECM remodeling. These interactions highlight the importance of considering both immune and non-immune components of the inflammatory microenvironment when exploring therapeutic targets for MDs [38].
Together, these interactions not only sustain chronic inflammation but also create a dysregulated immune environment that impairs muscle regeneration, highlighting the need for therapies targeting the crosstalk between macrophages and other immune cells in monogenic MDs.
Macrophages as therapeutic targets in muscular dystrophy
Macrophages represent promising therapeutic targets in MDs due to their central roles in driving inflammation, fibrosis, and impaired muscle regeneration. Modulating macrophage activity offers the potential to mitigate chronic inflammation, reduce fibrosis, and restore a pro-regenerative muscle environment. Advances in understanding macrophage biology and their molecular signaling pathways have paved the way for the development of targeted interventions to address the pathological processes mediated by these immune cells.
One key approach is to shift the macrophage polarization balance from the pro-inflammatory M1 phenotype to the anti-inflammatory and pro-regenerative M2 phenotype. Strategies to achieve this include the administration of anti-inflammatory cytokines, such as IL-4 and IL-13, which promote M2 polarization [71]. Similarly, IL-10 therapy has shown potential in reducing chronic inflammation by suppressing pro-inflammatory cytokine production and enhancing tissue repair mechanisms [72]. Targeting signaling pathways that regulate macrophage activation, such as NF-κB, can also be effective. Pharmacological inhibitors of NF-κB have demonstrated the ability to dampen inflammation and improve muscle repair in preclinical models [73].
Another promising strategy involves targeting chemokine signaling to modulate macrophage recruitment to the site of injury. Inhibition of the CCL2-CCR2 axis, which is critical for monocyte and macrophage recruitment, has been shown to reduce macrophage infiltration and alleviate inflammation in dystrophic muscle [74].
Macrophage metabolic reprogramming is an emerging area of interest for therapeutic intervention. The distinct metabolic states of M1 and M2 macrophages provide a potential target for shifting their activity. For example, promoting oxidative phosphorylation and mitochondrial biogenesis in macrophages has been shown to enhance M2 polarization, improving their ability to support muscle regeneration [75, 76]. Agents such as PGC-1α activators or AMPK agonists could be explored to modulate macrophage metabolism in dystrophic conditions [77, 78].
Macrophage-targeted therapies offer complementary benefits to existing treatments, such as corticosteroids, which focus on inflammation suppression and dystrophin restoration. However, these traditional therapies fail to fully address the interconnected issues of inflammation, fibrosis, and impaired muscle regeneration.
For instance, while corticosteroids broadly suppress inflammation, they often lead to immune suppression-related side effects [79]. Macrophage-targeted therapies, on the other hand, allow for localized modulation of inflammation through M1 to M2 polarization, reducing systemic immune suppression while simultaneously enhancing muscle regeneration. This synergy between macrophage-focused and traditional therapies provides a holistic approach to managing MDs.
Therapeutics targeting macrophage-mediated fibrosis have also shown potential. Modulating TGF-β signaling, a central driver of fibrosis, can reduce ECM deposition and improve muscle architecture. Small molecule inhibitors of TGF-β or SMAD signaling pathways, as well as neutralizing antibodies against TGF-β, have demonstrated efficacy in preclinical studies by reducing fibrosis and preserving muscle function [80–82]. Additionally, targeting the activity of FAPs influenced by macrophages may prevent excessive ECM deposition and mitigate fibrosis progression [83].
By addressing the underlying immune dysregulation, macrophage-focused interventions can complement these therapies to provide a more comprehensive approach to managing muscle degeneration.
In conclusion, macrophages are pivotal targets for therapeutic intervention in monogenic MDs. Modulating their activity and key molecular pathways could help reduce inflammation and fibrosis while enhancing muscle repair. By combining macrophage-targeted therapies with existing treatments, a more holistic approach to managing MDs is achievable. However, practical challenges, such as high costs and technical barriers, must be overcome to translate these strategies into clinical success.
Conclusions
Dystrophinopathy and dysferlinopathy are devastating MDs marked by chronic inflammation, progressive fibrosis, and impaired muscle regeneration, all of which are closely linked to macrophage dysregulation. Throughout disease progression, macrophages play a pivotal role, transitioning from mediators of acute repair to drivers of chronic pathological processes. This dual role highlights macrophages as key players in inflammation, fibrosis progression, and immune signaling disruption in dystrophic muscles [7, 10, 12].
Inflammatory macrophages contribute to the exacerbation of muscle damage by perpetuating the release of pro-inflammatory cytokines and chemokines. Key signaling pathways, such as NF-κB, IL-1β, and TGF-β, have been identified as crucial regulators of macrophage activity and their downstream effects on tissue inflammation and fibrosis [11, 17, 18]. Additionally, macrophages are implicated in shaping the fibrotic environment by interacting with FAPs, thereby promoting excessive ECM deposition and hindering effective muscle repair [1, 23].
Given their multifaceted roles, macrophages serve as promising therapeutic targets in MDs. Strategies to modulate macrophage activity include polarizing macrophages toward an M2 phenotype using cytokine therapies like IL-4 and IL-13 [71] and blocking macrophage recruitment through chemokine signaling inhibition, such as targeting the CCL2-CCR2 axis [74]. Emerging approaches focusing on macrophage metabolic reprogramming also hold promise, particularly by enhancing mitochondrial function to support a regenerative environment [75, 78]. Furthermore, targeting TGF-β signaling has demonstrated efficacy in mitigating fibrosis, emphasizing the potential for therapies addressing both inflammatory and fibrotic components of these diseases [80–82].
A comprehensive therapeutic strategy incorporating macrophage modulation alongside current treatments, such as corticosteroids or exon-skipping therapies, could significantly improve patient outcomes. By reducing inflammation, limiting fibrosis, and promoting muscle regeneration, macrophage-focused interventions have the potential to address the multifactorial challenges of dystrophinopathy and dysferlinopathy.
As our understanding of macrophage biology and its interactions with the dystrophic microenvironment deepens, the development of innovative and targeted therapies continues to offer hope for improving the quality of life for individuals affected by these debilitating conditions. Future research should focus on translating preclinical findings into clinical applications while exploring the synergistic potential of combining macrophage-targeted therapies with existing treatment paradigms.
Abbreviations
| Arg1: | arginase-1 |
| DAMPs: | damage-associated molecular patterns |
| DGC: | dystrophin-associated glycoprotein complex |
| DMD: | Duchenne muscular dystrophy |
| ECM: | extracellular matrix |
| FAP: | fibro-adipogenic progenitor |
| HMGB1: | high-mobility group box 1 |
| HSCs: | hematopoietic stem cells |
| iNOS: | inducible nitric oxide synthase |
| LGMD: | limb-girdle muscular dystrophy |
| LGMD2B: | limb-girdle muscular dystrophy type 2B |
| MDs: | muscular dystrophies |
| MM: | Miyoshi myopathy |
| MMPs: | matrix metalloproteinases |
| NO: | nitric oxide |
| PRRs: | pattern recognition receptors |
| ROS: | reactive oxygen species |
| TGF-β: | transforming growth factor-beta |
| TIMPs: | tissue inhibitors of metalloproteinases |
| TLR4: | Toll-like receptor 4 |
| TSP1: | thrombospondin-1 |
Declarations
Acknowledgments
We thank Minseo Hong for creating and providing Figures 1 and 2, which have been confirmed by both of the authors.
Author contributions
JHK: Investigation, Writing—original draft. JHB: Conceptualization, Validation, Writing—review & editing, Supervision. Both authors read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Research Foundation of Korea (NRF) through the Ministry of Education [2021R1I1A3059820] (to Jea-Hyun Baek). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2025.
Publisher’s note
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.