
Abstract
The coronavirus disease 2019 (COVID-19) results from the infection of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) and primarily affects the respiratory tissue. Since first reported from Wuhan, China in December 2019, the virus has resulted in an unprecedented pandemic. Vaccination against SARS-CoV-2 can control the further spread of the ongoing pandemic by making people immunised to SARS-CoV-2. Several vaccines have been approved for use in clinics, a lot many are in different stages of development. Diligent interpretations from the preclinical evaluation are crucial to identify the most effective and safest vaccine candidates. Multiple vaccine candidates/variants have been tested in small animal models with relative ease and further in non-human primate models before being taken into clinical development. Here, we review the state-of-the-art strategies employed for a thorough preclinical evaluation of COVID-19 vaccine candidates. We summarise the methods in place to identify indicators which make the vaccine candidate effective in controlling SARS-CoV-2 infection and/or COVID-19 and are safe for administration as inferred by their (1) biophysical/functional attributes (antigen expression, organization, functionality, and stability); (2) immunogenicity in animal models and protective correlates [SARS-CoV-2 specific binding/neutralising immunoglobulin titer, B/T-cell profiling, balanced T-helper type-1 (Th1) or type-2 (Th2) response (Th1:Th2), and anamnestic response]; (3) protective correlates as interpreted by controlled pathology of the respiratory tissue (pulmonary clinical and immunopathology); and finally, (4) strategies to monitor adverse effects of the vaccine candidates.
Keywords
Coronavirus disease 2019, severe acute respiratory syndrome coronavirus-2, World Health Organization, vaccines, antibody, virus, immune response, animal modelsIntroduction
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection in humans results in the coronavirus disease 2019 (COVID-19) first reported in Wuhan, China in December 2019 [1]. Since then, COVID-19 has spread to every geographical location, declared as a pandemic, and is the most challenging healthcare situation of recent times. This pandemic has affected approximately 200 million people and claiming close to 4.2 million lives at the time of writing this manuscript [2].
SARS-CoV-2 is an enveloped virus belonging to the genus Betacoronavirus of the family Coronaviridae with single-strand positive RNA genome encoding major structural proteins, spike (S), envelope (E), membrane (M), and nucleocapsid (NC) [3]. The S surface glycoprotein comprises of surface glycoprotein domain 1 (S1) and S2 domains; S1 via its C-terminal receptor binding domain (RBD) engages human angiotensin-converting enzyme 2 (hACE2) as a receptor while S2 allows SARS-CoV-2 fusion with the host cell membrane [4–7]. Blocking this interaction between the RBD and hACE2 can neutralise SARS-CoV-2 infectivity [4, 6]. Several academic and industrial research groups isolated RBD-specific SARS-CoV-2 neutralising antibodies (nAbs) which are effective blockers of RBD-hACE2 interaction and protected cells/animals from virus infection [8–15]. Therefore, an effective vaccine capable of eliciting a high titer of RBD-specific SARS-CoV-2 nAbs can protect from COVID-19. As of 10th November 2021, 130 vaccine candidates are in various phases of preclinical or clinical development, and 194 more are under preclinical studies [16]. In total, 24 vaccines are granted emergency use approval in clinics by various national and international drug regulatory agencies [17].
Although several novel vaccine formats for COVID-19 are under development, standard evaluation strategies and extensive clinical understanding make a few suitable for quick deployment [18–21]. For affordable mass vaccination, inactivated vaccines formulated from SARS-CoV-2 strains would be ideal for triggering the host immune system upon administration without establishing an active infection. Inactivated vaccines primarily raise humoral immune response, crucial for recovery from COVID-19 [18, 22–25]. Next, the SARS-CoV-2 subunit vaccines employing S or RBD protein antigen/immunogenic fragment in recombinant form can drive a protective immune response upon administration [18, 22, 26–28]. Subunit vaccines modified as virus-like particle to display S trimer or RBD on nanoparticles in a conformation/configuration mimicking that of the native virus are also under development [18, 22, 29–32]. Besides, viral-vector vaccines, where the genetic information of the SARS-CoV-2 S glycoprotein is delivered as a part of the adenoviral vector genome by live virus administration are available [18, 22, 33–38]. These viral vectors have reduced virulence but still able to infect host cells. Also, nucleic acid vaccines are available, encoding S glycoprotein or RBD antigen either as DNA or modified mRNA protected under a liposomal nanoparticle shell for administration [18, 22, 39–45]. The genetic material expresses S glycoprotein upon delivery into the cells, secreted/inserted into the host cell membrane or/and displayed on major histocompatibility complex class I or II (MHC-I/II) molecules to be recognized by immune elements to elicit potent immune responses (Figure 1).
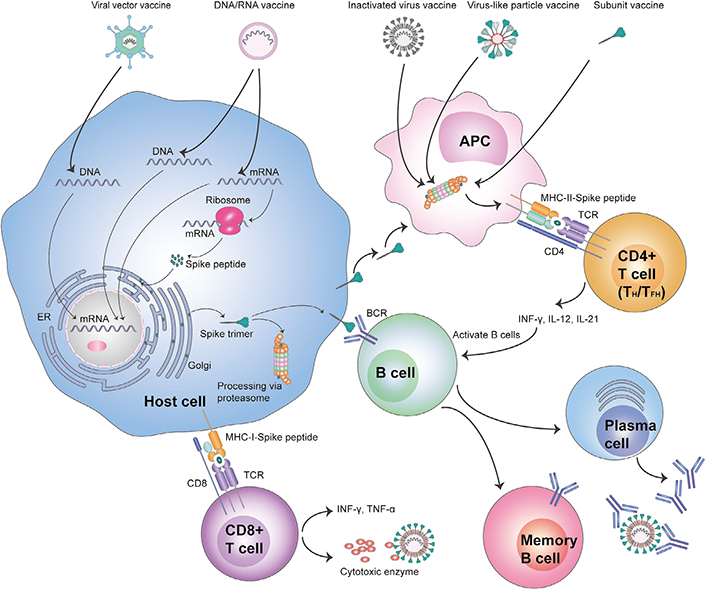
Diagrammatic view of COVID-19 vaccine recognition by host cells and activation of innate and adaptive immunity. ER: endoplasmic reticulum; APC: antigen presenting cell; TCR: T-cell receptor; INF-γ: interferon gamma; IL-12: interleukin 12; BCR: B-cell receptor; TNF-α: tumour necrosis factor alpha
Based on the clinical investigations on SARS-CoV-2 convalescent sera, S glycoprotein seems to be the primary antigenic determinant and the target of binding/nAbs that arise due to a natural infection [46–48]. Besides, interaction studies between SARS-CoV-2 S glycoprotein and hACE2 established the role of RBD in virus docking and S2 in virus fusion/entry into the host cell [4–7]. Interestingly, reported mutations among circulating SARS-CoV-2 genomes encompass S glycoprotein which implicate their role in either an enhanced binding to hACE2 or imparting stability under extreme conditions for establishing a successful infection or an immune escape phenotype [49–52]. These studies suggested that vaccine preparations capable of eliciting antibodies specific for RBD would be effective.
Evaluation of vaccine candidates and other therapeutic modalities needs well-established SARS-CoV-2 animal models. Animals like hamsters, ferrets, mice, rats, guinea pigs, rabbits, chicken/ducks, fruit bats, mink, cats, dogs, pigs, and non-human primates (NHPs) are not efficiently infected by SARS-CoV-2 and cause only mild to moderate COVID-19 [53–64]. However, they do present traits relating to (1) virus replication; (2) clinical signs; (3) pneumonia; (4) transmission; (5) immune activation; and (5) age/gender-related disease [53–64]. Another difficulty is that not all animal models show each of these traits after SARS-CoV-2 infection, making it challenging to select a particular model for testing SARS-CoV-2 intervention strategies [53–64]. Nonetheless, mice and NHPs show human-like virus infection/replication in the upper/lower respiratory tract and other organs, signs of pneumonia, immune activation, and age-related effects [55]. Therefore, mice and NHP are the models of choice for preclinical evaluation of vaccine efficacy and safety [23–45, 65, 66].
COVID-19 vaccine evaluation data has established RBD to be the main target of nAbs generated by natural infection/vaccination and their COVID-19 moderating attribute as observed in mice, NHP models, and humans [19–41, 49, 50]. COVID-19 vaccine candidates referred in this review are listed (Table 1). In the light of existing evidence, RBD specific nAbs titers, their efficacy in neutralising live SARS-CoV-2, a balanced T-helper type-1 (Th1) or type-2 (Th2) response (Th1:Th2) cellular response, and Fc-mediated immune enhancement could be the possible measurable correlates of protection (CoP) for COVID-19 vaccine candidates [14–17, 19–41, 49, 50]. In this article, we review the methodological strategies for evaluating COVID-19 vaccine candidates in mice and NHP models of SARS-CoV-2 infection encompassing biological/physical properties, immunogenicity, and indicators of pulmonary pathology with emphasis on the possible CoP from infection/disease (Figure 2).
Referred COVID-19 vaccine candidates
| Candidate/Animal model/Reference | Format | Organization | Immunization route/Dose/Schedule/SARS-CoV-2 challenge | S or RBD titer/nAb titer/T-cell profile | Protection in upper/lower respiratory tract | ADE effect |
|---|---|---|---|---|---|---|
| BBIBP-CorV/Macaca fascicularis/ [23] | Inactivated SARS-CoV-2 strain 19nCoV-CDC-Tan-HB02 | Beijing Institute of Biological Products, China. | IM/8 μg/(D0/ D14)/1 × 106/mL TCID50 (D24; IT) | NA/log2 ~1:256 (D21)/NA | CP/CP | None |
| BBV152/M. mulatta/ [24] | Inactivated SARS-CoV-2 strain NIV-2020-770 | ICMR-Bharat Biotech, India. | IM/3 μg/(D0/D14)/1 × 106.5/mL TCID50 (D35; IT + IN) | ~1:1,600 (D35)/1:209–1:5,217 (D35)/Th1 | CP/CP | None |
| PiCoVacc/M. mulatta/ [25] | Inactivated SARS-CoV-2 strain CN2 | Sinovac Biotech, China. | IM/6 μg/(D0/D7/D14)/1 × 106 TCID50 (D22; IT) | ~1:12,800 (D21)/~1:50 (D21)/Th1:Th2 | CP/> 95% | None |
| NVX-CoV2373/BALB/c mice transduced with 2.5 × 108 PFUs Ad/CMVhACE2 (D52; IN)/[26] | Subunit | Novavax, USA. | IM/1 μg/(D0/D14)/1 × 105 PFUs (D56; IN) | log10 ~5 (D28)/CPE100 log10 ~4 (D28)/Th1 | NA/CP | None |
| RBD Vaccine/M. mulatta/[27] | Subunit | Sichuan University, China. | IM/40 μg/(D0/D7)/5 × 105 PFUs (D28; IN) | 1:400–1:6,400 (D14)/EC50 1:200 102 (D14)/ Th1:Th2 | CP/CP | None |
| S-Trimer/M. mulatta/[28] | Subunit | Clover Biopharmaceuticals, China. | IM/30 μg/(D0/D21)/2.6 × 106 TCID50 (D35; IN + IT) | EC50 ~1:10,849 (D40)/ EC50 ~1:35,040 (D40)/Th1:Th2 | NC/CP | None |
| S-Fer/BALB/c mice/[29] | Subunit/Nanoparticle | Stanford University, USA. | SC/10 μg/(D0/D21) | EC50 ~1:80,000 (D21)/ EC50 ~1:30,000 (D21)/Th2 | NA/NA | None |
| RBD-I53-50/BALB/c & Darwin mice (Kymab)/[30] | Subunit/Nanoparticle | University of Washington, Seattle, USA. | IM/5 μg/(D0/D21)/1 × 105 PFUs (W9; IN) | EC50 ~4–8 × 106 (W3)/EC50 1:300–1:6,000 (W3)/NA | CP/CP | NA |
| S-I53-50NP/M. fascicularis/[31] | Subunit/Nanoparticle | University of Amsterdam, the Netherlands. | IM/50 μg/(W0/W4/W10) 1 × 106 PFUs (W12; IN + IT) | ~1:2,190 (W12)/~1:3,942 (W12)/NA | SP/SP | None |
| RBD-Ferritin/BALB/c & human ACE2 TG mice/[32] | Subunit/Nanoparticle | Sun Yat-sen University, China. | SC/10 μg/(D0/D21)/4 × 104 PFUs (D35; IN) | log10 ~5 (D35)/log10 ~4 (D35)/Th1 | NA/CP | None |
| ChAd-SARS-CoV-2-S/M. mulatta/[33] | Vector [Simian (Chimpanzee) Adenovirus (ChAd)] | University of Washington, St Louis, USA. | IM/2.5 × 1010 particles/D0/1 × 106 TCID50 (D28; IN + IB) | log2 ~2-fold (D21); log2 ~1.5-fold (D21)/Th1 | CP/SP | None |
| MVA/S/M. mulatta/[35] | Vector [Modified Vaccinia Ankara Virus (MVA)] | Emory University, USA. | IM/1 × 108 PFUs/ (W0/W4)/5 × 104 PFUs (W8; IN + IT) | ~1:24,000 (W8)/~1:177 (W6)/Th1 | NC/SP | None |
| ChAdOX1nCoV19/M. mulatta/[37] | Vector [Simian (Chimpanzee) Adenovirus (ChAd)] | The University of Oxford, Vaccitech, & AstraZeneca, UK. | IM/2.5 × 1010 particles/(D0/D28) 2.6 × 106 TCID50 (D28; IN + IT + Or + Oc) | 1:400–1:19,200 (D28)/1:10–1:160 (D56)/Th1:Th2 | NC/CP | None |
| Ad26-S.PP/M. mulatta/[38] | Vector [Adenovirus serotype 26 (Ad26)] | Harvard Medical School, USA & Janssen Vaccines and Prevention BV, the Netherlands. | IM/1 × 1011 particles/W0/1 × 105 TCID50 (W6; IN + IT) | ~1:5,000 (W4)/~1:113 (W4)/Th1 | CP/CP | NA |
| BNT162b/M. mulatta/[39] | Nucleic acid (mRNA) | BioNTech, Germany & Pfizer, USA. | IM/100 μg/(D0/D21)/ 1.05 × 106 PFUs (D41-45; IN + IT) | ~26,170 (U)·mL−1 (D28)/~1:283 (D56)/Th1 | CP/CP | None |
| LION/repRNA-CoV2S/M. nemestrina/[41] | Nucleic acid (Alphavirus replicon-based RNA) | University of Washington, Seattle, USA. | IM/50 μg/(D0/D28) | ~27.5 μg·mL−1 (D42)/~1:176 (D42)/Th1 | NA | None |
| mRNA1273/M. mulatta/[43] | Nucleic acid (mRNA) | Moderna, USA. | IM/100 μg/(W0/W4)/ 1 × 106 TCID50 (W8; IN + IT) | ~1:36,815 (W6)/~1:1,862 (W6)/Th1 | CP/CP | None |
| mRNA-RBD/BALB/c & human ACE2 TG mice/ [45] | Nucleic acid (mRNA) | Institute of Microbiology, CAS, China. | IM/15 μg/(W0)/ 1 × 105 FFUs (W6; IN) | ~1:100,000 (W4)/~1:920 (W4)/Th1 | CP/CP | None |
| S.dTM.PP/M. mulatta/[42] | Nucleic acid (DNA) | Harvard Medical School, USA & Janssen Vaccines and Prevention BV, the Netherlands. | IM/5 mg/(W0/W3)/1.1 × 104 PFUs (W6; IN + IT) | NA/NA/Th1 | SP/SP | None |
ADE: antibody-dependent enhancement; IM: intramuscular; SC: subcutaneous; IT: intratracheal; IN: inhalation; Or: oral; Oc: ocular; IB: intrabronchial; D: day; W: week; TCID50: half-maximum tissue culture inhibitory dose; PFUs: plaque forming units; FFUs: focus-forming units; EC50: half-maximum inhibitory concentration; CPE100: 100% cytopathic effect; TG: transgenic; CP: complete protection; SP: significant protection; NC: no correlation; NA: not available; mL−1: per milliliter
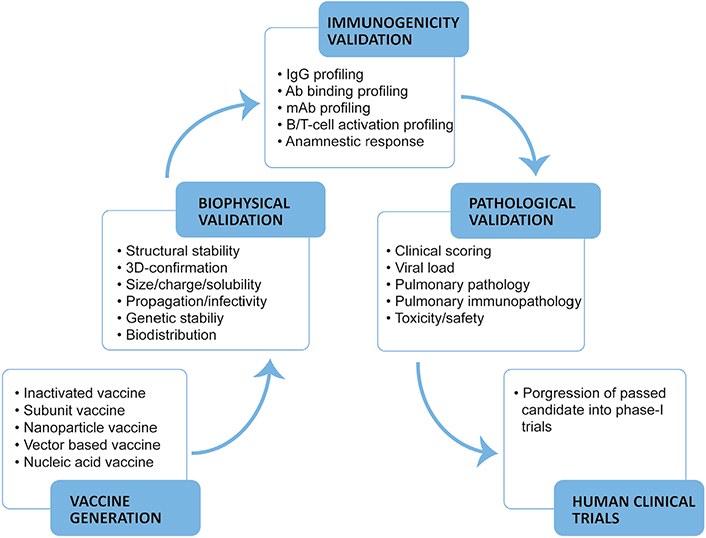
Evaluation COVID-19 vaccine constructs
COVID-19 vaccines are either comprised of expressed S glycoprotein or its domains as antigenic determinants in varying formats. Early evaluation of vaccine candidates involves multiple biophysical and functional tests before immunogenicity profiling.
Biophysical evaluation
The S glycoprotein or its domains are resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to infer the correct size, purity, and stability of the antigenic determinants. Further, Western blot using S-specific antibodies isolated from convalescent samples and size-exclusion chromatography (SEC), or size-exclusion chromatography multi-angle light scattering (SEC-MALS) for absolute molar mass and size determination are employed to remove candidates with the propensity to aggregate. The samples for such analyses are subunit vaccines by themselves and/or antigen extracted/secreted from model cell lines like HEK293 transfected/transduced with the viral strain (for an inactivated vaccine), vector-based or nucleic acid vaccine. SEC in combination with gel-shift assays is also used to confirm the multimeric display of antigens (like S trimer) and self-assembly of the nanoparticle vaccines [23, 25–32, 35, 38, 39, 42, 45]. The monodispersity of the subunit/nanoparticle and/or nucleic acid vaccines correlates with vaccine stability as determined by dynamic light scattering (DLS) and SEC-MALS to differentiate between a monomer, dimer, and oligomer [26, 29, 30, 41, 45]. In addition, nucleic acid vaccines are checked for their length, purity, and integrity of DNA/mRNA fragments before lipid encapsulation by microfluidic capillary or agarose gel electrophoresis, thus ensuring a contaminant-free (including protein, genomic DNA, nucleases, and enzymatic inhibitors) nucleic acid sample [39, 43–45]. Besides, the efficiency of encapsulating mRNA in lipid nanoparticles is a measure of mRNA vaccine stability usually interpreted in terms of particle size using DLS or ribogreen-based fluorescence assay [43–45]. Further, zeta potential measurements of mRNA vaccines give insight into the effect of varying pH on the vaccine stability or/and endosomal release of mRNA [45] and confocal imaging to study vaccine localization in endoplasmic reticulum/Golgi [39].
Functional evaluation
For a stable and effective vaccine, the antigenic determinants must have a physiologically relevant configuration to drive an immune response. The conformation of S glycoprotein expressed/displayed by vaccine candidates is studied structurally to confirm the prefusion state of S glycoprotein using electron microscopy (EM) and/or negative staining cryo-EM [23, 25, 26, 29–32, 39, 45]. The conformational flexibility (like up/down state of RBD) and multimeric display (like S trimer) of S glycoprotein mimics that of the native virus. Further, the correctly folded S glycoprotein on/expressed by vaccine preparations should bind hACE2 with high-affinity like SARS-CoV-2, which is studied using surface plasmon resonance (SPR) or biolayer interferometry (BLI) [26, 27, 29, 30, 32, 35, 39]. Further, the vaccine expressed S glycoprotein or its domains with correct folding and conformation alike native virus, should bind to the SARS-CoV-2 nAbs. SARS-CoV-2 nAbs from convalescent serum or a panel of recombinant nAbs help to evaluate functionally active S glycoprotein expressed/displayed by vaccine candidates [23, 29–31, 39, 41]. Further, N-linked glycans on S glycoprotein have an important role proper folding of trimers and modulating interaction with host proteases and nAbs [4, 67–69]. Analysis of site-specific N- and O-linked glycosylation on expressed S glycoprotein and glycoform distribution for subunit vaccines is analysed using glycoproteomics approach by recording mass spectra [27, 28, 30]. Another important aspect is the COVID-19 vaccine biodistribution upon administration which is a quantification of S glycoprotein in various organs and tissues of vaccinated animals [45]. Further, immune response against vector/nanoparticle can be detrimental to the vaccine stability upon administration. It must be characterised by test immunisations in a small group of animals [37].
Evaluation of inactivated vaccines
Central to an effective and broadly neutralising immune response from an inactivated whole virus vaccine is the shortlisting of most prevalent SARS-CoV-2 viral strain(s) in a phylogeny-based analysis using all available circulating genomes in Global Initiative on Sharing All Influenza Data (GISAID, https://www.gisaid.org/) [23–25]. The shortlisted viral strains are amplified in Vero cells for 7–10 passages to monitor the proliferation efficiency, high rate of infection, and genetic stability of the progeny for several generations by high throughput next-generation sequencing (NGS) methods [23–25]. This process ensures that no amino acid change should occur in the candidate strain compared to the parent and seed strain. Suitable candidate strain is assigned a clade using GISAID classification. The beta-propiolactone-mediated viral inactivation process is standardised. Complete elimination of infectivity and repeatability of the inactivation process for candidate strains is desirable [23–25]. Further, contaminating viruses or mycoplasma in the vaccine preparations are tested by NGS and/or infecting cells to isolate/characterise contaminating microorganisms [23–25].
Evaluating COVID-19 vaccine immunogenicity
Animal models for vaccine efficacy study
The WHO Research & Development Blueprint Team established a multi-location research consortium for COVID-19 modeling in animals, the initiative known as World Health Organization-COVID-19 Disease Modelling (WHO-COM) [70]. An overview of COVID-19 animal models established under WHO-COM initiative is available elsewhere [55]. Here, we only focus on mouse and NHP models useful for vaccine evaluation. Mice are the most sought-after animal models, which can be housed in a smaller space to make a relatively larger experimental group for meaningful statistical analysis of therapeutic efficacy. Although mice do not get efficiently infected by SARS-CoV2 due to a low-affinity interaction between S glycoprotein and mouse ACE2 (mACE2), they are the primary choice for COVID-19 vaccine evaluation [23, 25–32, 34, 35, 37, 39–41, 44, 45]. Mice susceptible to SARS-CoV-2 infection and mimicking severe COVID-19 could be more beneficial in such evaluation studies. Several approaches can make mice more susceptible to SARS-CoV-2 infection like (1) mutating SARS-CoV-2 RBD for enhanced binding to mACE2 [71]; (2) forced passaging of SARS-COV-2 in mouse lung tissue to allow enrichment of mutant virus capable of causing severe COVID-19 [72]; (3) infecting mouse with engineered adenovirus (Ad5-hACE2) capable of transducing hACE2 gene to sensitise for SARS-CoV-2 infection [54, 73]; (4) genetic modification of mouse to express hACE2 using tissue-specific promoter (K18-hACE2), universal promoter (CMV enhancer and chicken β-actin promoter), and endogenous mouse ACE2 promoter [73–75]; and (5) developing humanised mouse with hACE2 gene supporting SARS-CoV-2 replication in respiratory and brain tissue [76]. Although these strategies allow enhanced viral replication in mouse tissue, COVID-19 symptoms are still mild to moderate, similar to the wild-type animals.
Before ascertaining vaccine efficacy in humans, protective ability and adverse effects are evaluated in NHPs. The majority of COVID-19 vaccine candidates reviewed here have been evaluated in rhesus macaques (Macaca mulatta), cynomolgus macaques (Macaca fascicularis) and African green monkeys (Chlorocibus aethiops) [23–28, 31–33, 35–39, 41–43, 53, 56, 62, 64, 66]. During COVID-19 modeling studies, NHPs exposed to SARS-CoV-2 through either mucosal, intrabronchial, or aerosol exposure showed early radiographic abnormalities in lungs, high level of viral replication in both upper and lower respiratory tract for up to 2 weeks, virus shedding in respiratory and gastrointestinal tracts, and pathological features associated with viral pneumonia [53, 55, 56, 62, 64]. However, clinical disease in these models was still mild, and infection usually self-resolved within 11–14 days. Importantly, older macaques typically presents high viral load in the lungs, enhanced viral shedding, and more prominent radiological and histopathological changes [62]. Further, the SARS-CoV-2 challenge elicited natural protective immune responses, which offered robust protection against the virus rechallenge [53]. Alike mouse models, NHPs also do not recapitulate COVID-19 disease as in humans, although pathological manifestations have still allowed vaccine evaluation with the emergence of meaningful data [23, 24, 26–28, 31–33, 35–39, 42, 43, 66].
COVID-19 is usually a mild to moderate disease [77, 78]. However, lymphopenia and higher levels of the pro-inflammatory cytokine among older patients can cause severe disease. These conditions may lead to uncontrolled viral infection, severe pneumonia, acute respiratory distress syndrome (ARDS), septic shock, and multiple organ failure [77–79]. Further, clinical data show a high fatality rate among males (7.3%) compared to females (4.4%) due to COVID-19 [80, 81]. Pre-existing comorbidities like hypertension (16%), cardiovascular disease (12.11%), and diabetes (7.87%) among hospitalized COVID-19 patients can further predispose individuals to a greater risk of death from COVID-19 [82, 83]. Therefore, vaccine candidates must be evaluated in animal models closely mimicking the physiology linked to severe COVID-19 related fatality. Most vaccine candidates referred here were evaluated in normal mouse or NHP models, which may not be very useful in knowing vaccine effectiveness in the individuals with age-related dysfunctions or comorbidities.
COVID-19 vaccine-induced S1/RBD binding serum antibodies
An effective COVID-19 vaccine must trigger a robust serum antibody response against RBD and a complete seroconversion [23–45, 54, 66]. Therefore, all COVID-19 vaccine strategies strive to elicit high titers of RBD binding serum antibodies. The first evaluation strategy determines the immunogenic strength of COVID-19 vaccine candidates based on two prime indicators: (1) serum immunoglobulin G (IgG)/IgM/IgA titers (represented as arithmetic and/or geometric mean titers (GMTs) which are an estimate of S1/RBD-specific antibody class/IgG subclass/isotype in serially diluted serum samples [23–45, 54, 66] and (2) seroconversion rate implicating the percentage of vaccinated animals, which S1/RBD-specific serum antibodies [23–45]. The most prevalent method employed to determine antibody titers in serum samples is an enzyme-linked immunosorbent assay (ELISA) using IgG/IgM/IgA standards; serum antibody titers can also be determined by flow cytometry using anti-IgG/IgM/IgA beads. Among referred COVID-19 vaccine candidates, serum IgG titers were estimated generally every week after prime and boost vaccinations, and at an interval of two days post-virus infection. Both in mice (BALB/c, C57BL/6J, B6C3F1/J strains) and monkeys, serum IgG titers against S1/RBD and seroconversion rate depend on the vaccine dose and immunisation program (“prime only” and “booster”) without exception [23–45]. A booster dose in most cases further expands the serum IgG titers against S1/RBD and is responsible for complete seroconversion. During SARS-CoV-2 infection, the immune system also generates IgM response which peaks within three weeks of infection and protects against the virus in nasal tissue [84–87]. COVID-19 vaccines (RBD subunit, S-Fer nanoparticle, S-I53-50 NP, ChAdOx1nCoV19, and Ad26-S.PP) show elicitation of IgM antibodies recognising RBD in serum of BALB/c and CD1 mice and NHPs in second or third week after prime immunisation [27, 29, 31, 37, 38]. Besides, RBD-specific secretory IgA response generated by S-I53-50 NP, ChAd-SARS-CoV-2-S, and MVA/S vaccinations may be responsible for protecting the mucosal epithelia in respiratory, intestinal, and urogenital tracts against SARS-CoV-2 invasion [31, 33–35]. SARS-CoV-2 specific IgA remains neutralising in the saliva of infected subjects [88].
Most COVID-19 vaccine candidates referred to in this review were administered through the intramuscular (IM) route, which predominantly induces humoral immunity (IgG-directed) and limited mucosal immunity (IgA-directed) against the antigenic composition [89]. It can compromise the recognition/inhibition of SARS-CoV-2 in the nasopharyngeal tract, which is the initial site of virus contact. Therefore, IM administration might not provide sterilizing immunity against SARS-CoV-2 [33, 34, 89–91]. The potential of few vaccine candidates in generating mucosal immunity via intranasal (IN) administration in preclinical studies is known. Vaccine candidates like ChAdOx1 nCoV-19/AZD1222, ChAd-SARS-CoV-2-S, and lentiviral vector (LV) encoding full length, membrane anchored S [LV: :SFL] via IN administration enhances protection against SARS-CoV-2 in the upper respiratory tract [33, 34, 90, 91]. Besides eliciting systemic immunity, mucosal IgA response from IN-administered vaccines (ChAdOx1 nCoV-19/AZD1222 and ChAd-SARS-CoV-2-S) reduces virus shedding in the nasopharynx and can halt further spread [33, 34, 91]. Intranasal administration of booster dose can mediate a robust expansion of systemic and mucosal immunity in mice primed via the intraperitoneal (IP) route [90]. It may imply that additional IN administration in IM/IP vaccinated individuals can expand protective immunity or even provide sterilizing immunity. These studies were performed in hACE2 transduced mice, golden hamsters, and rhesus macaques [33, 34, 90, 91].
Previously, severe symptoms of vaccine-associated enhanced respiratory disease (VAERD) were present in measles and respiratory syncytial virus infected subjects vaccinated with a non-protective formulation [92]. Of some concern in the current scenario is that SARS-CoV vaccines like vaccinia virus-based Lc16mOrVV-N either alone or in combination with Lc16mOrVV-S, modified Vaccinia virus-based ADS-MVA, and double-inactivated vaccine (DIV) induce VAERD-like pathology (Th2 dominated inflammatory infiltrates and increased numbers of eosinophils) in animal models [93–95]. Therefore, the second evaluation strategy focuses on the physiological nature of the COVID-19 vaccine-induced serum IgG response concerning possible VAERD-like pathology. These involve analysing the (1) serum IgG isotypes; (2) Th cell subset; (3) eosinophil counts; (4) proinflammatory cytokines; or (5) anti-inflammatory cytokines after vaccination [92]. Non-neutralizing fraction of serum IgG can lead to ADE [92]. Serum IgG isotyping in COVID-19 vaccinated animals suggests a VAERD free immune response characterized by a balanced Th1/Th2 or slightly Th1 biased responses even with higher dose as inferred by a higher ratio of RBD binding serum IgG2a to IgG1 [41, 43, 96].
COVID-19 vaccine-induced nAbs in serum
Elicitation of nAbs upon SARS-CoV-2 exposure can protect from reinfection. SARS-CoV-2 nAbs preferentially recognise the epitopes within S1/RBD, [4, 9, 11, 23–35, 37–46, 48, 54, 66] while antibodies binding to the S2 domain and/or NC are non-protective [25, 27]. The nAb response against SARS-CoV-2 is an outcome of multiple clinical/immunological components which must be carefully evaluated. These include (1) optimisation of the vaccine dose since nAb titers are known to increase with a higher vaccine dose [23, 25, 27, 28, 39, 41, 43–45]. However, higher dose of BBV152 resulted in weaker neutralising response which was capable of resolving the lung pathology completely at the experimental endpoint [24]. It is difficult to interpret outcome as both low and high doses were formulated using the same adjuvant. Lower dose induced stronger IL-8 response which seems to be responsible for immune cell recruitment and vaccine efficacy [24]. However no such dose effect on neutralising antibody titers was evident in BBIBP-CorV inactivated vaccine [23]; (2) administration of booster dose as it has been widely proven that booster dose increases nAb titers and render a clear therapeutic benefit with complete seroconversion [23–35, 37–45]. In a particular case, a third vaccine dose resulted in maximal humoral response in animals [23]; (3) dosing interval as increasing the gap between a primary and booster dose can also enhance nAb titers in animals [25]; (4) presence of RBD binding antibodies since a strong correlation exists between the titers of anti-RBD binders and serum neutralising activity in comparison to S2 or N-specific antibodies, affirming the role of RBD in humoral immune response [23, 25]; (5) a higher ratio of “neutralising” to “binding only” RBD serum antibodies which lowers the risk of ADE upon vaccination, which has been previously reported in related infections caused by other members of the Coronaviridae family [13, 45, 97, 98]; and (6) immune effector responses which is the ability of nAbs to activate effector responses via FcγR2A-3, FcγR2A-1, IgM, and complement engagement to further enhance anti-SARS-CoV-2 immunity [31, 35, 38, 42]. The neutralising activity increases via activation of several effectors functions including antibody-dependent complement deposition (ADCD), antibody-dependent cellular phagocytosis (ADCP), antibody-dependent neutrophil phagocytosis (ADNP), and antibody-dependent natural killer cell activation (ADNKA) [31, 35, 38, 42, 99]. Therefore, all these parameters are crucial in finding the most effective vaccines.
The protective efficacy of nAbs is validated on (1) SARS-CoV-2 pseudo-typed virus (PSV) [27–32, 35, 38, 40, 42, 44, 45] or/and (2) live wild type (WT)/engineered SARS-CoV-2 isolates [23–27, 30–35, 37–39, 41–45, 54] produced/amplified in HEK293T and Vero-E6/CCL-81 cells respectively.
SARS-CoV-2 PSV neutralisation assay
PSVs are advantageous for the preliminary screening of nAb response if a BSL-3 laboratory is not available. Multiple systems are available to produce SARS-CoV-2 PSVs and are used to estimate neutralising strength of the COVID-19 vaccines (1) lentivirus-based system, with plasmids expressing the luciferase reporter or green fluorescent protein (GFP), the packaging machinery, and SARS-CoV-2 S gene [27, 29, 31, 32, 35, 38, 40, 42–45]; (2) murine leukemia virus (MLV)-based system comprising of the luciferase reporter in MLV transfer vector, MLV Gag-Pol packaging plasmid, and SARS-COV-2 S gene coding plasmid [30, 41]; and (3) recombinant vesicular stomatitis virus (VSVΔG)-based PSV with VSVΔG firefly luciferase plasmid and SARS-CoV-2 S gene coding plasmid [27, 28, 39, 44]. The titers of nAbs are determined by infecting hACE2 expressing cells with/without transmembrane serine protease 2 (TMPRSS-2, host cell protease) or Vero-E6/CCL81 monolayers with amplified PSV (infectious particles/mL) and measuring the reduction in pseudoviral infection of cells as relative light units (RLUs) [4, 28, 29, 31, 32, 35, 38, 40–45] or by fluorescence microscopy [27, 39].
Live SARS-CoV-2 neutralisation assay
Live SARS-CoV-2 clinical isolates or engineered live SARS-CoV-2 (possess luciferase/nanoluciferase or GFP gene inserts) have been widely used to determine neutralization potential of vaccinated serum samples. The titers of nAbs are determined by infecting hACE2 expressing cells with/without TMPRSS-2 (host cell protease) or Vero-E6/CCL81 monolayers at a predetermined median half-maximum tissue culture inhibitory dose (TCID50) mixed with dilutions of sera from vaccinated animals. The methods in use to determine the reduction in live viral infection of cells are (1) agar or carboxymethylcellulose (CMC) overlay method to quantify plaques by plaque reduction neutralisation test (PRNT) [24, 41, 43–45] or staining of fixed plaques by anti-SARS-CoV-2 S or N-specific antibody or crystal violet dye to estimate the immunospots by focus reduction neutralisation test (FRNT) [32–35]; (2) monitoring the cytopathic effect using microscope to directly visualise cell death upon virus infection [23, 25–28, 36, 37]; and (3) RLUs or fluorescent viral foci measurements [50% viral neutralisation titer (VNT50)] when PSVs or engineered live SARS-CoV-2 (possessing luciferase/nanoluciferase or GFP gene inserts) [28–31, 34, 35, 38–42, 44, 45].
Neutralising ability/effectiveness of vaccine candidates must also be validated against rapidly emerging SARS-CoV-2 lineages, which may have increased affinity for ACE2, transmissibility, and can evade immune response [49, 52, 100, 101]. The great diffusion of newer SARS-CoV-2 variants of concern (VOC) suggests an evolutionary advantage due to the mutations, which could lead to an immune escape phenotype [102, 103]. Two widely studied VOC [first isolated from South African (B.1.135 beta variant—K417N/E484K/N501Y) and Brazil (P.1 gamma variant—K417T/E484K/N501Y)] can render SARS-CoV-2 breach humoral immunity [49, 52]. Screening against existing/emerging VOC can be performed using live VOC isolates in neutralisation assays widely adopted to evaluate neutralising strength of COVID-19 vaccines and monoclonal antibodies. However, accessing/handling live VOC can be challenging. In such cases, SARS-CoV-2 PSVs can be generated rapidly by incorporating desirable mutations in the S protein by site-directed mutagenesis. SARS-CoV-2 inoculum may be contaminated with (e.g., V367F in the S protein and G251V in ORF3a) [31] or still evolving into variants that must be characterised. Similarly, SARS-CoV-2 variants which may or may not be immune escape mutants can emerge in infected/ inoculated animals before or after vaccination [31]. RNA extracted from inoculum, nasopharyngeal swabs, and bronchoalveolar larvage (BAL) fluid of animals can be sequenced to typify emerging variants. In addition, convalescent monoclonal antibodies/plasma may give rise to immune escape mutants [104]. Ability of serum antibodies to restrict emergence of such mutant can be determined by incubating with replication-competent PSVs, challenging ACE2 expressing cells, and RNA sequencing. This approach led to the discovery of S protein mutations in replication-competent PSVs upon exposure to potent anti-SARS-CoV-2 monoclonal antibodies and plasma isolated from convalescent samples [104].
COVID-19 vaccine-induced cellular response
Cellular immune components resulting from COVID-19 vaccination can provide long-term immunity against SARS-CoV-2 infection. Upon viral exposure, lung epithelial cells, alveolar macrophages, and neutrophils activate innate immunity, which in turn triggers adaptive immunity against the virus via activation of B lymphocytes (B-cell), helper T-cells (CD4+), and cytotoxic T-cells (CD8+) [18–22, 25, 30, 32, 34, 38, 39]. COVID-19 vaccines are shown to generate an immune response which is Th1 biased with neutralising serum IgGs [18–22, 26, 35, 40, 41, 44], enhanced drainage of B-cells into the lymphoid compartments [39], activation of T follicular helper (Tfh) cells, long-lived plasma cells (LLPCs), memory B-cells (MBPs), germinal center (GC) maturation in secondary lymphoid tissues [26, 32, 34, 35, 39, 43, 66], and S-specific INF-γ+/IL-2+/TNF-α+ CD4+/CD8+ T-cells [23, 24, 26–28, 31–35, 37–45].
COVID-19 vaccine-induced B-cell profiling
The frequency of B-cell subsets in circulation and lymphoid compartments can be a measure of COVID-19 vaccine efficacy. Vaccinated animals may show fewer circulating S-specific B-cells than controls indicating draining into secondary lymphoid tissues [39]. Booster dose gives a quick rise to circulating S-specific IgG/IgA secreting plasmablasts which become undetectable within three weeks. However, S-specific GC B-cells and plasmablasts remain detectable for up to 12 weeks in draining lymph nodes [105]. A multi-week window of increasing B-cell persistence in vaccine draining lymph nodes (VDN) producing antibodies against vaccinated antigen is known [106]. Therefore, GC maturation may have a role in COVID-19 vaccine efficacy; a booster dose can induce an enhanced GC reaction and could further increase neutralising antibody titers [66]. Detailed studies on GC maturation for most vaccine candidates are lacking. However, S/RBD-specific GC B-cells are Fas+GL7+ which co-localises with CD21+/35hi FDCs (follicular dendritic cells), IgD+ naive B-cells, and infiltrations of CD3+ T-cells [32, 35, 66]. Besides, COVID-19 vaccination can impact the immune cell population, and therefore the frequency of plasma cells, LLPCs, class-switched IgG1+, IgG2a+, and GC B-cells in lymph node and spleen may be characterised [26, 31, 32, 34, 35, 39].
COVID-19 vaccine-induced T-cell profiling
COVID-19 vaccine-mediated T-cell stimulation generates robust and high affinity neutralising IgG response against SARS-CoV-2. Splenic orcirculating T-cells are challenged with a pool of overlapping peptides constituting SARS-CoV-2 S glycoprotein and/or NC to characterise the T-cell phenotype [25, 27, 28, 32–35, 37–45]. Stimulated T-cells can be biased for Th1 or Th2 phenotype as determined by either interferon-γ (IFN-γ) enzyme-linked immune absorbent spot (ELISPOT) assays or multiparameter intracellular cytokine staining flow cytometry [24, 25, 27, 28, 30–33, 35, 37–45]. The frequency of S-specific IFN-γ+, IL2+, TNF-α+ (Th1) or IL-4+, IL-5+, IL-13+ (Th2) CD4+/CD8+ T-cells gives further information on the nature of protective immunity against SARS-CoV-2 [26, 37, 39–45]. A direct correlation exists between COVID-19 vaccine efficacy and Th1-biased T-cell response against S glycoprotein [26, 27, 37, 39–44, 66]. T-cell profiling can also help to characterise the effect of immunosenescence and the dynamics of COVID-19 vaccine response in aged animals. Aged mice are known to have a lower frequency of IFN-γ+ T-cells and a delayed IgG response compared to younger animals [41]. In addition, T-cell profiling in draining lymph nodes can give insight into vaccine efficacy, for example, COVID-19 vaccines induce an increased frequency of CD8+ and specialized subset of CD4+ T-cell [Tfh cells characterised by inducible T cell costimulator (ICOS+; CD278), CXC chemokine receptor type 5 (CXCR5+), and programmed cell death-1 (PD-1+)] which are vital for GC maturation and promotes antibody response to viral antigen [26, 31, 32, 39, 41, 43].
Anamnestic immune response after SARS-CoV-2 challenge
The anamnestic response is a natural reaction by the host against infecting pathogen (SARS-CoV-2 in this case) involving the memory compartments such as circulating memory B/T-cells capable of protecting from infection and/or disease [53, 54, 100, 102]. Such anamnestic responses can lead to low titers of binding/nAbs and CD4+/CD8+ T cells specific to S1, S2, NC, and non-structural (NS6, NS7a, and NS8) proteins of SARS-CoV2 within a short period of virus challenge [35, 38, 43]. Anamnestic responses can occur in sham control/unvaccinated and a minority of vaccinated animals after the live SARS-CoV-2 challenge [35, 38, 43]. Vaccinated animals (e.g., with S-I53-50NP, MVA/S, Ad26-S.PP, and mRNA1273) show no anamnestic expansion of nAbs and only low titers of S2 and NC specific CD4+/CD8+ T-cells for a shorter duration post-challenge compared to control animals [35, 38, 43]. Lack of NC-specific CD8+ T-cells expansion and nAbs in vaccinated animals suggests the nonavailability of NC protein produced by actively replicating virus [35]. A much lower viral replication in vaccinated animals after SARS-CoV-2 challenge points towards a protective vaccine-induced immune response. A low level of anamnestic response in vaccinated animals after the SARS-CoV-2 challenge indicates reduced viral replication [31, 35, 38, 43]. Weak anamnestic response after AD26-S.PP vaccination correlated with the immunophenotype of BAL cells indicating vaccine effectiveness [38]. An inefficient or even non sterilising vaccine candidate can also give rise to anamnestic response, and vaccine non-responders may also mount such a response [35, 38, 42, 43].
Therefore, anamnestic responses must be differentiated from a vaccine-induced immune response by accurately profiling the precise components of the SARS-CoV-2 immune response. Such analysis incorporates the determination of neutralising antibody titers against S1, S2, S, and NC protein in the serum from control and vaccinated animals before and after the SARS-CoV-2 challenge. Further, the neutralising antibody titers must be evaluated at frequent intervals after the virus challenge to understand the kinetic of anamnestic antibody response [31, 43]. For example, antibody levels in mRNA1273 vaccinated animals after the SARS-CoV-2 challenge remained stable while control animals showed increase within two weeks after the challenge [43]. Similarly, in S-I53-50NP vaccinated animals, nAbs continued to wane following SARS-CoV-2 challenge suggesting vaccine-induced immunity compared to controls [31]. Anamnestic expansion of CD8+/CD4+ T-cells recognising S1, S2, S, and NC in the spleen, lymph nodes, and BAL fluid of vaccinated and control animals after SARS-CoV-2 challenge must be accurately determined [31, 35, 38, 43]. As mentioned, anamnestic antibody response can correlate with the cellular subsets in BAL fluid; such correlations may help in unravelling the immune protective mechanism of vaccine candidates [38]. Further, a correlation between anamnestic CD8+/CD4+ T cells response and viral subgenomic RNA (sgRNA) may suggest effective vaccine candidates [35].
Evaluating disease protection in animal models
COVID-19 vaccine-induced protection/pulmonary pathology
A less severe lung pathology is an accepted indicator of vaccine efficacy. Most COVID-19 vaccines result in lower pulmonary pathological score in mice and NHP models of SARS-CoV-2 challenge. Pulmonary pathological score is the cumulative outcome of (1) thoracic/chest radiograph with evidence of pneumonia; (2) the number of upper, middle, lower lobes of lung affected; (3) extent of tissue lesions by necroscopy; (4) extent of bronchointerstitial pneumonia; (5) type 2 pneumocyte hyperplasia; (6) peribronchiolar hyperplasia; (7) alveolar septal thickening; (8) alveolar macrophage infiltration; (9) haemorrhages; (10) pulmonary edema (increased lung: body weight ratio) and fibrin deposits; and (11) formation of perivascular cuffs due to infiltration of lymphocytes [23–28, 31–35, 37, 39, 43–45]. A lower lung pathology score correlates with disease protection [23–28, 31–35, 37, 42–45]. The protective outcome of COVID-19 vaccine candidates correlates with significantly lower viral load in lung tissue and/or BAL fluid as measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) using primers specific for SARS-CoV-2 genomic RNA (gRNA; indicative of inoculated virus) and sgRNA; indicative of replicating virus) transcripts [24, 27, 28, 31–35, 38, 39, 42, 43, 45]. Lower sgRNA transcripts indicate incapability/reduced capability of SARS-CoV-2 replication and protection from COVID-19. Protective immunity can also be assessed by quantifying sgRNA in nasal turbinates/oropharyngeal swabs [35–37, 39, 43, 44]. Further, lung histopathology score and pulmonary viral load both correlate with vaccine-induced protection from COVID-19 [24–28, 31–35, 42–45]. However, discrepancies also exist, for example, in a study involving Macaca mullata, vaccination completely protected animals from lower lung infection with no detectable SARS-CoV-2 transcripts in BAL fluid but similar lung histopathology as in control animals [39]. Besides, in a few studies, extrapulmonary tissues from the heart, spleen, brain, lymphoid organs, GI tract, testes, etc., are also used for histopathological analysis and viral load quantification in vaccinated and control animals [24, 32, 34, 35, 37].
COVID-19 vaccine-induced protection/pulmonary immunopathology
SARS-CoV-2 infection alters the balance of innate and adaptive immunity in pulmonary tissues leading to a compromised immune status in controls compared to vaccinated animals [35, 43]. The balance between the kinetics of innate and adaptive immune responses in pulmonary tissues of vaccinated animals correlate with the protection from COVID-19 as inferred by lower viral load [35, 43]. The immunopathology status of the pulmonary tissue is analysed by monitoring various parameters such as (1) increased frequency of immune cells in BAL fluid and in the GC of draining lymph nodes of vaccinated animals suggests unstable kinetics in control animals [35]; (2) increased cytokine/chemokine levels (transcriptomics analysis by singe-cell RNA seq of IFN-stimulated genes (ISGs) in lung macrophages of BAL fluid in controls compared to vaccinated animals indicates lesser infection [35, 107]; (3) immune cell infiltration markers, for example, analysis of population shift in macrophage receptor with collagenous structure (MARCO+) resident alveolar macrophages compared to MACRO− interstitial macrophages/infiltratory monocytes in vaccinated animals suggest reduced recruitment of immune cells in pulmonary tissues and correlates with protection/reduced viral load [35, 107]; (4) generation of local lymphoid tissues as vaccines (for example, MVA/S) can also induce the formation of local lymphoid tissue like inducible bronchus-associated lymphoid tissue (iBALT) [35]. Bronchus-associated lymphoid tissues (BALTs) like areas induced by natural infection or vaccination are in direct contact with mucosal epithelium and involved in lymphocyte priming and B/T-cell proliferation to respond to infection in lung and lower respiratory tract [98, 108]. These can help in the expansion of lung immunity following virus infection [35, 97, 98, 107, 109]. Extent of BALT induction may depend on antigenic composition of the vaccine (for example, full length S in MVA/S was more effective in BALT induction compared to S1-subunit in MVA/S1) [35]. Such iBALT formation is evident upon infection with respiratory viruses in the absence of peripheral lymphoid organs and protein cage nanoparticles instillation in lungs [98, 107, 109].
Evaluating COVID-19 vaccine-induced adverse effects
Serious adverse effects of vaccine candidates are unwarranted and can compromise the safety of humans in clinical trials. Strategies to identify adverse effects must be carefully designed considering the vaccine type, administration route, dosage, and appropriate animal models and should match the proposed use in humans. Such monitoring must be performed for a minimum of two weeks after vaccine administration to conclusively evaluate any adverse effects or safety issues before clinical progression [110].
Vaccine administration can cause local adverse effects like skin erythema, swelling, discharge, rash, ulceration, or local inflammatory reactions at the site of injection. In published preclinical studies, no serious adverse effects of COVID-19 vaccine administration were observed relating to appetite, water intake, urine/fecal output, pyrexia, attitude/activity, serum biochemical parameters, body temperatures, complete blood counts, etc., [23, 25, 41]. Besides, systemic adverse effects can affect the administration route, draining lymph nodes, systemic immune system, and organs exposed to the vaccine. Extensive serum biochemistry and serum cytokine levels are also determined to identify possibility of vaccine-associated cytokine storm [23–25, 27, 28, 37, 41].
Toxicology studies like dose-dependent acute toxicity are important and can be performed in Sprague-Dawley rats and cynomolgus monkeys. Toxicity analysis comprises monitoring (1) signs of health deterioration, death, or impending death; (2) change in weight/feeding state; (3) lymphocyte subgroup distribution; (4) cytokines, interferons, interleukins, c-reactive protein, and complement levels; (5) histopathology of internal organs by analysis by systemic anatomy of the euthanized animal. Further, to infer vaccine safety, the maximum tolerated dose in animals without toxicity must be compared with the proposed maximal dose in humans and safe limits must be determined [23]. Similarly, systemic anaphylaxis studies to find any severe allergic reaction due to the vaccine candidate can be evaluated using the guinea pig model. Animals in the experimental group are sensitised with a low and high dosage of vaccine compared to placebo and are compared to positive control group administered with human blood albumin before intravenous excitation by foot to determine primary and secondary stimulation scores [23].
Conclusions
The preclinical vaccine efficacy protocols are thoroughly laid out for COVID-19 vaccine candidates by referred preclinical vaccine studies. The main component of preclinical efficacy studies is immunogenicity analysis. Immunogenicity of the vaccine candidate is inferred by quantifying serum antibodies against SARS-CoV-2 S-RBD, potent neutralisation activity of serum antibodies against live or/and PSV SARS-CoV-2, and elicitation of cellular immunity. Immunogenicity evaluations are extensive in terms of dose standardization, use of adjuvants, and vaccination schedule. Few studies have also focussed on the nature of anamnestic responses on SARS-CoV-2 challenge in controls and vaccinated animals which contribute vital information on optimum vaccine efficacy related to evoking specific and protective immune responses against SARS-CoV-2. However, except for very few studies, adverse effect analysis of vaccine administration, systemic adverse effects, ADE, and immunopathological analysis of pulmonary tissue are lacking. Authors suggest that though immunogenicity forms the core of the vaccine efficacy, adverse effects validation should be an essential part of preclinical vaccine efficacy and safety against SARS-CoV-2. Furthermore, adopted preclinical evaluation strategies represent a standard set of assays and assessments which are currently state-of-the-art for COVID-19 vaccines. It would make comparison among vaccine candidates easier. Most vaccine candidates are administered via IM route and generates SARS-CoV-2 specific humoral, mucosal, and cellular response free from anamnestic expansion of immunity in animals. Vaccine-induced serum antibodies and cellular subsets are directed towards the receptor-binding domain (RBD) of SARS-CoV-2, which is the protective immune determinant. ELISA-based assays and fluorescence-activated cell sorting (FACS)-based immunophenotypic profiling of cellular components can accurately assess the immunogenic strength/immuneprotective mechanisms of the vaccine candidates. Although vaccines clear replicating virus from the lungs of the infected animals, it is argued, these animal models do not reflect the phenotype of severe COVID-19 disease. Mild disease in such animals makes it difficult to predict actual effectiveness in subjects at greater risk of COVID-19 related fatality (aged > 60 years, immune dysfunction, and comorbidities). However, immunisations resulted in a strong antiviral response against the infectious challenge with SARS-CoV-2. Clinical trials for several approved vaccine candidates (BBIBP-CorV, BBV152, PiCoVacc, NVX-CoV2373, ChAdOX1nCoV19, Ad26-S.PP, BNT162b, and mRNA1273) were conducted based on the referred preclinical studies suggesting the accuracy and specificity of the preclinical evaluation strategies described in this review. Collectively, these studies support the development of COVID-19 vaccine candidates for clinical trials.
Abbreviations
| ADE: | antibody-mediated disease enhancement |
| BAL: | bronchoalveolar larvage |
| BALTs: | bronchus-associated lymphoid tissues |
| COVID-19: | coronavirus disease 2019 |
| CP: | complete protection |
| EC50: | half-maximum inhibitory concentration |
| GC: | germinal center |
| GFP: | green fluorescent protein |
| hACE2: | human angiotensin-converting enzyme 2 |
| iBALT: | inducible bronchus-associated lymphoid tissue |
| IFN-γ: | interferon-γ |
| IgG: | immunoglobulin G |
| IM: | intramuscular |
| IN: | intranasal |
| IT: | intratracheal |
| M: | membrane |
| MLV: | murine leukemia virus |
| nAbs: | neutralising antibodies |
| NC: | nucleocapsid |
| NHPs: | non-human primates |
| NS: | non-structural |
| PFU: | plaque forming unit |
| PSV: | pseudo-typed virus |
| RBD: | receptor binding domain |
| S: | spike |
| S1: | surface glycoprotein domain 1 |
| SARS-CoV-2: | severe acute respiratory syndrome coronavirus-2 |
| SEC: | size-exclusion chromatography |
| sgRNA: | subgenomic RNA |
| SP: | significant protection |
| TCID50: | half-maximum tissue culture inhibitory dose |
| Th1: | T-helper type-1 |
| TNF-α: | tumour necrosis factor alpha |
| VAERD: | vaccine-associated enhanced respiratory disease |
| VOC: | variants of concern |
Declarations
Author contributions
AK and AS conceptualised the manuscript; DG and QJ organised literature for review; DG, AK, and AS wrote the first draft; BB prepared figures; BB, QJ, SP and GY wrote the parts of manuscript; AK and AS revised the final draft; GY is mentor and senior authors on the manuscript. All authors read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2021.