 Review
Review

Affiliation:
Pneumology and Pulmonary Cell Research, Departments of Biomedicine & Internal Medicine, University and University Hospital Basel, 4031 Basel, Switzerland
ORCID: https://orcid.org/0000-0002-2568-730X
Affiliation:
Pneumology and Pulmonary Cell Research, Departments of Biomedicine & Internal Medicine, University and University Hospital Basel, 4031 Basel, Switzerland
Email: michael.roth@usb.ch
ORCID: https://orcid.org/0000-0002-8139-2821
Explor Immunol. 2022;2:303–319 DOI: https://doi.org/10.37349/ei.2022.00052
Received: February 18, 2022 Accepted: April 05, 2022 Published: May 30, 2022
Academic Editor: Bernhard Ryffel, CNRS-University of Orleans, France
Lower respiratory tract infections caused over 4 million deaths per year worldwide, especially in low-income countries. Viral respiratory infections often occur as rapidly spreading seasonal endemic or epidemic, and sometimes due to new respiratory viruses including corona viruses. The first level of host defense against viral infection is based on the innate immune system and intracellular killing mechanisms. The latter is activated by the release of viral DNA or RNA into the cytosol of the infected cells during the initial phase of virus replication. Viral DNA and RNA are recognized by the cyclic guanosine monophosphate (cGMP)-adenosine monophosphate (AMP) synthase (cGAS)–stimulator of interferon (IFN) genes (STING) sensing pathway, leading to the activation of type-I and -III IFN synthesis, with the aim to limit viral replication. However, the efficacy of the cGAS-STING sensing mechanism seems to vary with different viruses, and therefore, so is the efficacy of the host defense mechanism. Viral DNA can be sensed by different proteins including DNA-dependent activator of IFN regulating factor (DAI), cGAS, and toll-like receptor-9 (TLR-9). Viral RNA is recognized by retinoid acid-inducible gene 1 (RIG-1), TLR-7 and TLR-8. The question if cGAS also recognizes viral RNA remains unclear. The activation of IFN synthesis by cGAS is initiated by the recognition of purines and pyrimidines and their enzymatic conversion into cGMP and cyclic AMP (cAMP), followed by the activation of STING. In addition, it is indicated that several viruses can evade the cGAS-STING signaling and escape the host defense. This review aims to summarize the role of cGAS-STING as a host defense mechanism against viral respiratory tract infections.
Viral infections of the respiratory tract frequently occur most often seasonally and are commonly recognized as cold or flu. In 2015, a United Nations summit adopted the aims of the Global Alliance Against Respiratory Diseases (GARD), which was initiated by the World Health Organization in 2006. One of the goals was to reduce the impact of non-communicable diseases until 2030, which includes chronic respiratory diseases [1]. In order to achieve this, it has to be considered that bacterial and viral infections often initiate and exacerbate chronic inflammatory lung diseases. Thus, without improving hygienic conditions, this goal is not achievable. Respiratory tract infections cause more deaths than human immunodeficiency virus (HIV), tuberculosis and malaria combined [1]. Especially in children younger than 5 years, infections with Respiratory Syncytial Virus (RSV) and influenza viruses are the most frequent cause of death [2]. Regarding lower respiratory tract infections, eight virus types have been identified including Adenovirus, Bocavirus, Coronavirus, Influenzavirus, Metapneumovirus, Parainfluenza, Rhinovirus, and RSV. In addition, other viruses including Cytomegalovirus, Herpesvirus, Measles virus, and others can affect the lung structure and function [2].
Viral infections are the major cause of exacerbation in chronic inflammatory lung diseases in adults, including asthma and chronic obstructive pulmonary disease (COPD). In children, the most frequent viral infections are RSV and rhinoviruses [3]. It is indicated that viral infections during childhood may predispose the lung to develop asthma or COPD later in life [4, 5]. There is also evidence that viral infections can be transmitted across the blood-placenta barrier from the mother to the embryo, leading to impaired lung development [6]. Later in life, viral infections can either worsen chronic inflammatory lung diseases or induced exacerbation [7–9]. In adult asthma patients, the effects of rhinovirus infection on disease staging and exacerbation may depend on disease-specific factors that yet to be identified [10].
Most viruses infect the respiratory tract through the epithelium, from which they spread to body fluids and sub-epithelial cells [11, 12]. In COPD, the epithelium response to rhinovirus infection is impaired and therefore the immune response through interferons (IFNs) is deregulated [13]. There is also evidence for the modified function of bronchioalveolar stem cells after infection by respiratory viruses, which impaired the capacity of the lung to repair and regenerate at any age [14]. Thus, it is important to understand how viral infections are defeated by the human host response and innate immunity, which includes the immune response against nucleic acids [15].
Innate immune response is the first step of protecting an organism against infection by microorganisms including viruses. The airway epithelium provides a physical barrier against inhaled pathogens and plays an important role in the activation of the innate and adaptive immune response. Bronchial epithelial cells express a variety of secreted and membranous proteins with direct anti-viral and anti-bacterial properties including mucins and β-defensins [16]. In addition, epithelial cells express trans-membrane and intracellular pattern recognition receptors (PRR), which sense bacterial and viral proteins and activate the innate immune response [17, 18]. These groups of PRR proteins include toll-like receptor-2 (TLR-2) and TLR-4, expressed as membranous proteins. Intracellular PRR are TLR-3, TLR-7, retinoic acid-inducible gene 1 (RIG-1)-like receptors, cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) synthase (cGAS) and IFN-γ inducible protein 16 [19]. However, the same epithelial cells express proteins that help viruses to adhere and infect [20].
In recent years, evidence is increasing that the initial step activating innate immunity response is the intracellular recognition of viral DNA or RNA by the host cells [21, 22]. In regards to viral infections of the respiratory system, it has also been shown that rhinovirus and severe acute respiratory syndrome (SARS) coronavirus 2 (SARS-CoV-2) can activate the host response through DNA or RNA sensing [23, 24]. Viral DNA and RNA are recognized through the cGAS, which activates the stimulator of IFN genes (STING) signaling [25]. It should be mentioned that the cGAS-STING signaling is not specific to infectious diseases; it can also be activated by tumors, senescence, autophagy, and autoimmune diseases [26, 27]. A summary of the cGAS-STING signaling is shown in Figure 1.
The sugar phosphate backbone of the double-stranded viral DNA is recognized by cGAS [28]. This interaction of cGAS with the viral DNA is independent of the DNA sequence, while retrovirus DNA is recognized by cGAS through a sequence-specific interaction [29]. In the cytosol, double-stranded DNA of any source activates cGAS. This converts ATP and guanosine tri-phosphate to form the second messenger cGAMP, which binds to STING, and induces its dimerization and translocation to perinuclear proteins [30]. Subsequently, STING phosphorylates tank binding kinase 1 (TBK1), which in turn activates the IFN regulatory factor (IRF) and nuclear factor κB (NFκB), leading to the synthesis of IFN type-I and -III [31, 32]. It had been described earlier that TLR is an independent inducer for the host defense mechanism against viral infections. However, a recent study on HIV-1 suggested that TLR-2 and TLR-4 also activate IFN production through cGAS-STING signaling [33]. An overview of respiratory viruses and their targets in the cGAS-STING signaling is provided in Table 1.
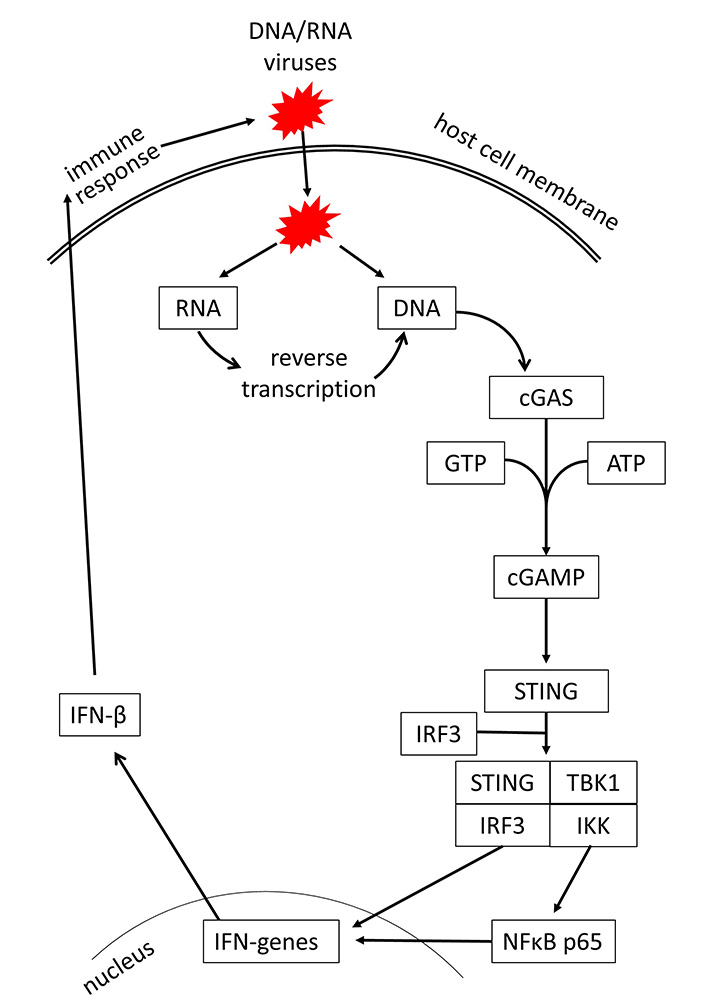
Mechanism of cGAS-STING signaling by DNA/RNA viruses and induction of IFN-mediated host defense. GTP: guanosine triphosphate; IKK: inhibitor of κB kinase
Most frequent viruses causing respiratory tract infections
| Virus | Sub-types | Effect on cGAS-STING and host defense |
|---|---|---|
| Adenovirus | 55 serotypes | Inhibited by viral proteins activated when bound to antibodies |
| Bocavirus | 1 serotype | No direct effect reported inhibited TNF-α, NFκB, IFN-β |
| Coronavirus | α (NL63, 229E)β (HKU1, MERS, SARS) | Cell type specific epithelium: inhibited STING, IRF3 T-/B-cells: activated cGAS via cGAMP |
| Influenzavirus | A (H1–3, N1–2), B, C | Activated cGAS, STING, IFN-β |
| Metapneumovirus | A, B | No report |
| Parainfluenzavirus | 4 serotypes | Activated STING |
| Rhinovirus | A, B, C (> 100 serotypes) | Activated cGAS, STING, IFN-β |
| RSV | A, B | Activated cAMP, IFN-β |
TNF-α: tumor necrosis factor-alpha; MERS: Middle East respiratory syndrome; cAMP: cyclic adenosine monophosphate
In regards to tumor-induced response, cGAMP can be secreted by tumor cells, entering into macrophages or dendritic cells and thereby activating an immune response [34]. To date, it is unknown if viral or bacterial infections are also able to stimulate the secretion of cGAMP and thereby induce an immune response of non-infected cells. Such a mechanism would be important during the very early phase of infections.
The capsid of Adenoviruses has a diameter of 70–90 nm and contains a double-stranded DNA, which is released into the host cell cytosol after the infection. Adenoviruses are the major cause of the common cold in humans [35]. In human cells, cGAS is sensing DNA released from Adenoviruses, which activates anti-viral type-I IFN synthesis and the activation of transcription factors signal transducer and activator of transcription 1 (STAT1) and STAT2 [36]. In murine macrophages and endothelial cells, knockdown of cGAS, STING, or TBK1 significantly reduced the IFN response to Adenovirus infection [36]. In mice, the loss of cGAS or STING genes significantly lowered the secretion of IFN-β and pro-inflammatory cytokines, indicating an impaired early anti-viral innate immune response. However, silencing cGAS-STING signaling had no effect on the long-term clearance of the virus [37]. In another study, it was indicated that Adenovirus specific protein E1A acts as an inhibitor of the cGAS-STING pathway, and would thereby reduce the host IFN response [38]. In young chickens, the inhibition of cGAS-STING signaling resulted in more potent infections with Fowl Adenovirus serotype IV [39]. In a variety of human cells including embryonic kidney cells, fibroblasts, and HeLa cells, Adenovirus induced cGAS-STING signaling was counteracted by the activation of oligoadenylate synthase-like (OASL) [40]. In COPD patients, infection with Adenovirus leads to exacerbation, which might be explained by the inhibition of STING and IFN-β signaling by cigarette smoke [41].
However, there are other DNA sensing proteins such as DNA-dependent protein kinase (DNA-PK), which initiate human host defense upon Adenovirus infection [42]. DNA-PK is activated by the viral E1A protein and leads to the phosphorylation of heat shock cognate 71kDa protein (Hsc70/HSPA8), which is a member of the heat shock protein (HSP) 70 family, and is known to control apoptosis and autophagy [43]. Adenovirus infection also suppresses the innate immune response and IFN synthesis by activating the ubiquitination of STING through the expression of MYSM1 (Myb-like SWIRM and MPN domains 1) [44]. There is also evidence that the immune response of a human to Adenoviruses requires the interaction of several anti-viral sensing mechanisms. In humans who have antibodies to recognize adenoviruses, the first response is the capture of the virus by the antibodies, which activates nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing-3 (NLRP3) and subsequently interleukin-1β (IL-1β). Antibody-captured Adenoviruses might still be entering the host cells and are then recognized by tripartite motif-containing protein 21 (TRIM21) and cGAS, leading to IFN synthesis [45], as summarized in Figure 2.
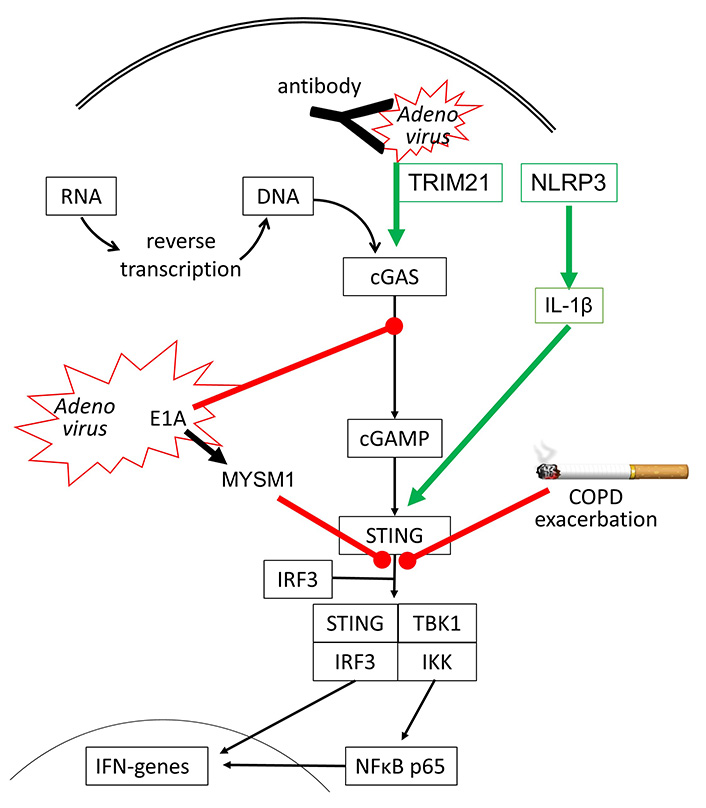
Inhibitory (red line) and stimulating (green arrow) effects of Adenovirus infection on cGAS-STING signaling. It has been reported that cigarette smoke can inhibit STING activation in COPD, thereby reducing IFN synthesis and leading to exacerbation
Bocaviruses have only recently been described as pathogenic for humans and were linked to upper respiratory tract infections and the common cold [46]. Bocavirus has been suggested to cause severe asthma exacerbation in children [47]. An increasing number of reports implies that bocaviruses are found worldwide, and can infect both children and adults [48, 49]. Unfortunately, Bocaviruses can re-infect adults and may cause neurological damage or death [50]. In addition, it has been shown that Bocavirus infection can become persistent at least in tonsillitis [51]. From China, there are reports that Bocavirus infection can be transmitted through rats [52]. Like SARS-CoV-2, high dose glucocorticoids have been successfully used to prevent the death of Bocavirus lung infections [53]. Bronchial epithelial cells are the first target cell type for Bocaviruses and allow the virus to replicate and spread [54, 55].
So far, there is no direct evidence that bocaviruses affect the cGAS-STING signaling, but indirectly data suggest its suppression. The viral load of Bocavirus correlated with the expression of STING but was not correlated with COPD or smoking [56]. In human cells, Bocavirus non-structural protein 1 (NP1) inhibited IFN-β synthesis by suppressing IRF3, which is also a part of the cGAS-STING signaling [57]. This may occur through inhibition of TNF-α and NFκB, which are both needed for IFN synthesis through cGAS-STING signaling [58], as shown in Figure 3.
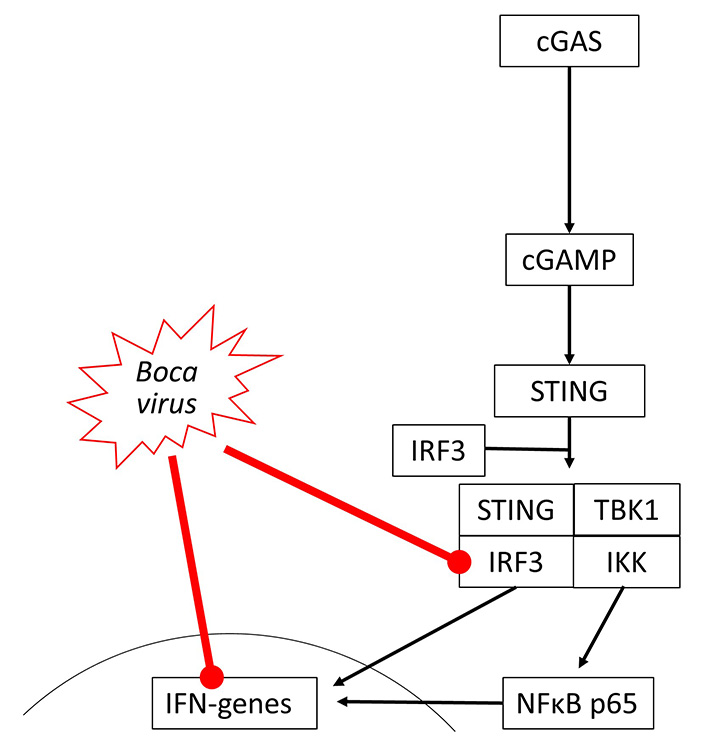
Bocavirus has been reported to inhibit IRF3 activity and reduce IFN gene expression in different studies, but there is no evidence of the direct link between these two events
Human Coronaviruses are single-stranded RNA viruses with a diameter of 60–220 nm and an RNA genome consisting of 15–20,000 nucleotides. All human Coronaviruses infect preferentially host cells carrying a protein that interacts with the Coronavirus spike protein [59], such as angiotensin converting enzyme 2 (ACE2) for SARS-CoV-2.
Despite the current SARS-CoV-2 pandemic, human Coronaviruses occurred seasonally causing a variety of lung diseases ranging from the common cold to pneumonia [60, 61]. The latter study suggested that the production of antibodies to human Coronaviruses declines with age [61]. Such an age-dependent decline of anti-coronavirus antibodies might in part explain the observed increased severity of SARS-CoV-2 infections in the elderly and be a feature of all Coronaviruses [62].
The effect of coronaviruses on host defense is incompletely understood. Transcriptome analysis of patients who died of acute respiratory distress syndrome during the first SARS-CoV outbreak, indicated that IFNs and IRF3 were upregulated, thus, suggesting the activation of cGAS-STING signaling [63]. In human kidney (HEK-293T) and epithelial (HeLa) cells, transfection with SARS-CoV-2 open reading frame 9b (ORF9b) suppressed type-I and -III IFN synthesis by interfering with IRF3, TBK1, RIG-1 and STING [64]. The interference with RIG-1 might suggest that SARS-CoV-2 modifies STING signaling by activating damage-associated molecular pattern (DAMP) regulated mechanisms. Similarly, SARS-CoV-2 ORF3a interfered with cGAS-STING signaling by disrupting the complex formed by STING and IKK, thereby inhibiting the activation of NFκB [65].
However, recent studies showed that STING activation blocked the infection of various human cell lines with coronaviruses including SARS-CoV-2 [66, 67]. Furthermore, SARS-CoV-2 infection has also been reported to activate cGAS-STING signaling by different mechanisms. Micronuclei formation induced by SARS-CoV-2 spike protein binding to ACE2 activated cGAS-STING signaling [68]. In human epithelial cells, knockout of different cGAS-STING components implied that the interaction of the SARS-CoV-2 spike protein with ACE2 host cells is sufficient to initiate the formation of cytoplasmic chromatin by cell fusion, thereby inducing the host defense [24]. This observation is in line with the induction of pro-inflammatory cytokines through cGAS-STING and NFκB activation in SARS-CoV-2 infected immortalized human epithelial cells [69]. SARS-CoV-2 infection stimulates immune defense by type-I IFN in lung-on-chip models and skin samples of coronavirus disease 2019 (COVID-19) patients [70]. It should be mentioned that extended type-I IFN in response to SARS-CoV-2 infection might explain the tissue damage reported in the lungs of COVID-19 patients [70]. Furthermore, SARS-CoV-2 binding to ACE2 over-activated STING signaling and resulted in symptoms characteristic of Kawasaki syndrome [71].
Interestingly, T- and B-cells respond to SARS-CoV-2 infection by the activation of the cGAS-STING signaling, but these cells do not carry ACE2, and would not be infected by the virus [72]. The authors suggested that the activation of lymphocytes might occur through the import of cGAMP released by other SARS-CoV-2 infected cells. Such a mechanism has been reported for macrophages activated by secreted cGAMP from tumor cells [34]. In this context, it is interesting to note that the addition of cGAMP to virus-like particles improved the anti-viral response to SARS-CoV-2 spike protein-triggered immune response, and was suggested as an additive to vaccination [73]. The interactions of coronavirus proteins on cGAS-STING signaling are summarized in Figure 4.
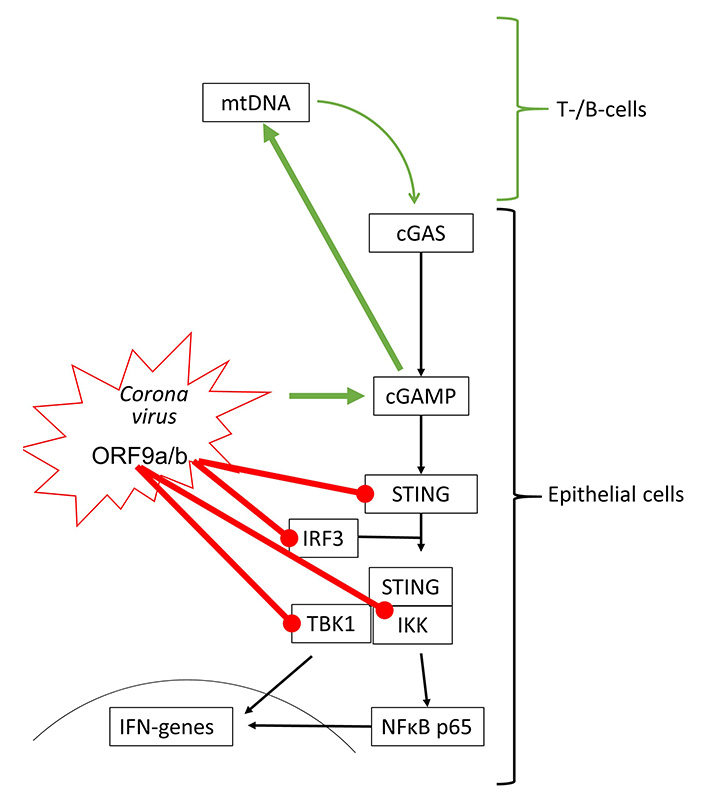
Coronaviruses can activate (green arrow) and inhibit (red line) cGAS-STING signaling. All inhibitory actions have been shown to involve direct interaction of ORF9 proteins with specific signaling proteins of the STING protein complex. mtDNA: mitochondrial DNA
Influenza viruses type-A and type-B are the most frequent cause of the common cold and belong to the RNA viruses. Their interaction with the cGAS-STING signaling has not been well investigated. In 2016, it was reported that the Influenza A virus targets cGAS-independent, but STING-controlled immune response, leading to type-I IFN synthesis [74]. STING was activated by two components of the Influenza A virus, namely the hemagglutinin-fusion-peptide and the enveloped virus. In a mouse model, infection by Influenza Virus M2 activated the cGAS-STING signaling indirectly by causing mtDNA release into the cytosol of the host cells. Therefore, one can argue that Influenzavirus may activate cGAS-STING signaling through DAMP receptors including TLR-2 to TLR-9 and RIG-1.
Moreover, the STING-dependent anti-viral effect was mediated to neighboring non-infected cells through gap junctions, suggesting a role of cGAMP [75]. In another mouse model, it was reported that fever/hyperthermia promoted influenza virus-induced cGAS signaling, leading to IFN-β dependent virus defense [76]. Viral defense to RNA viruses, such as Influenza viruses, seems to be independent of cGAS but involves the activation of STING. Such a mechanism has been shown for Influenza A virus activating STING by directly binding to one of its complex members synaptotagmin binding cytoplasmic RNA interacting protein (SYNCRIP) [77]. In human bronchial epithelial cells, the replication of Influenza A virus varied with the cell line used. Higher resistance to viral infection correlated with the expression of IRF-7, STING, and IFNs, which was controlled through Janus-activated kinase [78].
STING activation through synthetic agonists reduced the replication of human Parainfluenza Virus 3, but there is no direct evidence for activation of the cGAS-STING by this virus type [79]. The known effects of Influenza and Para Influenza viruses on cGAS-STING activation are shown in Figure 5.
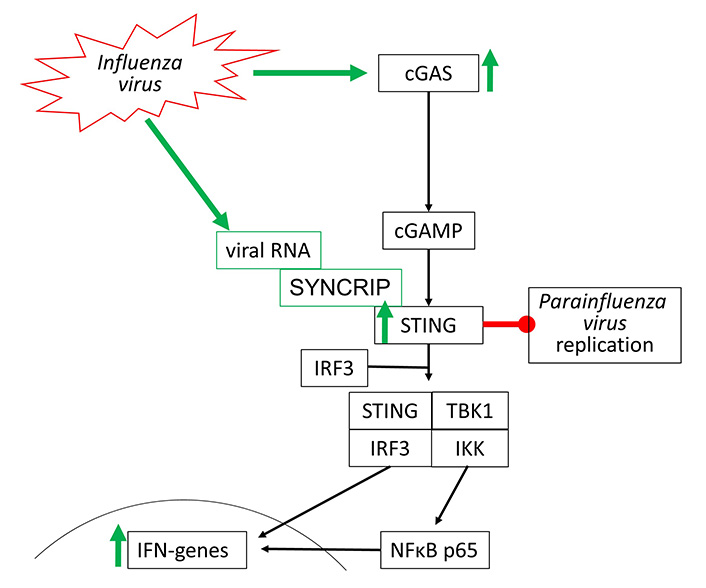
Influenza viruses activate cGAS-STING signaling through two different independent mechanisms. Parainfluenza virus replication can be directly inhibited by STING
The replication of human Rhinovirus 16 was inhibited by activating STING through a synthetic agonist [79]. Rhinovirus A and Rhinovirus C have been reported to activate STING signaling, which however, seems to be independent from cGAS, followed by the synthesis of IFN-β [80]. This observation confirms the earlier hypothesis that the activation of cGAS-STING signaling and type-I IFN suppresses Rhinovirus A replication [81]. The same mechanism seems to apply to the inhibition of human Rhinovirus C in the human airway epithelium [82].
Indirect evidence in asthma and COPD patients implies the impaired function of cGAS-STING signaling and therefore reduced anti-viral defense in both diseases [83–85]. In asthma, the modification of IRF3 and NFκB controlled the immune defense against rhinovirus infection [86]. As described above, IRF3 and NFκB are essential to initiate the expression of IFN genes, and therefore the viral defense (Figure 6).
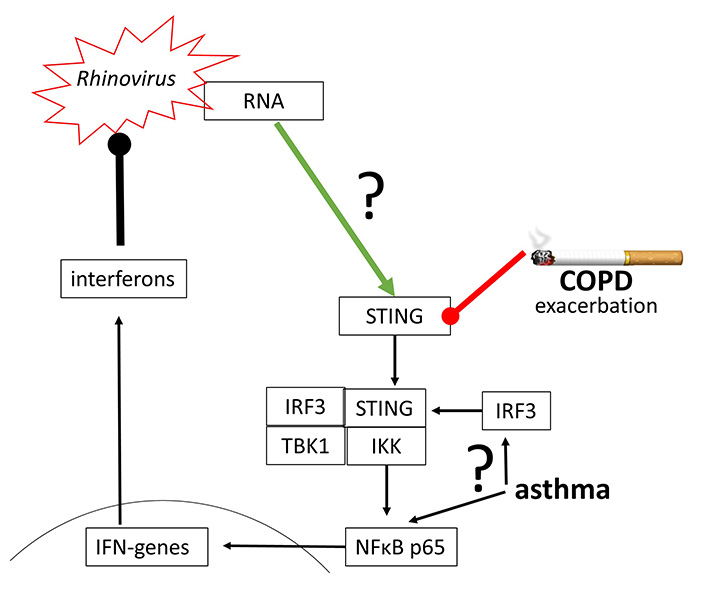
Rhinovirus may activate STING directly, but the underlying mechanism has not been proven. Furthermore, STING and IRF3 action has been reported to be impaired in COPD and asthma, thus leading to exacerbation
RSV is the most common cause of respiratory tract infection in, but not limited to, young age children. RSV is an RNA virus and consists of a double-layered capsid with intrusive glycol-proteins and one adhesion regulating G-protein. The major host cell type in humans is ciliated epithelial cells [87]. In COPD patients, it was shown that bronchial epithelial cells secret IFN-β when infected with RSV [88]. In a mouse model of RSV infection, it was reported that cAMP plays a role in the activation of host immune defense through the transfer protein Epac2 (exchange protein directly activated by cAMP 2) [89]. However, there was no report describing the activation of host defense by RSV through cGAS-STING signaling.
Despite the most frequent viruses causing respiratory tract infections, Cytomegalovirus and Herpesvirus have been reported to affect cGAS-STING signaling. Several independent studies reported in 2016 that Herpesvirus and Cytomegalovirus can activate the cGAS-STING signaling and thereby host defense [90–92]. However, both viruses can escape the host defense by reducing the acetylation of lysine 198 in the cGAS protein, thereby downregulating the activation of IFNs in various human lung cell lines [93]. Several proteins of Cytomegaloviruses can inhibit cGAS-STING signaling, by direct interference with specific human proteins. Two Cytomegalovirus proteins namely UL31 and pUL83 directly bind to cGAS and block its action [94, 95]. The Cytomegalovirus protein UL94 inhibits cGAS and mediator of IRF3 activation (MITA) [96]. In addition, three Cytomegalovirus proteins, namely UL82, US9, and IE86 directly inhibit STING [97–99]. A detailed overview of how Herpesviruses escape DNA sensing and even use cGAS-STING signaling for their replication has been provided by Stempel et al. [100].
Measles Virus might also affect lung function [2], and triggers host immune defense through indirect activation of cGAS-STING signaling, which depends on mitochondria degradation and release of mtDNA into the cytosol [101].
Several of the proteins that can activate STING signaling are known as receptors for DAMP, including TLR-2 through to TLR-9, RIG-1, and several HSPs [102]. The ligands for these DAMP receptors span over a wide range of bio-active factors including extracellular matrix components such as fibronectin, fibrinogen, hyaluronan (low molecular weight), decorin, heparan-sulfate, as well as HSPs, histones, DNA, RNA, mtDNA, and secreted proteins such as defensins, neurotoxins, and others [102]. Some of these factors can be activated by cGAS such as RIG-1, DNA, mtDNA, and RNA. Thus, the role of DAMP on virus-dependent STING activation might be indirect. It would be beyond the scope of this review to summarize the role of the different DAMPs and their receptors for each of the lung-relevant viruses. A few examples may show how interconnected DAMP and cGAS-STING signaling might be in the context of viral lung infections. Rhinovirus was reported to activate RIG-1 [103], NLRP3 [104], and defensin-β [20]. RSV activated TLR-4 by degradation of fibronectin [105], and hyaluronic acid [106]. Another DAMP receptor, NLRP3, was activated by Bocavirus [55], SARS-CoV-2 [107], Influenza A virus [108], and human Parainfluenzavirus HPIV3 [109].
Furthermore, some DAMP receptors might act as binding proteins for viruses. TLR-4 has been reported to play a role in the entry of different respiratory viruses into the host cells [110]. Such a contribution of different TLRs to infection success of respiratory viruses was further supported by reports on TLR polymorphisms and the severity of viral infections [111, 112]. Another DAMP, heparan sulfate, might help Rhinoviruses to infect the host cells [113]. These few examples might illustrate the difficulty to distinguish between virus-induced and DAMP-induced cGAS-STING signaling.
Glucocorticoids, long-acting β2-agonists (LABA), and long-acting muscarinic antagonists (LAMA) are the most prescribed therapeutic drugs for asthma and COPD [114, 115].
In a mouse model of a neuro-degenerative disease, steroids have been shown to inhibit cGAS-STING regulated cell death [116]. In a mouse model of acute lung injury induced by paraquat, the upregulation of STING, IRF3, and IFN-β was inhibited in animals treated with dexamethasone [117]. In regards to COPD, the loss of the barrier function of the epithelium by cigarette smoke might be resulted from the activation of cGAS-STING signaling and type-I IFN secretion, leading to cell damage [118].
In an asthma glucocorticoid resistance mouse model and in human bronchial epithelial cells, the interaction between the glucocorticoid receptor-interacting protein 1 and IRF3 was demonstrated [119]. In contrast, fluticasone did not inhibit the activation of IRF3 of NFκB in human bronchial epithelial cells (BEAS-2B), suggesting that this steroid does not reduce viral defense [120]. In normal human bronchial epithelial cells, it has been reported that NFκB and IRF3 were inhibited by fluticasone [121].
No report is available regarding the effect of LABA or LAMA on the cGAS-STING signaling.
Host defense against viral infections depends on the synthesis of IFN, which can be achieved by a variety of human cell types including epithelial cells, fibroblasts, macrophages, and immune cells. Sensing of viral infection occurs intracellularly by specific proteins recognizing either single or double-stranded RNA or DNA. One of the major sensings of nucleotide strands occurs through the cGAS-STING signaling, which leads to the activation of NFκB and IRF proteins, finally inducing the synthesis of IFN [122]. However, different viruses can activate cGAS-STING signaling and IFN synthesis on different levels of this signaling cascade, merging in the activation of STING and IRF3. IRF3 binding sites are found in the gene-regulating promoters of most IFNs, which has been summarized by others [123–125]. Unfortunately, evolution has enabled viruses to avoid or inhibit the cGAS-STING signaling and thereby escape or prevent host defense.
The effect that IFN regulation requires activation of NFκB might explain why anti-inflammatory medications support viral respiratory tract infections [126, 127]. However, the effect of anti-inflammatory medication, especially glucocorticoids on viral infections is indicated to be dependent on the time point of treatment. Early application of glucocorticoids increased the severity of viral infections, while later application reduced the inflammation and did not increase the severity of Influenza infection [128]. Regarding the use of glucocorticoids to treat SARS-CoV-2 infection [129, 130], it would be of interest to study the modulation of cGAS-STING signaling by these drugs.
The exact mechanism regulating cell type- or virus-specific cGAS-STING signaling is not fully understood. Future studies should investigate the interaction of viruses with DAMP receptors and their role in cGAS-STING signaling and innate immunity. In regards to virus-induced exacerbation in asthma and COPD patients, it would be of interest to study the effect of asthma and COPD triggers such as pollutants, allergens, and cigarette smoke on cGAS-STING signaling, and on the expression of DAMP receptors. In addition, it should be investigated how to achieve the optimal cGAS-STING activation level, which is sufficient to defend viral infections, but not leading to tissue damage or over-reactive immune response. This improved understanding of the regulation of the cGAS-STING signaling on the context of viral lung infections will hopefully lead to the development of new diagnostic and therapeutic strategies allowing personalized medicine.
ACE2: angiotensin converting enzyme 2
cAMP: cyclic adenosine monophosphate
cGAMP: cyclic guanosine monophosphate-adenosine monophosphate
cGAS: cyclic guanosine monophosphate-adenosine monophosphate synthase
cGMP: cyclic guanosine monophosphate
COPD: chronic obstructive pulmonary disease
DAMP: damage-associated molecular pattern
HSP: heat shock protein
IFNs: interferons
IKK: inhibitor of κB kinase
IRF: interferon regulatory factor
mtDNA: mitochondrial DNA
NFκB: nuclear factor κB
NLRP3: nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing-3
ORF9b: open reading frame 9b
PRR: pattern recognition receptors
RIG-1: retinoid acid-inducible gene 1
RSV: Respiratory Syncytial Virus
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2
STING: stimulator of interferon genes
TBK1: tank binding kinase 1
TLR-2: toll-like receptor-2
LF and MR both wrote the first draft, edited, read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

