 Review
Review

Affiliation:
Cell and Gene Therapy Program, Department of Pediatrics, Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta, Emory University School of Medicine, Atlanta, GA 30322, USA
ORCID: https://orcid.org/0000-0003-3862-0689
Affiliation:
Cell and Gene Therapy Program, Department of Pediatrics, Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta, Emory University School of Medicine, Atlanta, GA 30322, USA
Email: sraikar@emory.edu
ORCID: https://orcid.org/0000-0003-2903-9542
Explor Immunol. 2022;2:334–350 DOI: https://doi.org/10.37349/ei.2022.00054
Received: December 25, 2021 Accepted: March 28, 2022 Published: June 07, 2022
Academic Editor: Zhinan Yin, Jinan University, China
The article belongs to the special issue Interplay of γδ T cells and Tumor Cells
Cancer immunotherapy, especially T-cell driven targeting, has significantly evolved and improved over the past decade, paving the way to treat previously refractory cancers. Hematologic malignancies, given their direct tumor accessibility and less immunosuppressive microenvironment compared to solid tumors, are better suited to be targeted by cellular immunotherapies. Gamma delta (γδ) T cells, with their unique attributes spanning the entirety of the immune system, make a tantalizing therapeutic platform for cancer immunotherapy. Their inherent anti-tumor properties, ability to act like antigen-presenting cells, and the advantage of having no major histocompatibility complex (MHC) restrictions, allow for greater flexibility in their utility to target tumors, compared to their αβ T cell counterpart. Their MHC-independent anti-tumor activity, coupled with their ability to be easily expanded from peripheral blood, enhance their potential to be used as an allogeneic product. In this review, the potential of utilizing γδ T cells to target hematologic malignancies is described, with a specific focus on their applicability as an allogeneic adoptive cellular therapy product.
Gamma delta (γδ) T cells, a unique population of lymphocytes that mature in the thymus, account for 1–10% of circulating human T cells in the peripheral blood and up to 20% of intraepithelial T cells in the intestinal mucosa [1–5]. This T-cell subset has the distinctive ability to interact and display qualities of both the innate and adaptive immune systems. The robust properties of γδ T cells, allow it to polarize its immune response between anti- or pro-inflammatory, anti- or pro-tumorigenic, as well as between regulatory and effector functions in the immune system depending on the situation [5, 6]. A sought-after attribute of γδ T cells is their inherent cytotoxic properties against malignant and infected cells. A study involving the molecular profiling of ~5,000 tumors showed that infiltrating γδ T cells were the strongest favorable leukocyte predictor of survival [7]. The major contributor towards cytotoxicity is the γδ T-cell receptor (TCR), which can identify antigens independent of major histocompatibility complex (MHC) presentation, in stark contrast to αβ T cells, which respond predominantly to antigens bound and restricted to MHC molecules. This singular property amplifies the potential of developing γδ T cells into an allogeneic product, given the minimal risk of graft-versus-host disease (GvHD). γδ T cells also express multiple activating natural killer (NK) cell surface receptors such as NK group 2D (NKG2D), NK protein 30 (NKp30) and NKp44 as well as the Fas ligand (FasL) and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) leading to the release of lysing mediators such as perforin and granzymes. Additionally, activating DNAX accessory molecule-1 (DNAM-1) receptors, leukocyte function-associated antigen-1 (LFA-1) and costimulatory receptor CD27, all lead to T-cell activation and enhanced cytotoxicity [8–12]. Tumor cell death can also occur by antibody-dependent cellular cytotoxicity (ADCC) in a CD16-dependent manner, binding to the Fc region of immunoglobulin G (IgG) deposited on tumor cells. Finally, γδ T cells can also act like antigen-presenting cells (APCs), thereby playing an important role in the adaptive immune system. Thus, γδ T cells facilitate direct-targeted cell death, aiding in tumor and pathogen clearance while releasing immune-modulatory cytokines such as interferon-γ (IFN-γ), interleukin-17 (IL-17) and TNF-α. The multimodal approach γδ T cells that utilize in directing their natural cytotoxicity, make them an attractive tool for development into an anti-cancer cellular immunotherapeutic. Hematologic malignancies, which include leukemias, lymphomas and myeloma, are more suited to be targeted by cellular therapies, given the direct access to tumor cells through blood vasculature and lymphatics. Additionally, these cancers typically have a less immunosuppressive tumor microenvironment compared to solid tumors, further enhancing potential therapeutic effects. Here, we will explore the use of allogeneic T cells in targeting hematologic malignancies by examining the properties that make allogeneic γδ T cells an attractive immunotherapeutic candidate and reviewing all reported preclinical and clinical studies investigating the use of γδ T cells against blood cancers.
Classically, T cells can be divided into two broad categories based on the structure of their TCR, alpha beta (αβ) T cells, the majority subset and γδ T cells, the minority subset. In γδ T cells, TCR loci encode for the gamma chain [TCR gamma locus (TRG)] and the delta chain [TCR delta locus (TRD)]. The TCRs expressed in γδ T cells can rearrange depending on the expression of recombination-activating genes (RAGs) [5, 13]. The heterodimer of γδ V(D)J gene segments is restricted by the γ gene TRG locus only having 12 variables (6 of which are functional) and the δ gene TRD locus only having eight functional variable genes [6]. In γδ T cells, the γ chains most frequently used are Vγ1, Vγ7, Vγ4, Vγ5, Vγ6, and Vγ9 while frequently used δ chains are Vδ1, Vδ2, Vδ3 and Vδ5. This is in comparison to the more dominant αβ T cells, which have 52 variable β and 10 variable α loci [6, 14, 15]. Vγ and Vδ genes tend to delineate a particular organ/location in the body in abundance. Vδ1 is specifically found in the thymus, skin, lungs and intestines [16] with Vγ5 present in the skin and Vγ7 in the intestine, Vγ6 in the reproductive mucosa and Vγ1/Vγ4 in secondary lymphoid organs [15]. Vδ2 is found primarily in peripheral blood alongside Vγ9 and Vδ3 is mainly found in the liver [15, 16]. Both Vδ1 and Vδ2 are associated with Vγ9-recognizing phosphoantigens (pAgs), such as non-peptide prenyl-pyrophosphate metabolites, which in turn are associated with stress-related antigens and selective expansion of specific γδ TCR clonotypes [17–19]. As stated before, the γδ TCR recognizes antigens in an MHC-independent manner, and thus unlike its αβ counterpart does not require antigen presentation by APCs.
In peripheral blood, Vδ1 T cells are typically a minority population compared to the more dominate Vγ9Vδ2 [5, 18]. However, Vδ1 T cells paired with Vγ8 and Vγ9 chains, enriched in tissues, have targeted a variety of host and microbial antigens [5, 18–21]. Some studies have also shown Vδ1 T cells, through the γδ TCR, recognizing class 1b MHC-like proteins such as CD1 proteins similar to other unconventional T cells such as NKT or mucosal-associated invariant (MAIT) cells [22, 23]. Current studies have also indicated great benefits of Vδ1 T cells following allogeneic hematopoietic stem cell transplantation (allo-HSCT) and cytomegalovirus (CMV)-infections in patients with leukemia [3]. Some data also imply a balancing ratio between Vδ1 and Vδ2 T cells in tumor cells necessitates γδ T cell to either pro- or anti- tumorigenic responses [24, 25]. Despite multiple benefits indicated by Vδ1 T cells in targeting malignancies and post-transplantation survival, the prior inability to expand this small subset of T cells had hindered its clinical therapeutic benefits as an adoptive cellular product [3]. Notably, two recent publications have challenged that narrative by successfully expanding Vδ1 T cells with an anti-Vδ1 antibody [26, 27]. This new expansion method has now opened avenues for allogenic Vδ1 T cell therapy in clinical settings.
Vγ9Vδ2 T cells, the majority population in peripheral blood, have been directly implicated in both anti-viral and anti-tumor immunity. Their TCR is specifically reactive to pAgs, such as isopentenyl pyrophosphate (IPP), which are upregulated in certain stressed, infected and tumor cells. Butyrophilins (BTNs) have also emerged as an essential tool in γδ T-cell activation. BTNs are a large family of proteins and members of the extended B7 family of costimulatory molecules [28, 29]. BTN3A1 and BTN2A1 have been identified as crucial molecules indispensable for activation of Vγ9 T cells by pAgs. Vγ9Vδ2 TCR recognizes the BTN3A1/BTN2A1 complex in the membrane presenting IPP leading to activation [29–33]. IPP expression can also be artificially induced via inhibition of farnesyl pyrophosphate synthetase (FPPS) in the mevalonate pathway by amino-bisphosphonates. This unique property of the γδ TCR has been exploited by several groups, including ours, to isolate and expand Vγ9Vδ2 T cells from peripheral blood mononuclear cells (PBMCs) using bisphosphonates such as zoledronic acid [34–38]. This ability to easily activate and expand Vγ9Vδ2 T cells from peripheral blood makes them an attractive candidate to develop into a cellular immunotherapeutic product.
Along with the γδ TCR, expression of the NKG2D receptor plays a significant role in the cytotoxic ability of γδ T cells. The NKG2D receptor recognizes markers of cellular stress, which include the unique long 16 binding proteins (ULBPs) 1–6, and the MHC class I chain-related protein A and B (MICA/B) ligands. The NKG2D receptor-ligand interaction results in increased granzyme and perforin expression leading to target cell killing [39–45]. Along with NKG2D receptors, γδ T cells also express other activating NK cell receptors such as NKp30 and NKp44 to augment anti-tumor activity and cell signaling [46, 47]. The DNAM-1 receptor can trigger cytotoxicity upon interaction with its ligands CD112 (nectin) and CD155 (PVR), which are commonly expressed on hematologic malignancies [48–50]. Additionally, upregulation of FasL and TRAIL through TCR activation can also lead to enhanced tumor killing by interaction with Fas and TRAIL-R1/R2 respectively expressed on target cells. Other activating receptors include LFA-1 and the costimulatory receptor CD27 [8–12]. Cytokines such as IL-2, IL-15, IL-12, IL-18, IL-21 and IL-36γ also aid in γδ T-cell mediated cytotoxicity against malignant cells [51–53]. Finally, γδ T cells can mediate ADCC through the upregulation of CD16. γδ T cells can trigger cytotoxicity by recognizing the Fc regions of specific monoclonal antibodies (mAbs) bound to target cells, resulting in expression of CD107a, IFN-γ and TNF-α [54–56]. Apart from its innate-like direct cytotoxic mechanisms, γδ T cells also participate in the adaptive immune system by functioning as APCs, analogous to dendritic cells [5, 57, 58]. Vγ9Vδ2 T cells can process a wide range of microbial and tumor antigens for presentation to CD4+ and CD8+ T cells, and can also induce dendritic cell maturation through TNF-α production [57–59]. Thus, in addition to their TCR-dependent cytotoxicity, γδ T cells can employ several different killing mechanisms to target malignant cells (Figure 1).
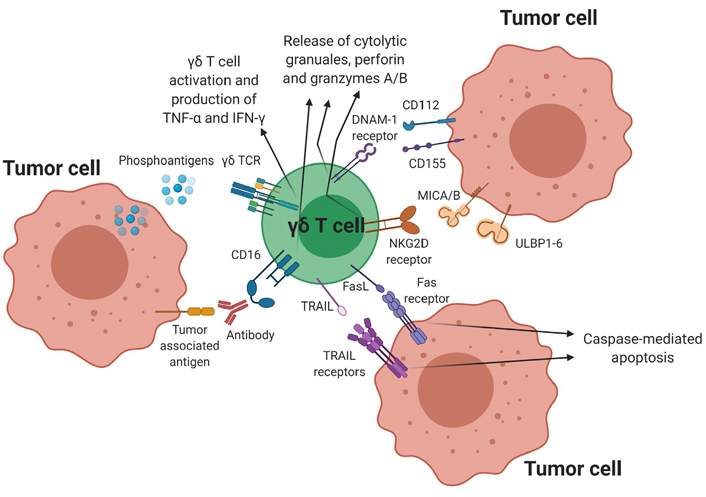
γδ T cell-mediated cytotoxicity against tumor cells. γδ T cells have several direct cytotoxic mechanisms against tumor cells as shown above. Binding of the pAg to the γδ TCR triggers activation resulting in target cell lysis and also stimulates the release of TNF-α and IFN-γ, which enhances the anti-tumor activity of other immune cells. Additional cytotoxic mechanisms include ADCC through CD16 expression, NKG2D and DNAM-1 receptor-ligand interactions as well as the activation of the TRAIL-TRAIL receptor and FasL-Fas receptor pathway
A critical factor in the manufacturing of a cellular therapy product is the ability to expand the product to reach desirable cell numbers in a robust efficient manner. The ability to expand Vγ9Vδ2 T cells from peripheral blood by taking advantage of the unique properties of the γδ TCR coupled with its non-MHC target recognition makes it an ideal candidate to develop into an allogeneic cellular therapy product. The γδ TCR is specifically reactive to pAgs, such as IPP, which are upregulated in infected and tumor cells. As mentioned before, it is now known that BTN3A1 and BTN2A1 are essential for the presentation of pAgs to the γδ TCR [29–33]. Expression of IPP can be artificially induced via inhibition of FPPS in the mevalonate pathway by amino-bisphosphonates such as zoledronic acid, pamidronate and risedronate. Several groups, including ours, have utilized this strategy to isolate and expand Vγ9Vδ2 T cells from PBMCs [39–43]. We have further characterized the variability in γδ T cell expansion among different donors, and have shown that IL-21 can be used to improve expansion in donors with poor ex vivo γδ T cell expansion [60]. Furthermore, we successfully depleted αβ T cells on day 6 of the expansion, providing a better environment for the γδ T cells to expand, while confirming that the αβ T cell population remains below clinically acceptable standards for T cell-depleted allogeneic stem cell products [60, 61]. Two recent studies have shown successful expansion of Vδ1 T cells from peripheral blood [26, 27]. These studies performed by Adicet Bio, Inc. (Boston, USA) utilize a proprietary agonist anti-Vδ1 mAb that selectively activates and expands Vδ1 T cells from healthy donor derived PBMCs. Similar to our studies, an αβ T cell depletion step is utilized before the final product formulation [26].
Allo-HSCT can be an effective treatment option for patients with high-risk leukemia and other hematologic malignancies, which are refractory to conventional treatments. The success of an allogeneic transplant depends on several different factors, such as disease status prior to hematopoietic stem cell transplantation (HSCT), type of hematologic malignancy, and donor characteristics such as human leukocyte antigen (HLA) match status, age, and stem cell source. GvHD remains the most significant toxicity in patients undergoing allo-HSCT, and the pathogenesis of GvHD is primarily driven by donor αβ T cells. While several measures are taken to reduce potential GvHD, the graft-versus-leukemia (GvL) effect seen in the setting of allo-HSCT is known to be beneficial to patients. Given that γδ T cells identify antigens in an MHC-independent manner, they can provide therapeutic GvL effects without the risk of GvHD; hence there is a growing interest in the role γδ T cells play in the success of allo-HSCT [46]. Indeed, high γδ T cell immune reconstitution after allo-HSCT of αβ T cell and CD19+ depleted grafts has been shown to result in overall higher survival rates and decreased rate of acute GvHD [62–64]. In a large cohort of patients with leukemia undergoing allo-HSCT that received a T-cell depleted bone marrow graft from partially mismatched HLA donors, patients in which γδ T cells accounted for greater than 10% of circulating lymphocytes had superior disease-free 30 months after treatment [64]. There was no significant difference in acute and chronic GvHD, suggesting a superior GvL effect without GvHD. Two subsequent long term follow-up studies for this population, at 42 months and then 8 years, confirmed there was a significantly better disease-free survival (DFS) and overall survival for patients with higher proportion of γδ T cells [65, 66]. A more recent pediatric study of 102 patients from St Jude Children’s Research Hospital analyzing the γδ T cell reconstitution after allo-HSCT, showed that a significantly better event-free survival and overall survival was seen in patients with increased γδ T cells at a median follow-up of 2.7 years [67]. Additionally, the patients with higher γδ T cells had a lower incidence of bacterial and viral infections, emphasizing the anti-microbial properties of γδ T cells [68]. Based on the superior GvL effects of γδ T cells without causing GvHD, combined with their ability to fight infections, several clinical studies are now exploring the utility of the adoptive transfer of allogenic donor-derived γδ T cells in the post-transplant setting, as we discuss in a later section in this article [46].
Hematologic malignancies, which include leukemias, lymphomas and myelomas, have become an attractive target for cellular therapies over the past decade, especially with the advent of chimeric antigen receptor (CAR) based T-cell therapies. In this innovative therapy, T cells are genetically modified to express a receptor, called a CAR, which can identify target tumor antigens with the specificity of an mAb, thereby enabling the T cell to directly kill its tumor target [69]. CAR T-cell therapy has been very successful in hematologic malignancies, especially B-cell malignancies and more recently multiple myeloma (MM), compared to solid tumors [70–72]. Hematologic malignancies are more suited to be targeted by cellular therapies, given the direct accessibility to tumor cells through the blood vasculature and lymphatics. Additionally, blood cancers typically have a less immunosuppressive tumor microenvironment compared to solid tumors, further enhancing potential therapeutic effects [73].
However, most current cellular therapies use autologous patient-derived αβ T cells. T cells are first collected from the patient through a process called leukapheresis and then genetically modified using a viral vector encoding the CAR. Cells are then expanded to the desired numbers and finally given back to the patient after lympho-depleting chemotherapy [69]. While there has been significant progress in the manufacturing process over the past few years, the production and administration of an autologous cellular therapy product are still very complex and time-consuming, taking at minimum between 2–4 weeks from collection to infusion. Although this strategy has been successful in B-cell malignancies, the delay in delivering the therapeutic product may not be feasible in more aggressive cancers such as acute myeloid leukemia (AML) and T-cell malignancies [74, 75]. Furthermore, we are now learning that poor T-cell fitness is a major factor in the failure of these therapies, especially when cells are collected from patients heavily pre- treated with chemotherapy [70].
To overcome this challenge, there has been a concerted effort to develop “off-the-shelf allogeneic cellular therapies using healthy donors as the effector cell source. However, given the severe risk of GvHD using αβ T cells from non-HLA-matched donors, certain genetic modifications are necessary to make allogeneic αβ T cells a safe and feasible therapeutic. The most common approach has been to knock down the expression of the αβ TCR by gene editing of the TCR alpha constant (TRAC) and/or TCR beta constant (TRBC) locus. The different gene editing tools that have used in this setting include Zinc finger nucleases (ZFN) [76–78], transcription activator-like effector nucleases (TALEN) [79–81], and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 genome editing [82, 83]. While gene editing of αβ T cells is an exciting approach, the translatability to a clinical product can be challenging and expensive. γδ T cells, given their non-MHC dependence for antigen recognition, provide an excellent alternative as the effector cell source, and do not require any further genome editing to be developed into an allogeneic product. Furthermore, given their inherent anti-tumor properties, they form a promising candidate to move forward as an “off-the-shelf cellular therapeutic.
In this section, we will review the different approaches that have been tested in the preclinical setting to enhance the cytotoxic effect of γδ T cells against hematologic malignancies. These include the use of γδ T cells in combination with amino-bisphosphonates, checkpoint inhibitors, chemotherapeutic drugs, mAbs and bispecific T-cell engagers (BiTEs) as well as the use of γδ T cells genetically modified to express CARs.
As stated before, most studies have used amino-bisphosphonates to selectively expand γδ T cells ex vivo. Several studies have shown that γδ T cells are cytotoxic towards AML blasts. Gertner-Dardenne et al. [48] showed that Vγ9Vδ2 T cells efficiently killed autologous AML blasts via the perforin/granzyme pathway utilizing both TCR and DNAM-1 dependent mechanisms. Vγ9Vδ2 T cells also killed AML blast in a xenograft mouse model improving survival. More recently, Benyamine et al. [84] showed that anti-BTN3A1 antibodies have the ability to mimic pAg stimulation, which in turn selectively activates Vγ9Vδ2 T cells. Incubation of AML blasts with anti-BNT3A1 triggered BTN3A1 on the blasts, resulting in enhanced Vγ9Vδ2 T cell-mediated killing, while also sensitizing resistant blasts to Vγ9Vδ2 T cell lysis. They further validated their results in an AML xenograft model demonstrating that the agonistic anti-CD277/BTN3A1 antibody mAb 20.1, enhanced the therapeutic efficacy of adoptively transferred Vγ9Vδ2 T cells [84]. Vγ9Vδ2 T cells have also been shown to be effective against lymphoma cell line Daudi and MM cell lines RPMI8226 and U266 [85, 86]. Interestingly, the Vδ1 T cell subset appears to play a more important role in targeting chronic lymphocytic leukemia (CLL) blasts, with increased number having been reported in patients with CLL [87]. Siegers et al. [88] developed an expansion protocol using the mitogen concanavalin A (Con A) that selectively expanded Vδ1 cells over the Vδ2 subset when combined with IL-2 and IL-4. They subsequently showed that these were more cytotoxic against the CLL cell line MEC1 compared to Vδ2 cells [88]. A subsequent Vδ1 T cell expansion protocol developed by Almeida et al. [89], resulting in a cellular product called Delta One T (DOT) cells, showed impressive efficacy in CLL xenograft models. The DOT cells have also been tested in an AML xenograft model and were shown to have impressive efficacy [90].
Several studies have taken advantage of the NKG2D receptor-ligand axis as a means to effectively target hematologic malignancies using γδ T cells. Expression of the NKG2D ligands ULBP1–6 and MICA/B can be upregulated in leukemia and myeloma cells by pre-treatment with chemotherapeutic agents. Different classes of chemotherapeutics have been. The proteasome inhibitor, bortezomib, has been tested in MM, AML and T-cell acute lymphoblastic leukemia (T-ALL) [61, 91, 92]. Niu et al. [92] showed treatment of MM cells with low-dose bortezomib resulted in enhanced killing by γδ T cells and NK cells, through increased NKG2D and DNAM-1 ligand expression. Story et al. [61] showed that bortezomib increased the ULBP2/5/6 expression in both AML and T-ALL cell lines, enhancing γδ T cell-mediated killing. Importantly, both studies showed that bortezomib had minimal inhibitory effects on γδ T cell proliferation and function. The other class of chemotherapeutics that have been tested in this setting are epigenetic drugs. These include histone deacetylase inhibitors such as valproic acid and romidepsin as well as demethylating agents such as azacitidine and decitabine [93–95]. de Weerdt et al. [96] showed that treatment of CLL patient-derived Vγ9Vδ2 T cells with the tyrosine kinase inhibitor irutinib restored its functional phenotype and improved cytotoxicity against CLL cells.
γδ T cells mediate ADCC through expression of the Fc-γ receptor III CD16. Multiple studies have evaluated the combination of γδ T cells with anti-CD20 mAbs such as rituximab, ofatumumab and obinutuzumab (GA101) to target B-cell malignancies. Tokuyama et al. [56] demonstrated the rituximab enhanced the ADCC effect of γδ T cells against CLL and follicular lymphoma cells. Braza et al. [97] showed that the highest ADCC effect against follicular lymphoma cells was seen when using obinutuzumab, compared to rituximab and ofatumumab. Similarly, Gertner-Dardenne et al. [98] found that alemtuzumab, an anti-CD52 antibody, also increased γδ T-cell dependent ADCC against lymphoma cell lines. Another new category of antibody-based drugs is BiTEs which consist of two single chain variable fragments (scFvs) binding domains, one typically specific to CD3 present on T cells, and another to a tumor associated antigen on cancerous cells [99–101]. Concurrent binding of a BiTE combination results in forming a lytic immune synapse between the cytotoxic T cell and the cancerous target cell. A recent preclinical study by Chen et al. [102] showed that combining the CD19-directed BiTE blinatumomab with γδ T-cells improved overall survival in a murine B-ALL model. Previously, Seidel et al. [54] had tested both CD19-CD3 and CD19- CD16 using the CD19 antibody 4G7SDIE as its backbone, and showed that the dual antibody constructs could induce cytotoxic reactions from γδ T cells. Schiller et al. [103] then created a single chain triple antibody (CD19-CD19-CD16) called SPM-1 and showed it had higher NK and γδ T-cell mediated killing when compared to 4G7SDIE. More excitingly, a new γδ T-cell specific BiTE against AML has now been created, TRGV9/CD123, which binds the Vγ9 chain of the Vγ9Vδ2 T cell, thereby enabling it to selectively target CD123-expressing AML cells [104, 105].
CAR T-cell therapy has revolutionized cancer immunotherapy over the past decade, with great success seen in hematologic malignancies. However, given the complexity and cost involved in manufacturing autologous CAR T cells, there is a growing need to develop “off-the-shelf allogeneic CAR T-cell therapeutics. Given the lack of an MHC-dependent TCR, γδ T cells have not been implicated in GvHD pathogenesis, and are ideal candidates to be developed into allogeneic CAR T-cell therapeutics. While several companies are now developing CAR γδ T cells, only limited preclinical data has been published so far. A few reported studies have demonstrated effective preclinical CAR-γδ T cell cytotoxicity against hematologic malignancies. Deniger et al. [106] used a sleeping beauty transposase system to develop CD19 CAR γδ T cells, which showed efficacy against CD19 positive tumor cell lines in vitro and reduced leukemia burden in xenograft models. Similarly, Rozenbaum et al. [107] recently showed the CD19-directed CAR γδ T cells generated from lentiviral transduction were effective in both in vitro and in vivo studies. Multiple doses of γδ T cells were given to attain the desired effect given their short lifespan. Additionally, CD19 negative leukemia cells when primed with zoledronate were also killed. In an interesting study by Fleischer et al. [108], non-signaling CARs (NSCARs) were expressed in γδ T cells, to prevent fratricide while targeting T-cell leukemia. The NSCARs lacked signaling/activation domains, but retained the ability to interact with the tumor cell with antigen-specificity, thereby acting as an anchor. This then allowed the γδ T cell to use its inherent MHC-independent mechanism to lyse the tumors cells. They demonstrated that both CD5 and CD19-NSCAR modified γδ T cells had a significant increase in killing against T-ALL and B-ALL cell lines respectively. Importantly, as hypothesized, no increase in cytotoxicity was seen in the NSCAR approach when using αβ T cells [108]. Finally, a recent study by Nishimoto et al. [26] (Adicet Bio, Inc., Boston, USA) demonstrated successful large-scale manufacturing of anti-CD20 CAR γδ T cells utilizing healthy donor derived Vδ1 T cells. As mentioned before, Vδ1 T cells were expanded utilizing an agonist anti-Vδ1 mAb. The anti-CD20 CAR Vδ1 T cells exhibited effectively in vitro tumor cell killing and pro-inflammatory cytokine production, as well as in vivo tumor growth inhibition of B-cell lymphoma xenografts in immunodeficient mice. Interestingly, the CAR Vδ1 T cells exhibited a naïve-like T-cell memory phenotype and only a single dose of γδ T cells was used in mouse studies [26]. Based on these findings, a phase 1 clinical trial has been initiated in patients with CD20-positive B-cell malignancies (NCT04735471).
Clinical trials evaluating the use of γδ T cells to target hematologic malignancies fall into three separate categories: (a) in vivo stimulation of autologous γδ T cells, (b) adoptive transfer of ex vivo expanded autologous γδ T cells, and (c) adoptive transfer of ex vivo expanded allogeneic γδ T cells. Most studies using allogeneic γδ T cells having been the post-HSCT setting where cells are derived from the allo-HSCT donor; however, newer trials are now beginning to evaluate the use of allogeneic γδ T cells as a standalone cellular therapeutic. The results from all past completed trials utilizing γδ T cells to target hematologic malignancies are reviewed in Table 1.
Clinical studies utilizing γδ T cells against hematologic malignancies with published results
| Reference | Year | Disease | N | Intervention | Response |
|---|---|---|---|---|---|
| In vivo stimulation of γδ T cells | |||||
| Wilhelm et al. [109] | 2003 | NHL, CLL, MM | 19 | Pamidronate and IL-2 | 3/19 had objective response |
| Laurent et al. [110] | 2009 | Follicular lymphoma | 45 | Rituximab + BrHPP + IL-2 | 75% of first 12 patients had response |
| Kunzmann et al. [111] | 2012 | AML | 8 | Zoledronate and IL-2 | 2/8 had partial remission |
| Bertaina et al. [112] | 2017 | ALL and AML | 43 | Zoledronate post allo-HSCT | Improved DFS, higher circulating γδ T cells |
| Merli et al. [113] | 2020 | ALL, AML and MPAL | 46 | Zoledronate x 3 post allo-HSCT | Improved DFS, lower TRM, reduced GvHD |
| Adoptive transfer of γδ T cells | |||||
| Abe et al. [116] | 2009 | MM | 6 | Four infusions of ex vivo expanded autologous Vγ9γδ2 T cells | 4/6 had stable disease, no toxicity |
| Wilhelm et al. [117] | 2014 | T-NHL, AML, MM, plasma cell leukemia | 4 | Haploidentical γδ T cells, followed by zoledronate + IL-2 | 3/4 had complete response |
N: total number of patients; NHL: non-Hodgkin lymphoma; BrHPP: bromohydrin pyrophosphate; ALL: acute lymphoblastic leukemia; MPAL: mixed phenotype acute leukemia; TRM: transplant-related mortality; T-NHL: T-cell NHL
In one of the first completed trials, Wilhelm et al. [109] tested the effects of pamidronate and IL-2 on Vγ9Vδ2 T cell activation and anti-tumor activity in 19 patients with NHL, CLL or MM. Objective responses were seen in only three patients, and corresponded to the Vγ9Vδ2 T cell proliferation in vitro [109]. Laurent et al. [110] studied the combination of rituximab with IPH1101 (BrHPP, a Vγ9Vδ2 T cell agonist) along with IL-2 in 45 patients with follicular lymphoma. Only data for the first 12 patients were reported in an abstract with 75% showing a response; however, the final outcomes were never published. In another study, Kunzmann et al. [111] studied the use of zoledronate and IL-2 in patients with different advance malignancies, including eight patients with refractory AML. Two of the eight AML patients had a partial remission. In a pediatric study of patients undergoing allo-HSCT for acute leukemia, patients who received a zoledronate infusion post-transplant had improved DFS and a higher number of circulating γδ T cells [112]. A subsequent trial showed that three or more infusions of zoledronate lowered TRM, improved DFS and was associated with a reduced incidence of GvHD [113].
The adoptive transfer of γδ T cells has more commonly been tested in solid tumor malignancies. Published data from a recent study using allogeneic Vγ9Vδ2 T cells in 132 advanced stage liver and lung cancer patients showed that allogeneic Vγ9Vδ2 T cells produced no significant adverse effects (e.g., immune rejection, cytokine storm, or GVHD effects) [114]. A follow-up case report was published on one of these patients, who is a 30-year-old male with stage 4 cholangiocarcinoma, post liver transplantation, with recurrent mediastinal lymph node metastasis [115]. This patient successfully received 8 infusions of allogenic Vγ9Vδ2 T cells (4 × 108 cells total) and had a significant reduction in the size of his lymph nodes without any significant adverse effects, thereby confirming its potential as a therapeutic. Several current ongoing trials are now evaluating this approach in hematologic malignancies. All the current ongoing γδ T cell clinical trials targeting hematologic malignancies, including two CAR-based γδ T cell studies are listed in Table 2. Results of only two studies using adoptively transferred γδ T cells in hematologic malignancies have been published. Abe et al. [116] treated six MM patients with ex vivo expanded autologous Vγ9γδ2 T cells. Cells were expanded from PBMCs using zoledronate and IL-2, and each patient received four infusions of cells at 2-week intervals. The infusions were safely tolerated and disease remained stable in 4/6 patients [116]. In a pilot study, Wilhelm et al. [117] reported the successful transfer of haploidentical γδ T cells in four patients with refractory hematologic malignancies (one T-NHL, one AML, one secondary plasma cell leukemia, and one MM), followed by in vivo stimulation with zoledronate and IL-2. Three out the four patients achieved complete remission, with one patient having a sustained response for 8 months [117].
Active adoptive γδ T-cell immunotherapy clinical trials against hematologic malignancies
| ClinicalTrials.gov identifier | Sponsor | Disease | Intervention | Phase | Status |
|---|---|---|---|---|---|
| NCT04696705 | Institute of Hematology & Blood Diseases Hospital | NHL and PTCL | Ex vivo expanded allogeneic γδ T cells | Early Phase I | Recruiting |
| NCT05015426 | H. Lee Moffiff Cancer Center and Research Institute | AML | Ex vivo expanded donor γδ T cells post allo-HSCT | Phase I/Ib | Recruiting |
| NCT04764513 | Chinese PLA General Hospital | AML, ALL, MDS, lymphoma | Ex vivo expanded donor γδ T cells post allo-HSCT | Phase I/II | Recruiting |
| NCT03533816 | University of Kanas Medical Center | AML, CML, ALL, MDS | EAGD T-cell infusion following haplo-HSCT | Phase I | Recruiting |
| NCT05001451 | GammaDelta Therapeutics Limited | AML | GDX012 infusion–allogeneic Vδ1+ γδ T cells | Phase I | Recruiting |
| NCT04008381 | Wuhan Union Hospital | AML | Ex vivo expanded allogeneic γδ T cells from suitable donor | Phase I | Recruiting |
| NCT03790072 | TC Biopharm | AML | Ex vivo expanded allogeneic γδ T cells from suitable donor (OmnImmune®) | Phase I | Completed |
| NCT04702841 | PersonGen BioTherapeutics | CD7+ T-cell malignancies | CAR–γδ T cells | Early Phase I | Recruiting |
| NCT04735471 | Adicet Bio. Inc. | B-cell malignancies | Anti-CD20-CAR-T | Phase 1 | Recruiting |
PTCL: peripheral T-cell lymphoma; MDS: myelodysplastic syndrome; CML: chronic myeloid leukemia; EAGD: ex vivo expanded/activated γδ; haplo: haploidentical
As we have discussed here, γδ T cells provide an optimal platform for development into an “off-the-shelf” allogeneic cellular therapeutic. There continues to be a growing interest in the use of these unique multi-functional immune cells as reflected in the increasing number of companies developing γδ T-cell based immunotherapies (Table 3). While there has been remarkable success in utilizing CAR T-cell therapies for hematologic malignancies, the overall efficacy has somewhat been limited primarily to B-cell malignancies. In order to extend this therapy to other disease types, newer approaches need to be considered. Additionally, we now have a better understanding of the mechanisms of treatment failure, and a poor quality cellular product remains one of the major concerns. A shift to an allogeneic platform may be necessary to overcome these manufacturing challenges and increase the availability of the unique immunotherapy to the general population. γδ T cell therapies can be utilized in number of different approaches, with adjuncts such as zoledronic acid, in combination with chemotherapies to upregulate NKG2D ligand expression, enhancing ADCC when used with mAbs or BiTEs, on by genetic modification to express a CAR. Thus, allogeneic γδ T cells have great potential to be an effective cellular therapy option for hematologic malignancies. Continued efforts are needed to enhance and maximize the benefits of this unique cellular therapeutic.
Current companies developing γδ T cell-based immunotherapies
| Company | γδ T cell type | Allogeneic vs. autologous | Targeting strategy/engineering |
|---|---|---|---|
| GammaCell Biotechnologies | Vδ2 | Autologous/allogeneic | Unmodified |
| Hebei Senlang Biotechnology | Vδ2 | Autologous | CAR/αβTCR |
| Incysus Therapeutics | Vδ2 | Autologous | Engineered for chemo-resistance |
| Adicet Bio, Inc. | Vδ1 | Allogeneic | CAR |
| Beijing Doing Biomedical | Vδ2 | Allogeneic | Unmodified/CAR |
| Cytomed Therapeutics | Vδ2 | Allogeneic | CAR |
| GammaDelta Therapeutics | Vδ1 | Allogeneic | CAR |
| Immatics | Vδ2 | Allogeneic | αβTCR |
| PhosphoGam Inc. | Vδ2 | Allogeneic | Unmodified |
| TC BioPharm | Vδ1/Vδ2 | Autologous/allogeneic | Unmodified/CAR |
| Imcheck Therapeutics | Vδ2 | Autologous (in vivo) | Vδ2 activation with BTN3A |
| Lava Therapeutics | Vδ2 | Autologous (in vivo) | Activated Vδ2 with BiTE |
| PersonGen BioTherapeutics | γδ T | Allogeneic | TAA3-UCAR, CD7 UCAR |
| Expression Therapeutics | Vδ2 | Allogeneic | Unmodified/CAR |
UCAR: universal CAR
Note. Adapted from "Cancer immunotherapy with γδ T cells: many paths ahead of us", by Kabelitz D, Serrano R, Kouakanou L, Peters C, Kalyan S. Cell Mol Immunol. 2020;17:925–39 (https://www.nature.com/articles/s41423-020-0504-x). CC-BY.
ADCC: antibody-dependent cellular cytotoxicity
ALL: acute lymphoblastic leukemia
allo-HSCT: allogeneic hematopoietic stem cell transplantation
AML: acute myeloid leukemia
APCs: antigen-presenting cells
BiTEs: bispecific T-cell engagers
BTNs: Butyrophilins
CAR: chimeric antigen receptor
CLL: chronic lymphocytic leukemia
DFS: disease-free survival
DNAM-1: DNAX accessory molecule-1
GvHD: graft-versus-host disease
GvL: graft-versus-leukemia
HLA: human leukocyte antigen
HSCT: hematopoietic stem cell transplantation
IFN-γ: interferon-γ
IL-17: interleukin-17
IPP: isopentenyl pyrophosphate
mAbs: monoclonal antibodies
MHC: major histocompatibility complex
MM: multiple myeloma
NHL: non-Hodgkin lymphoma
NK: natural killer
NKG2D: natural killer group 2D
NKp30: natural killer protein 30
NSCARs: non-signaling chimeric antigen receptors
pAgs: phosphoantigens
PBMCs: peripheral blood mononuclear cells
T-ALL: T-cell acute lymphoblastic leukemia
TCR: T-cell receptor
TNF: tumor necrosis factor
TRAIL: tumor necrosis factor-related apoptosis-inducing ligand
TRG: T-cell rearranging gamma locus
ULBP: unique long 16-binding proteins
NJ and SSR both wrote the first draft, edited, read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by the K08CA248962 grant (S.S.R.) from the National Cancer Institute (NCI) of the National Institutes of Health (NIH). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Xiangjin Zhang ... Wei He
Huidi Wang ... Jia Yin
Chang Liu ... Wei He
Joseph M. McGraw, Deborah A. Witherden
Yue Wang ... Wei He
Rupert Handgretinger ... Manon Queudeville
Gabrielle M. Ferry, John Anderson
Juan-Pablo Cerapio ... Jean-Jacques Fournie
Alessandro Poggi, Maria Raffaella Zocchi
Jessica Da Gama Duarte ... Andreas Behren
