 Review
Review

Affiliation:
1Female Reproduction and Metabolic Syndromes Laboratory, Division of Molecular Reproduction, DBT-Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram 695014, Kerala, India
2Research Centre Kerala University, Senate House Campus, Palayam, Thiruvananthapuram 695034, Kerala, India
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-2633-2957
Affiliation:
1Female Reproduction and Metabolic Syndromes Laboratory, Division of Molecular Reproduction, DBT-Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram 695014, Kerala, India
†These authors contributed equally to this work.
Email: laloraya@rgcb.res.in; laloraya@gmail.com
ORCID: https://orcid.org/0000-0003-4985-8932
Explor Immunol. 2022;2:393–413 DOI: https://doi.org/10.37349/ei.2022.00058
Received: December 06, 2021 Accepted: April 27, 2022 Published: June 27, 2022
Academic Editor: Nathalie Lédée, Hôpital Pierre-Rouquès Les Bluets-Drouot, France
The article belongs to the special issue Human Reproduction: Involvement of the Immune System
Polycystic ovary syndrome (PCOS) is one of the most frequently observed endocrinopathies among women of reproductive age that redound to subfertility. The specific etiology of this heterogenic syndrome remains ambiguous. Metabolic complications, hormonal imbalance, deregulation in the immune system and their interrelationship make PCOS more complex. Hyperandrogenism and chronic low-grade inflammation modulate each other and enhance the self-perpetuation of PCOS. Even though there are many literature studies on PCOS and immune deregulation, this review focuses on the endocrine-immune nexus and how the altered endocrine system is embroiled in the immunopathology of PCOS.
Polycystic ovary syndrome (PCOS) is a frequently observed endocrine disorder in the female population during their fertile period. The syndrome is presented by a constellation of manifestations including chronic oligo/anovulation, multiple cysts, enlarged ovaries, hirsutism, and hyperandrogenism. Being the foremost reason for anovulatory infertility, PCOS also increases the risk for cardiovascular complications, insulin resistance (IR), obesity, and type-2 diabetes mellitus (T2DM) in reproductive-aged women. Consequently, the disorder has a considerable repercussion on the physical and mental well-being of these females. Since the disorder encompasses endocrine, metabolic, and reproductive dysfunction, PCOS is considered as a clinical model for understanding the interaction between these systems [1, 2]. As per the Global Burden of Disease Study conducted in 2010, PCOS is one of the utmost sequelae affecting females in their fertile period with a prevalence rate of 3.42% [3]. Based on National Institutes of Health (NIH)/National Institute of Child Health and Human Development (NICHD) criteria, the pooled incidence of PCOS was 7%, while pooled PCOS incidence rate using Rotterdam and androgen excess (AE)-PCOS criteria was 12% [4], and a series of reports demonstrated a range of 6–28% of the global PCOS incidence rate [5–8]. The pathology of PCOS is less understood but is a vastly studied area. Hence to date, there are several hypotheses on the etiology of this multifactorial disease which include hormonal imbalance, immune deregulation, and genetic and epigenetic factors, making it more complicated.
The primary role of sex hormones includes proper functioning of the reproductive system and sexual differentiation. Additionally, the involvements of steroid hormones in the functional activity of the immune system are also well explained. This is evident by the sexual dimorphism in immunity observed among females and males, specifically, the gender difference in rejection of graft and incidence of autoimmune diseases is attributed to sex hormones; females experience more occurrence of autoimmune disease than males [9–11]. More dynamic cell-mediated and antibody-mediated immunity, higher resistance to several bacterial, viral, and parasitic infections and increased response to vaccination by high antibody titers were also observed in females [12–14]. In comparison to males, females exhibit differential dynamics in the concentration of hormones that regulate the menstrual cycle phases and even pregnancy and parturition [11]. PCOS is a condition characterized by altered sex hormonal levels with significantly high levels of androgen [15]. In this review, we focus on endocrine deregulation and its impact on the immune etiology of PCOS.
To understand the labyrinth of altered hormones and immune deregulation in PCOS, we conducted a comprehensive and systematic literature search in databases such as PubMed, Scopus, Embase and Google. No restrictions in language, study subjects, type of study and date of publication were imposed during the search. We included original articles, reviews and meta-analyses in this study. Hormonal implications of female disorders other than PCOS were excluded from the study. We have incorporated all types of clinical studies including retrospective, prospective, randomized case control and cohort studies. The query terms used for the search were Medical Subject Heading terms (MeSH) or free terms including “Hyperandrogenism” AND “PCOS”, “Sex steroids” AND “Immune system”, “Hormones” AND “Immunity status”, “Immune system” AND “PCOS”, “Immune system” AND “Female infertility”, “Hormonal imbalance” AND “Cytokines in PCOS”, “Adipose tissue dysfunction” and “Sex steroids”, and “Autoimmunity” AND “PCOS”. Relevant hits obtained from each search result were included in the study. The initial selection was performed by rejecting irrelevant articles. Moreover, to avoid the missing of any relevant articles during the electronic database search, the bibliography list of retrieved publications was also reviewed.
In general, the action of estrogen is immunoenhancing while testosterone (T) is immunosuppressive (Figure 1) [12]. Immune modulatory actions of sex hormones have been well described in both humans as well as in animal models. The androgen receptor (AR) mediates the biological activity of androgens through two different signalings cascades. The genomic or canonical signaling is mediated by cytoplasmic or classical AR and the non-classical or non-genomic signaling is mediated by AR located in the cell membrane. Androgen-AR binding results in the dissociation of AR from intracellular chaperones and translocation of androgen-AR complex into the nucleus where it modulates the target genes transcription by binding to androgen-responsive elements (AREs) of DNA. Direct binding of androgens can activate non-classical AR by triggering intracellular calcium influx and by activation of downstream transcription factors [16–18]. Monocytes, B cells, macrophages, T cells, mast cells, neutrophils, and megakaryocytes are known to express AR [19]. Size of thymus and thymocyte selection is dependent on androgen [20] as well as naive T cell output [21]. B-lymphopoiesis in bone marrow can be modulated by androgens [22]. Production of cytokine by male cells prefers the T helper 2 (Th2) response cytokines [23] including interleukin (IL)-10 upregulation upon dihydrotestosterone (DHT) treatment [24]. Androgens are known to hinder Th1 differentiation [25, 26] by constraining interferon-gamma (IFN-γ) and IL-12 production [26–28].
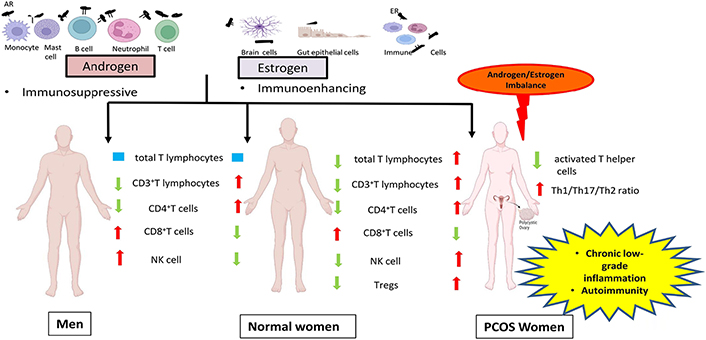
Schematic representation of the role of sex steroids in the immune system of normal women, men and PCOS by comparing the immunologic features of PCOS women and men with normal women. ER: estrogen receptor; NK: natural killer; Tregs: regulatory T cells
Modulation of B cell activity, negative selection of self-reactive B cells and T cell homing, and augmented Th2 response are major immunoenhancing actions of estrogen [29, 30]. Estrogen also mediates its signaling via both classical genomic and non-classical non-genomic pathways. The classical genomic pathway involves two cytoplasmic receptors: ERα and ERβ. ER upon transactivation by estrogen, translocates to the nucleus and interacts with estrogen response elements (ERE) for regulation of various cellular mechanisms. By binding to the ER on cell membrane (mER), estrogen triggers the non-genomic signaling by activating release of intracellular calcium and other transcription factors [31–33]. Bone marrow, hematopoietic cells, thymocytes and stromal cells of thymus, peritoneal macrophages and splenic dendritic cells of mice are known to express ERα, while ERβ expression is observed in human thymus and spleen during the mid-gestational stage, lymph nodal lymphocytes, murine thymus and bone marrow. Estrogen regulates number and function of neutrophil, chemokine and cytokine induction [34], superoxide and nitric oxide production in immune cells [35]. Estrogen also modulates dendritic cell differentiation [36] as well as toll like receptors (TLR) signaling which requires binding of ERα to ERE [37]. Also, estrogen mediated activation of both G protein-coupled ER (GPER) and mER signaling events are integrated with CD19, B cell receptor (BCR) mediated intracellular signaling to impact cellular activities in B lymphocytes [38].
By enhancing the anti-inflammatory mediators and reducing pro-inflammatory mediators, androgens impose an immunosuppressive state in various cell types of the adaptive and innate immune systems. It has been shown that besides the similar total lymphocyte count in both females and males, the proportion of CD3+T lymphocytes amidst the population of total lymphocytes in males is lower in comparison with females [39]. Similarly, another study also reported a higher percentage of CD3+ and CD4+T cells in females than males, and, on the flip side, a higher fraction of CD8+T cells and NK cells in males than females (Figure 1) [40]. T treatment resulted in up-regulation of protein tyrosine phosphatase non-receptor type 1 (Ptpn1) expression, a phosphatase in CD4 T cells and thereby inhibiting IL-12 signaling culminating in the suppression of CD4 Th1 cell differentiation in both mice and humans [25]. Treatment of monocytes with physiological concentrations of T resulted in enhancement of proportion of IL-1β and IL-12 producing monocytes, which was comparable with the in vivo observation of elevated frequency of monocytes that secrete IL-1β and IL-12 in males when compared to females [41]. Increased splenocytic IL-10 secretion was observed in female mice after DHT administration. Furthermore in vitro studies on CD4+T cells showed more IL-10 secretion on treatment with DHT [24] confirming that T accounts for the increased Th2 cytokine production in males [42].
Females and males vary in their vulnerability to infections [43, 44]. Males of human as well as non-human animals are more prone to fungal, protozoan, viral, and bacterial infections than females [45]. This notion is additionally aided by the observation of greater severity of novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in males when compared to females [46, 47]. Furthermore, elevated cell-mediated and humoral immune responses to herpes simplex viruses and cytomegalovirus were shown in females. This difference in host resistance could be attributed to sex steroids based on the observation that, males of rodents maintained in a controlled laboratory setting, still, exhibited more susceptibility to infections than females [48, 49]. Host immunity plays an essential role in deciding resistance and predisposition to infections. Additionally, other internal and external factors are modulated by sex steroid hormones which may lead to increased infection in males in comparison with females. Sex steroids can modulate the expression of disease resistance genes encoding the major histocompatibility complex (MHC) molecules, TLR and B cells immunoglobulins [50]. Increased systemic levels of basal immunoglobulins, elevated B cell number and antibody response were observed in women than men [51–53]. Antigen presentation by antigen-presenting cells of female mice demonstrated more efficacy than that of male mice, by expressing high levels of class II MHC and other co-stimulatory molecules. Androgen treatment in female mice decreased, while castration of male mice enhanced this antigen presentation capability of immune cells, pointing to the hormonal influence on immunoreactivity among males and conspecific females [54]. TLR are pattern recognition receptors that detect pathogens and commence the natural and acquired immune reactions. High concentration of serum lipopolysaccharide (LPS)-binding protein (LBP) levels along with increased cell surface expression of TLR4 and CD4 in male mice derived macrophages, subsequent to LPS challenge, contribute largely to the severe endotoxic shock as compared to that of females. This might explain the observed sexual dimorphism in LPS-induced inflammatory reaction [55].
Infertility is the illness of the reproductive system that affects males or females and is defined by the difficulty to attain a pregnancy without using any contraception after 12 or more months [World Health Organization (WHO), International Classification of Diseases, 11th Revision (ICD-11), 2018] [56]. The female reproductive system is a highly regulated and harmonized network of the hypothalamic-pituitary-ovarian axis and hence any defect or diseases in the neuroendocrine system, reproductive tract and even immune system can culminate in female infertility and associated diseases [57]. Either activation of the immune system in general or specific immune reactions such as those against ovaries comprise the immune etiologies associated with infertility in females [58]. Additionally, several studies have described and reviews have comprehensively discussed the involvement of autoimmune nature with heightened release of autoantibodies in infertility conditions like premature ovarian failure (POF), endometriosis, PCOS, unexplained infertility, and recurrent implantation failure. Production of autoantibody against ovarian antigens, infiltration of inflammatory cells during follicular development and atrophy of primordial follicles can be explained as pathogenic mechanisms in POF [59, 60]. Increased levels of proinflammatory cytokines [61], enhanced production of antiphospholipid antibodies (APAs), antithyroid antibodies (ATAs), and anti ovarian antibodies (AOAs) [62–64], along with altered levels of CD4+T cells subsets with increased Th1/Th2 ratio and Th17 cells, and reduced Tregs, with a defect in the uterine T cell to Treg differentiation [65–67], also accounts for unexplained infertility and repeated in vitro fertilization (IVF) loss in females. Higher concentrations of cytokines and the presence of antibodies specific to endometrium as well as AOAs, APAs, smooth muscle autoantibodies (SMA) and antinuclear autoantibodies (ANA) in endometriosis suggests the involvement of immune system deregulation and inflammatory reactions in infertility related to endometriosis [68–72]. PCOS women are often presented with multiple autoantibodies and altered humoral and cell-mediated immune status, which accounts for the putative pathogenic immune mechanism involved [73].
The major clinical manifestation of PCOS is excess androgen in the circulatory system. Sixty percent to eighty percent of PCOS women have hyperandrogenism [74–76]. Rather than being considered as a mere clinical feature, hyperandrogenism appears to contribute to several reproductive, metabolic and immunologic irregularities in PCOS. Dehydroepiandrosterone sulphate (DHEAS), androstenedione (A4), dehydroepiandrosterone (DHEA), DHT, and T are major androgens in the female body. A4, DHEA, and DHEAS are regarded as pro-androgens, while T and DHT are active in the form that binds to the AR. Zona reticularis of adrenals is the site of DHEAS production. Fifty percent of DHEA is from zona reticularis, while 20% is from ovaries and 30% is by DHEAS peripheral conversion. The ovary and adrenal glands take part equally in A4 synthesis. T is produced from the ovaries and zona fasciculata of adrenal glands [77]. 5α-reduction of T leads to DHT synthesis in peripheral tissues like adipose tissue, skin, liver, and even peripheral blood mononuclear cells (PBMC) (Figure 2) [77, 78]. Interestingly, serum levels of androgen precursors are more than potent androgens in women, while bioactivity shows in reverse order (Figure 3) [74]. The liver is the site of steroid metabolization and the wastes are removed by the renal system. DHT can bind to ARs more efficiently than T, hence DHT shows 3 times more activity in target cells [79]. In the circulatory system, lion’s share of T and DHT is bound to transport protein while only 1% is free, which is the biologically active form [80]. The androgens have important physiological roles including, enhancement of bone growth, calcium deposition, muscle mass buildup, development of signs of puberty such as axillary and pubic hair growth, and maintenance of female sexual desire [81–83]. Serum levels of DHEAS, T, DHEA, and free T were known to be elevated in PCOS patients. Ovary thecal cells of PCOS women exhibited an upregulated messenger RNA (mRNA) level of steroidogenic acute regulatory (StAR), cytochrome P450 family 11 subfamily A member 1 (CYP11A), cytochrome P-450c17alpha (CYP17A1), and luteinizing hormone (LH) receptors when compared to normal cycling women [84]. PCOS women also have an increased free DHT level and T to DHT ratio [85]. Hence, an imbalance of androgen metabolism can cause deleterious effects in the female body.
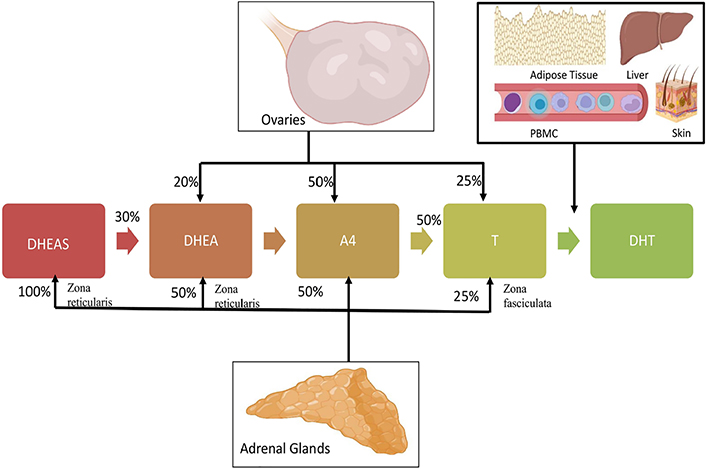
Relative contribution of adrenals and ovaries in circulating androgens in the female system
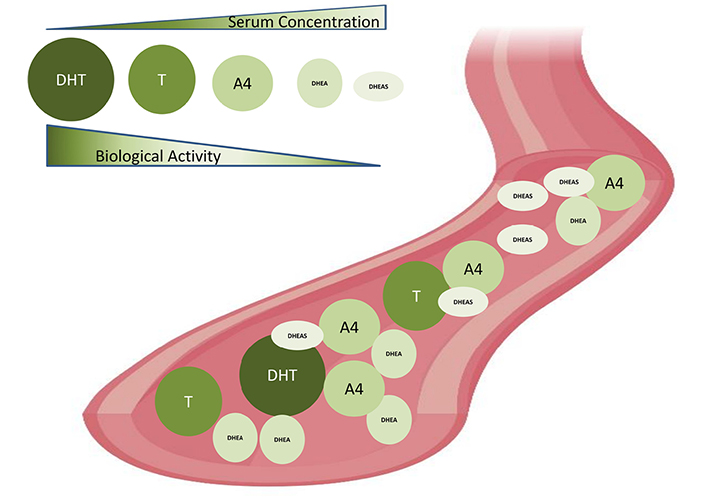
Biological activities and serum concentrations of androgens in women. Serum androgen concentration: DHT < T < A4 < DHEA < DHEAS. Biological activities: DHT > T > A4 > DHEA > DHEAS
An inherent aberration in the theca cells steroidogenic machinery might be accountable for the excess androgen synthesis in PCOS. Nelson et al. [86] reported that enhanced enzymatic activity of 3β-hydroxysteroid dehydrogenase (3βHSD), steroid 17 alpha-hydroxylase/17,20 lyase (P450c17), and 17βHSD, along with upregulated expression of CYP17A1 and CYP11A, culminates in increased synthesis of T, 17-hydroxyprogesterone (17-OHP), and progesterone in propagated theca cell of PCOS. Further research by the same group demonstrated that the main reason driving heightened T secretion in theca cells of PCOS is the increased activity of 3βHSD and P450c17 enzymes, resulting in augmented T precursors synthesis [87]. It has also been shown that isolated PCOS theca cells have enhanced promoter activity of CYP11A leading to increased mRNA expression [88]. Additionally, augmented transcript levels of CYP17A1, CYP11A, StAR, and LH receptors were also reported in PCOS theca cells [84]. The major signaling cascades regulating steroidogenesis like mitogen-activated protein kinase (MAPK) pathways [89], Wingless and int (Wnt) signaling [90] were also found to be defective in PCOS, imparting hyperandrogenism. Hyperandrogenism in PCOS exerts several cellular dysfunctions in PCOS. Specifically, it enhances apoptosis of granulosa cells [91] developmental arrest of preantral follicles [92], endoplasmic reticulum stress [93, 94], and mitochondrial dysfunction [95, 96]. In addition to this, AE has also been implicated in metabolic complications of PCOS such as obesity [97], T2DM [98, 99], and cardiovascular disease (CVD) [100, 101].
Follicle-stimulating hormone (FSH) and LH are the two pituitary gonadotropins secreted in a pulsatile manner in response to the gonadotropin-releasing hormone (GnRH) of GnRH neurons positioned in the hypothalamus [102]. Increased LH levels observed in PCOS are because of the additive effect of both increased basal hormone level as well as increased frequency in its pulsatile secretion [103–106]. Androgen production from theca cell of the ovary is regulated by LH which subsequently causes hyperandrogenism in PCOS patients [104]. In addition to this, polymorphism of LH/choriogonadotropin receptor (LHCGR), FSH receptor (FSHR) [107], and FSH subunit beta (FSHβ) [108] showed a positive association with PCOS predisposition.
PCOS women showed an elevated number of lymphocytes, white blood cells (WBC) and neutrophils, when compared with controls [109]. Another group could demonstrate an elevated CD4+T, total T lymphocytes and NK cell percentage as well as CD4/CD8 ratio in PCOS patients [110]. Increased leukocytes in women with PCOS correlated with CVD risk, but hormone levels were not shown as interpreters of leukocyte number by multiple linear regression analysis (Figure 1) [111]. A negative association of androgen levels and reduced count of activated Th cells and amount of IL-12 in follicular fluid (FF) of PCOS patients were also reported [112]. Increased WBC count showed an association with total T, A4, DHEAS concentrations in PCOS women [113] and an elevated number of NK cells was correlated with infertility in women with PCOS [110]. As yet, studies have reported the presence of ARs in peripheral blood cells including T and B cells, neutrophils, macrophages and monocytes [114]. Interestingly, androgen therapy emerges as a promising tool for leukemia [115]. These observations could explain the importance of androgens in the commencement and progression of leukocytosis and low-grade inflammation in PCOS. Additionally, a higher rate of H. pylori seropositivity, elevated antibody titer against Chlamydia infection [116] and increased severity of periodontitis [117] in PCOS women suggest a possible relationship between increased infection rate in PCOS subjects. Most importantly, the presence of IR, vitamin D deficiency, and increased ARs, make PCOS women more susceptible to the recent novel coronavirus disease [118]. A study conducted on a hyperandrogenic mouse model of PCOS demonstrated elevated expression of angiotensin-converting enzyme 2 (ACE2) protein, the receptor for SARS-CoV-2, in the heart, intestine, and kidney. Along with this, increased transcript levels of SARS-CoV-2 priming proteases like Cathepsin L Tmprss2, and Furin in the kidney of PCOS mice, suggest the possible risk of renal, gastrointestinal, and cardiac disorders in SARS-CoV-2 infected PCOS women [119]. Though, further studies on larger and more diverse populations are needed to envisage the mechanism and outcome of increased susceptibility to infection in PCOS.
It has been shown that serum IL-17F, IL-17A levels and IL-17A/F ratio which is predominantly pro-inflammatory were elevated, while IL-17E which has an anti-inflammatory activity reduced in the non-obese PCOS group [120]. Nasri et al. [121] reported an increased Th1/Th2 ratio and Th17/Th2 ratio in their follicular phase in the PCOS group in comparison with controls (Figure 1). Another study conducted by Krishna et al. [122] demonstrated a reduced number of Tregs in PBMCs of women with PCOS. They also reported that expansion of Treg in PCOS subjects was significantly reduced due to defective IL-2 signaling. Interestingly reduced percentage of CD8+T cells, and the levels of CD69 and IFN-γ were demonstrated in the FF of the PCOS group in comparison with control subjects. Elevated expression of programmed cell death protein 1 (PD-1) in both CD4+/CD8+T cells of FF showed a positive association with serum estradiol levels [123]. Cytokine analysis in pregnant PCOS women showed notable participation of tumour necrosis factor α (TNF-α) and IL-8 in all terms and IL-2, C-reactive protein (CRP), IL-1β, IL-12, and IL-6, in initial pregnancy stages, strongly points to the pregnancy complications observed in PCOS women [124]. Several studies conducted in different ethnic population have reported the involvement of pro-inflammatory cytokine in the immunopathogenic condition of PCOS. This was demonstrated by increased blood levels of various cytokines including chemokine (C-X-C motif) ligand 5 (CXCL5), TNF-α, IL-1 β, IL-6, CRP, IL-8, IL-1α, IL-18, CD163 and matrix metalloproteinase 9 (MMP9) in women with PCOS [125–127]. Increased mRNA and protein levels of TNF-α were observed in FF of PCOS subjects with metabolic syndrome [128]. TNF-α acts as a powerful enhancer of ovarian activity by enhancing the proliferation of steroidogenically active theca-interstitial cells and thus causative to AE, creating an additional level of the labyrinth and self-perpetuating nature of PCOS [129], while in vitro study conducted in bovine thecal cells showed suppressed androgen secretion on treatment with IL-6 and TNF-α [130]. Moreover, increased serum TNF-α was associated with IR and concentration of androgens in PCOS patients [131]. It has been observed that overexpression of muscle and adipose tissue TNF-α decreases the insulin receptor tyrosine kinase activity further aiding the IR development [132–134]. TNF-α contributes to follicular atresia and anovulation by promoting apoptosis in the rat ovary [135]. T treatment in rats induced heightened TNF-α production, which resulted in elevated ovarian A4 and decreased estradiol secretion [136]. Increased levels of TNF-α in FF of PCOS subjects showed a negative correlation with that of estradiol [137]. Thus elevated TNF levels would account for the perpetuation of PCOS by enhancing androgen production [138], IR [139], poor oocyte quality [140, 141] and defective folliculogenesis [128]. Higher oxidative stress is tightly associated with inflammation in PCOS which induced androgenesis in the ovary [142]. Peng et al. [143] showed that increased blood IL-6 levels were linked to IR and androgen levels rather than body mass index (BMI) in PCOS subjects. An increase in adrenal steroidogenesis was observed on exposure to IL-6 on human adrenal cells in vitro [144]. Additionally, hyperandrogenism in PCOS women intensifies the proatherogenic risk by altering the levels of inflammatory mediators, lipid profile, blood pressure and IR. The plasma concentration of monocyte chemoattractant protein-1 (MCP-1) was positively related to that of AR and intracellular mononuclear cells nuclear factor kappa B (NFκB) levels in women with PCOS [145]. This was supported by the observation of augmented reactive oxygen species production and NFκB activation following oral glucose intake in PCOS women, which was not dependent on obesity but on androgen levels [146, 147]. In addition to IR, PCOS is also linked with inflammation, oxidative stress and mitochondrial dysfunction [142, 148]. Blood levels of cytokines and CRP showed association with IR in PCOS patients [149], indicating the involvement of hyperandrogenism, IR, and obesity either individually or in the group, in systemic inflammation observed in PCOS. Excess androgens can be the progenitor of diet-induced inflammatory response in PCOS by enhancing the activation and sensitivity of mononuclear cells to glucose intake [150]. Moreover, pro-inflammatory mediators could enhance ovarian androgen synthesis by upregulating the expression of steroidogenesis-related genes in thecal cells in vitro [129, 151, 152] and thus supporting the argument that inflammation has direct action in the polycystic ovary, stimulating the production of excess androgens.
Another link between inflammation and hyperandrogenism in PCOS relies on adipocyte dysfunction, in which excess androgens induce hypertrophy of adipocytes, and chronic low-grade inflammation in effect promotes androgen production by cystic ovaries [153, 154]. Obesity causes impairment of adipose tissue function culminating in hypoxia, adipocyte hypertrophy, adipocyte inflammation, IR and associated metabolic difficulties [155]. Intriguingly, adipose tissue dysfunction has a robust impact on immunity and the immune system, which includes the disturbance of the architecture of lymphoid tissue, disruption of leukocyte expansion, activity and phenotypic distribution, loss of harmonization of natural and acquired immune reactions, thus resulting in a greater occurrence of chronic disease, increased predisposition to various infection infections, and vaccine inefficiency [156–158]. The proposed role of adiposity in chronic inflammation in PCOS is depicted in Figure 4. Hypertrophy of adipocytes results in hypoperfusion by constriction of blood vessels culminating in hypoxia and associated activation of NFκB. Activated NFκB stimulates adipose tissue macrophages recruitment by the release of inflammatory mediators like transforming growth factor-beta, IL-18, MCP-1, IL-1β, IFN-γ, IL-6, TNF-α, and IL-10, complement system factors, and soluble vascular cell adhesion molecule (sVCAM), sustaining the inflammatory condition, damaging adipocyte function, and triggering autolysis of the cell [159]. Hence hypertrophic fat cells are susceptible to increased cell death, fibrosis, inflammation, lipolysis and free fatty acids release [135]. The cytokines such as IL-6 and IL-1β released from foamy cell macrophages execute a crucial role in the pathology of atherosclerosis by enhancing liver CRP release and promoting lipid uptake in atherosclerotic plaques [160]. A genomic microarray study conducted in adipose tissue of PCOS women identified the involvement of insulin signaling, inflammation, Wnt signaling, immune function, oxidative stress and lipid metabolism suggesting the role of abdominal obesity in the pathophysiology of PCOS [161]. Visceral adiposity in hyperandrogenic women could be attributed to the involvement of androgens in pre-adipocyte differentiation, particularly in abdominal fat [162]. A recent report by Juan et al. [163] on the elevated expression of C-C chemokine receptor type 5 (CCR5) in adipocytes and peripheral blood cells and its association with AE and hyperinsulinemia in PCOS patients points to the role of chronic inflammation in adipose tissue dysfunction and AE of PCOS, aiding to the augmented metabolic aberrations manifested in this most common disorder of female population. There is ample evidence that obesity intensifies androgen production, an event characteristic of the associated hyperinsulinemia, which also includes the involvement of the adipose-derived hormones called adipokines [154]. Of note, contradictory reports on the levels and association of adipokines with sex steroids were also reported. Major adipokines and their observation in PCOS were briefed in Table 1. Adiponectin is one of the adipokines that have a role in insulin sensitivity, and inflammatory action, and also modulates ovarian follicular proliferation and steroidogenesis [164, 165]. Upregulation in the expression of adiponectin receptors was observed in adipocytes with androgen treatment [166]. Decreased serum adiponectin [167] and lower thecal receptors observed in PCOS women [168] suggest another link between chronic inflammation and hyperandrogenism in PCOS. Visfatin has insulin-mimetic action and has been linked to endothelial dysfunction in PCOS women [169]. Increased concentration of plasma visfatin in PCOS women correlated positively with T, and LH levels [170, 171]. Resistin is the adipokine that can hinder insulin action and has a potential linkage between diabetes and obesity [172]. Elevated levels of resistin showed a correlation with BMI and total T in women with PCOS. Moreover, exposure to resistin in combination with either forskolin or insulin rather than resistin alone, induced 17α-hydroxylase activity in human ovarian thecal cells, pointing to the synergic involvement of resistin and insulin in the propagation of ovarian hyperandrogenism in PCOS [173]. On contrary, another study reported no change in serum resistin levels in PCOS subjects in comparison with controls. Yet, they could demonstrate an upregulation of adipocytic resistin transcript levels in PCOS women [174]. Leptin is another adipokine found to be elevated in PCOS subjects. Leptin has a regulatory role in body mass, food intake, reproductive function, pro-inflammatory immune reaction, and lipolysis [175]. Estrogen has been implicated in the regulation of leptin levels in both animals and humans [176]. Vaspin is a serine protease inhibitor released by abdominal adipose tissue and has a role in insulin sensitivity. Adipose tissue transcript levels, as well as serum protein concentration of vaspin, were found to be elevated in women with PCOS when compared to control subjects [177, 178] while conflicting studies show decreased or unchanged serum vaspin concentrations [179, 180]. Increased levels of vaspin and its receptor: glucose regulated protein (GRP78) in FF and granulosa cells of PCOS women were found to be associated with steroidogenesis, propagation and viability of granulosa cells [181].
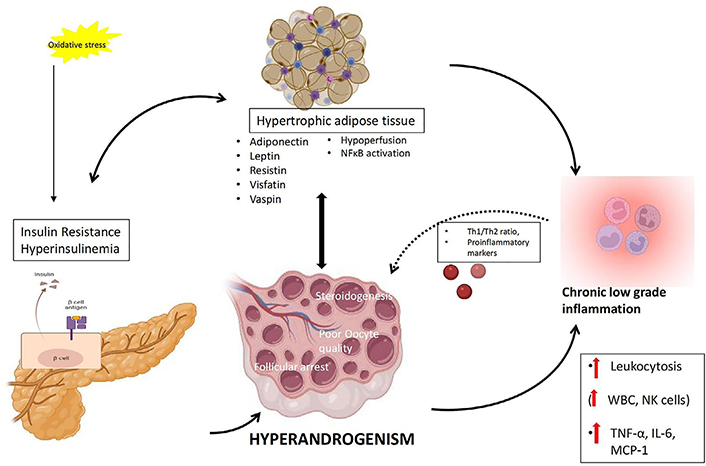
Proposed mechanism of interconnection between adiposity, hyperinsulinemia, chronic low-grade inflammation and hyperandrogenemia in PCOS
Adipokines and observations in PCOS
| Adipokine | Action | Serum levels in PCOS | References |
|---|---|---|---|
| Adiponectin | Insulin sensitivity, inflammatory reaction, ovarian folliculogenesis | Decreased in PCOS | [164, 165, 167, 168] |
| Visfatin | Insulin mimetic action | Increased in PCOS | [169–171] |
| Resistin | Resist insulin action | Increased/unchanged in PCOS | [172–174] |
| Leptin | Regulatory role in body mass, food intake, reproductive function, pro-inflammatory immune reaction, and lipolysis | Increased/unchanged in PCOS | [176, 195] |
| Vaspin | Insulin sensitivity | Increased/decreased/unchanged in PCOS | [177–181] |
Even though chronic AE and hyperinsulinemia attribute to the pathogenesis of this disorder, the real players in the etiology of PCOS are still under debate. Recent studies try to delineate the immune mechanism in PCOS and propose that the disease could be an autoimmune disorder [182]. Studies have also demonstrated the presence of inflammatory markers and autoantibodies in PCOS [183]. Reduced Tregs and altered sex hormone levels can be other causes of the autoimmune etiology of PCOS. Studies have already proposed the role of estrogen in the development of autoimmune diseases in females [184, 185]. The frequency of systemic lupus erythematosus (SLE) incidence is higher in women than in men [186]. Men with Klinefelter syndrome, having high estrogen, also have a greater occurrence of autoimmune diseases like SLE and rheumatoid arthritis (RA) [187]. High estrogen can induce Th cells for secretion of type 2 cytokine thus stimulating Th2 immune response characterized by increased antibody production [185] and inhibiting apoptosis of autoreactive B cells [188]. Additionally, estrogen reduces the immature T lymphocytes count [189] and enhances the inflammatory process by stimulating the release of ILs like IL-6, IL-10, IL-1 and IL-4 [9, 190, 191] and the synthesis of immunoglobulin M (IgM) and IgG immunoglobulins [190]. Estrogen is involved in the induction of Treg pool expansion and forkhead box P3 (FOXP3) expression in mice [192]. Moreover, in normal cycling women, Tregs showed an expansion in accordance with estrogen level during the follicular phase of the menstrual cycle [193]. This is supported by the finding that female mice exposed to estrogen demonstrated defective natural Treg production and later development of PCOS symptoms [194]. These diverse estrogen actions in the immune system oppose the protective role of elevated T against autoimmune reactions in PCOS.
Being complicated in its pathogenesis and etiology, PCOS remains chaotic to the scientific community. Chronic low-grade inflammation and hyperandrogenism interact and intensify each other, making this syndrome more complicated. Additionally, this might also account for the vulnerability of PCOS women to autoimmune diseases. Although studies are elucidating the role of androgens in the commencement and progression of low-grade inflammation in PCOS, controversial reports show the involvement of inflammatory mediators in the hyperandrogenic etiology of PCOS. Thus, further studies are warranted to clarify the hormonal and inflammatory knot of PCOS by assessing whether altered sex steroid status is the key culprit for immune aetiology in PCOS. Further research needs to be conducted on the altered immune status in PCOS to prove whether a defective immune response could perform a vital role in PCOS pathogenesis.
3βHSD: 3β-hydroxysteroid dehydrogenase
A4: androstenedione
AE: androgen excess
AR: androgen receptor
CRP: C-reactive protein
CYP11A: cytochrome P450 family 11 subfamily A member 1
CYP17A1: cytochrome P-450c17alpha
DHEA: dehydroepiandrosterone
DHEAS: dehydroepiandrosterone sulphate
DHT: dihydrotestosterone
ER: estrogen receptor
FF: follicular fluid
FSH: follicle-stimulating hormone
IFN-γ: interferon-gamma
IL: interleukin
IR: insulin resistance
LH: luteinizing hormone
LPS: lipopolysaccharide
MCP-1: monocyte chemoattractant protein-1
mRNA: messenger RNA
NFκB: nuclear factor kappa B
NK: natural killer
PBMC: peripheral blood mononuclear cells
PCOS: polycystic ovary syndrome
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2
T: testosterone
Th2: T helper 2
TLR: toll like receptors
TNF-α: tumour necrosis factor α
Tregs: regulatory T cells
WBC: white blood cells
ML contributed conception and design of the study, reviewing and editing; BSJ wrote the first draft of the manuscript and analyzed the literature. All authors contributed to manuscript revision, read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
ML is supported by Department of Biotechnology (DBT)-Rajiv Gandhi Centre for Biotechnology. BSJ is supported by a Senior Research Fellowship from ICMR [RBMH/FW/2020/2]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Gustaaf Albert Dekker, Pierre Yves Robillard
Kushaan Khambata ... Satish K. Gupta
Gursaran P. Talwar ... Krishna M. Ella
Julia Szekeres-Bartho
Shigeru Saito ... Sayaka Tsuda
Noémie Abisror ... Arsene Mekinian
Ruchi Sachdeva, Rahul Pal
Shibin Cheng ... Surendra Sharma
Alison McCallion ... Chandrakant Tayade
Pier Luigi Meroni ... Francesco Tedesco
Marijke M. Faas
Alaa Kazhalawi ... Nathalie Lédée
Chiara Agostinis ... Roberta Bulla
Mickey V. Patel ... Charles R. Wira
Thanh Luu ... Joanne Kwak-Kim
