Original Article
Original Article

Affiliation:
1Laboratoire des Sciences du Numérique de Nantes (LS2N), Inria, IMT Atlantique, F-44307 Nantes, France
2Centre de Recherche en Transplantation et Immunologie UMR1064, INSERM, Université de Nantes, CHU Nantes, ITUN, F-44000 Nantes, France
ORCID: https://orcid.org/0000-0002-2279-1435
Affiliation:
2Centre de Recherche en Transplantation et Immunologie UMR1064, INSERM, Université de Nantes, CHU Nantes, ITUN, F-44000 Nantes, France
ORCID: https://orcid.org/0000-0002-6762-4083
Affiliation:
2Centre de Recherche en Transplantation et Immunologie UMR1064, INSERM, Université de Nantes, CHU Nantes, ITUN, F-44000 Nantes, France
ORCID: https://orcid.org/0000-0002-3767-6210
Affiliation:
1Laboratoire des Sciences du Numérique de Nantes (LS2N), Inria, IMT Atlantique, F-44307 Nantes, France
Email: mario.sudholt@imt-atlantique.fr
ORCID: https://orcid.org/0000-0002-1855-1519
Affiliation:
2Centre de Recherche en Transplantation et Immunologie UMR1064, INSERM, Université de Nantes, CHU Nantes, ITUN, F-44000 Nantes, France
Email: pierre-antoine.gourraud@univ-nantes.fr
ORCID: https://orcid.org/0000-0003-1131-9554
Explor Immunol. 2022;2:749–759 DOI: https://doi.org/10.37349/ei.2022.00080
Received: May 23, 2022 Accepted: August 05, 2022 Published: November 01, 2022
Academic Editor: Narinder K. Mehra, Indian Council of Medical Research, India
Aim: Human leukocyte antigen (HLA) population genetics has been a historical field centralizing data resource. HLA genetics databases typically facilitate access to frequencies of allele, haplotype, and genotype format information. Among many resources, the Allele Frequency Net Database (AFND) is a typical centralized repository that allows users to research and analyze immune gene frequencies in different populations around the world. With the massive increase in medical data and the strengthening of data governance laws, the proposal for a new distributed and secure model for the historical centralization method in population genetics has become important. In this paper, a new model of HLA population genetic resources, an alternative distributed version of HLA databases has been developed. It allows users to perform the same research and analysis with other remote sites without sharing their original data and monitoring data access.
Methods: This new version uses the Master/Worker distributed model and offers distributed algorithms for the calculation of allelic frequencies, haplotypic frequencies and for individual genotypic calculations. The new model was evaluated on a distributed testbed for experiment-driven research Grid’5000 and has obtained good results of accuracy and execution time compared to the original centralized scheme used by researchers.
Results: The results show that distributed algorithm applied to HLA population genetics resources enables usage control and enables enforcing the security framework of the data-owning institution. It gives the same results for all counting methods in population immunogenetics. With the same frequencies’ estimations, it yields a much quicker computation time in many cases, in particular for large samples.
Conclusions: Distributing previously centralized resources is an interesting perspective enhancing better control of data sharing.
With the massive increase in the amount of biomedical data nowadays, the extraction of information for the improvement of clinical decision-making requires the development of new analysis techniques. The ever-larger volume of patient data makes providing effective and safe treatment decisions a great challenge for clinicians. This is the case, in particular, with human leukocyte antigen (HLA) analyses where the number of volunteer donors and HLA alleles to be considered increases exponentially. An allele is an alternative version of the same gene, which is distinguished by variations in its nucleotide sequence. HLA alleles are defined by DNA sequencing and associated with a name to identify their sequence. For each HLA gene, each individual has a maximum of 2 alleles per locus, one from its father and one from its mother. Frequencies of HLA allele in populations is very important information for both research and clinical applications. One of the largest databases of HLA phenotypes by the World Marrow Donor Association (WMDA) includes more than 38 million registered donors and cord blood units from 55 different countries [1]. This centralized “book” [Bone Marrow Donors Worldwide (BMDW)] is used for a clinical application: the search of suitable sources of unrelated hematopoietic stem cells. BMDW represents continuing efforts to collect HLA phenotypes of volunteer stem cell donors and cord blood units in a centralized and controlled way which could be easily distributed transferring the access control to the data owner and facilitating the update operations. Another large system, the centralized Immuno Polymorphism Database (IPD) was developed for the study of polymorphism in genes of the immune system, mostly describing the diversity of the polymorphisms rather than their frequencies in human populations. It currently contains over 30,000 HLA alleles as part of the IPD-IMGT/HLA database [2].
All of these databases provide a dictionary of HLA alleles and their frequencies in different populations. The interest of collecting all these data from different populations is to know globally whether people from a given population have HLA of certain frequencies. This helps a lot with histocompatibility problems as it allows to know if it is likely to find a donor matching the HLA of a recipient. The collection of all these data from several countries, in particular, is not easy because of the sensitivity and confidentiality of such data. In pharmacogenetic research and clinical practice there has been increasing interest in understanding the global distributions of HLA alleles for risk profiling. Indeed, HLA alleles are associated with various auto-immune and infectious diseases, such as multiple sclerosis [3]. The data of healthy populations in the Allele Frequency Net Database (AFND) can be of great help to evaluate those frequencies [4]. AFND [5] is a freely available resource for the storage of frequency data on polymorphisms of several immunity-related genes, including the HLA, killer immunoglobulin-like receptors (KIRs), Major histocompability complex (MHC) class I polypeptide-related sequence (MIC), and several cytokine gene polymorphisms [e.g., the interleukin 4 (IL4), transforming growth factor-beta 1(TGFB1), tumor necrosis factor (TNF)]. These loci, known to be among the most polymorphic regions in the human genome, play an important role in the immune system response. HLA population data from AFND, also often underpin anthropological studies, as well as in-silico analyses for vaccine development based on epitope prediction, among many other applications. The AFND has solved the problem of HLA frequencies access. Technically all the databases have always seen centralization as the only possibility, we here propose a different data storage and data query architecture, an architecture that is distributed.
The centralized architecture of the database has facilitated the collection of data from different populations in a common database: clinicians and researchers have been able to access a large set of data in order to broaden their analyses. Historically, the centralized data infrastructure of the AFND platform has shown its robustness for large-scale analyses. With the strengthening of data governance laws over the past years, distributed data infrastructures have gained in popularity. This is due to data security and confidentiality guarantees that must be ensured to minimize the risk of privacy breaches for research participants and the possibility of controlling the use (who accesses? what to do with?) of the distributed infrastructures.
We propose a distributed model working on different levels in the AFND framework, allowing secure distributed analysis between sites without sharing confidential data between them.
In this paper, we apply database and computation distribution to HLA datasets, providing an alternative to the historical centralization method in AFND. We also describe the different levels of analysis on centralized AFND and the challenges of a distributed analysis. It also presents the distributed model proposed for distributed AFND.
The AFND was set up in 2003 with few sections and frequencies of HLA alleles/allelic lineages. It has been enriched over the years by new tools integrated into a new major version in 2015 [6, 7]. In 2020, another update was carried out on the available datasets that enabled the submission and sharing of data using a data quality classification criterion (GSB: gold, silver, bronze) [5].
The AFND has been accessed by over 100,000 different users from 186 countries over the past few years. This reference contains information from more than 10 million healthy individuals from more than 1,600 populations which allow for the analysis of the most polymorphic regions of the human genome. These demographic data come from 141 countries all over the world with varied population coverage. With these data, users can perform analyses on allele frequencies, genes, genotypes, or haplotypes for HLA, KIR, MIC, and cytokines. A haplotype is a set of alleles of the same chromosome. The genes, being very close to each other on the chromosome, few recombination events, therefore, take place and these genes are transmitted as a “block”, which is called a haplotype. A genotype is always the result of a combination of 2 haplotypes (a haplotype inherited from each parent). A genotype corresponds to all the alleles of a locus (or gene) of an individual. There are billions of possible genotypic combinations of alleles of each HLA genes.
Data from the AFND site stems from (1) peer-reviewed publications, (2) population data from international HLA and immunogenetics workshops (IHWS), (3) submissions by laboratories around the world and, (4) short publication reports (SPR) in collaboration with the journal Human Immunology.
For data submission, AFND imposes some requirements such as allele name validation according to the official IPD-IMGT/HLA and IPD-KIR nomenclature guidelines; the homogenization of the population denomination; the attribution of the geographical region to which belongs the population; and frequency data validation by AFND’s SPR submission upload tool.
The AFND is available for storing frequency data on polymorphisms of several immunity genes. This resource provides an idea of the global distributions of HLA alleles. Users integrate the results of their work into a common (centralized) database and search the database for information already available. This repository currently contains data in allele, haplotype and genotype formats. Most of the data managed are tables of allele frequencies for a gene. These are evaluated by counting of individual genotyping data or by marginal summation on the haplotype frequency table.
As a major data repository, AFND relies on centralization of the data resources, that is, the actual transfer of aggregated data (allele or haplotype frequencies), rather than the original individual HLA genotype data. These three analytical levels are subject of various challenges in data governance; as presented in Table 1. The distributed version of AFND we propose addresses these 3 challenges.
Different levels of AFND and challenges for distributed AFND
| Analytical level | AFND | Challenges for data governance | ||
|---|---|---|---|---|
| Privacy risk | Usage control | Computational requirement | ||
| 1 | Allele frequency | X | ||
| 2 | Haplotype frequency | X | X | |
| 3 | Individual genotypic data | X | X | X |
Each AFND level tackles one or multiple challenges illustrated here by an 'X'. In order to respect confidentiality due to security-sensitive patient data and avoid the contractual agreement of data transfers between the data-owning institution and the data-hosting institution, individual data are often confined to data-owning institution. In addition, centralization implies a transfer of the privacy breach, a risk of data loss and an implicit loss of usage control. Risk of re-identification is not addressed by distribution
Currently, all allele frequency data are stored in a common (central) database. This database contains all alleles and the corresponding allele frequencies of several populations. To visualize or perform analyses on these data, AFND offers users a query tool to explore these allele frequencies in one or more populations based on given criteria.
Our distributed version of this level offers the same tools as AFND (classic/centralized) to users to explore allele frequencies. In addition, it allows centers to keep their data at their original sites without the need to submit or share them and preserving the knowledge who is using their data.
Regarding the same challenge as first-level allele frequencies, our new distributed version for analyzing haplotype frequencies allows users to consult a particular haplotype in a set of populations with two or more loci like the centralized case. At this second level, our approach enables performing distributed analyses without requiring the centers to share their data in a central database. Our new version of distributed AFND offers not only the control of who does what with the data, but also to perform calculations locally and simply send aggregated results to the server, which performs a global calculation of the haplotype frequencies at the end.
Finally, at the most sensitive level, the current version of AFND collects individual genotype data via population reports. The user at this level can view genotype frequencies from a given profile. Raising awareness of the sensitivity of population data motivates us to offer our new distributed version of AFND. Our method does not need to share individual genotype data. It allows users to perform allele frequency calculations at the sites of data providers and just share these frequencies with the server which then performs the global calculation. At this level, our new approach allows data providers to know who is doing what with their data, to perform calculations locally, to not overload the server by calculations and guarantee the confidentiality of individual patient data.
Currently, all sites participating in AFND send their allelic frequencies, haplotypic frequencies and genotypic data to the servers. Users who want to analyze data send their requests to a platform which is linked to the central server which hosts all the data. The server executes the query over the submitted data and presents the resulting data set based on well-determined criteria (see Figure 1a).
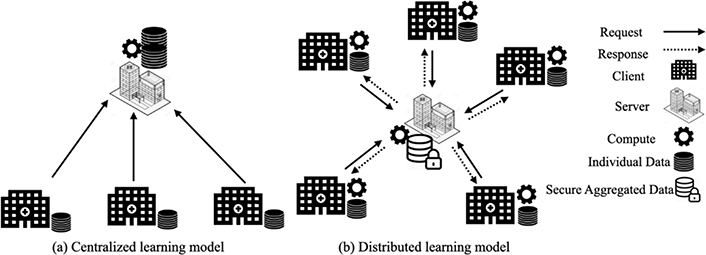
Centralized vs distributed data model applied to AFND (partially based on [8]).This figure shows two different models of operation of the AFND.net site calculations. Model (a) shows the current centralized calculation model, where the server collects all data from participating sites in the calculation and performs collaborative calculations on the server in a centralized manner. Model (b) presents our new distributed model proposed for AFND.net which allows each site participating in the collaborative analysis to perform the calculations locally without moving its data to the server. After that, they send calculation results that allow the server to just safely perform aggregate calculations and generate the result
In order to support large-scale distributed analyses and ensure data governance, several distributed Information Technology (IT) models/architectures can be harnessed that allow analyses to be distributed without sharing data between sites. The use of such distributed infrastructures is a central element of multi-stakeholder data governance.
We strive to meet the challenge of enabling data mining while keeping sensitive data on-premises and ensuring strong data protection when data moves. Our approach is based on the architecture Master/Worker (see Figure 1b). In this model, calculations are performed on distributed (client) sites linked to a calculation aggregator (server) that allows a site to interact and access certain data elements from remote sites. Each center collects, stores, analyses and controls the data of its own patients. When a user sends a request to the server, the server sends appropriate requests to the concerned sites so that they can carry out their calculations locally. The sites respond with the results of calculations or requested parameters. Then the server collects these results, performs the aggregation locally, and provides the result to the user.
The founding principle of our architecture is that no individual data circulates outside the centers (sites). However, this sharing paradigm offers the possibility of locally controlling who accesses data, what are the uses of these data and also guarantee the data confidentiality.
The allele frequency y of an A allele is defined in the centralized case as follows:
where N is the number of individuals and g are the genotypes of individuals.For the distributed case, the allele frequency is calculated as follows:
where S is the number of sites and ni is the number of individuals on each site.To test the different levels of our distributed genetic computations, we used a dataset of 1,000 samples (alleles) that are available from the allele frequency.net website. This HLA data stems from the population of “Northern Ireland”. They were submitted at the end of 2019 [9]. In order to perform large-scale analyses (and measure accuracy and execution time) to assess the stability of our new distributed model, we also used another public HLA dataset [10] which contains 2,504 individuals with 16,979 allelic frequencies per 1,000 genomes.
We used the Grid’5000 platform to carry out our experiments on a distributed environment. Grid’5000 [11] is a European platform, involving clusters at eight different sites, for research in the field of large-scale distributed systems and high-performance computing.
For our experimentation, we booked a machine on a server running a Python program to manage analysis, client interactions, and generation of the result. We have also booked machines distributed over different sites to function as client machines for our model. These machines contain the client data to carry out the experiments of our distributed algorithm.
Our model and our algorithms are available on the following Gitlab site [12].
To evaluate our proposed approach, we carried out experiments for each level. The Figure 2 shows an example of level 3 distribution of individual genotypic data by showing the genotypic data at each site, the distribution model, and the resulting allele frequency data. The figure shows the distribution scenario of HLA-B alleles. Instead of sharing data with the server, clients perform calculations on their data locally and then distribute calculation results that allow them to aggregate results at the server side without accessing local data of each site.
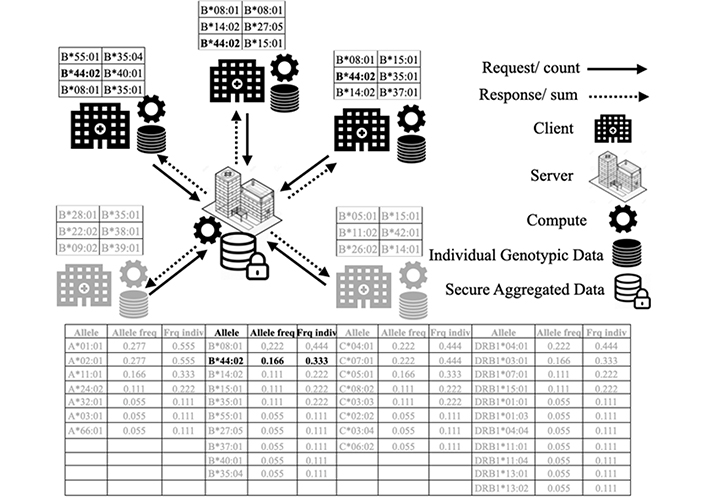
Distributed individual genotypic data example of HLA-B*44:02. This figure shows a scenario of distributed computation of individual genotypic data of HLA-B alleles. This represents a request of distributed computing of the HLA-B*44:02 allele among the different data sites. Hospital sites and their data in black show sites that contain the HLA-B*44:02 allele. Hospital sites and data in gray do not contain the relevant allele for the calculation. The table shows the results of a distributed calculation of all the alleles in gray and the results of HLA-B*44:02 allele which concerns our query in black. This analysis is performed in a distributed manner without moving data from their sites. A user of a site sends a count request on an affected allele. The server performs a summation on the allele frequencies received from the other sites and broadcasts the result at the end
The principle shown in Figure 2 applies to all alleles of all loci for allele frequencies (level 1) and haplotype frequencies (level 2) as well as genotypes frequencies (level 3).
We have also compared the classical centralized calculation model of AFND against our new distributed model with regard to the precision of the results and different performance criteria.
We used the accuracy as a first comparison criterion between the centralized and distributed cases. The Figure 3 is a diagram showing a linear regression to compare the allele frequency estimates for several cases of the centralized and distributed methods. Each point represents an experiment of an allele. This figure is a visualization of method 2 (distributed case) as a function of method 1 (centralized case). As expected, there is no dispersion of the data, the values are concordant, we obtain the same values for the two methods with a correlation coefficient r2 = 1.
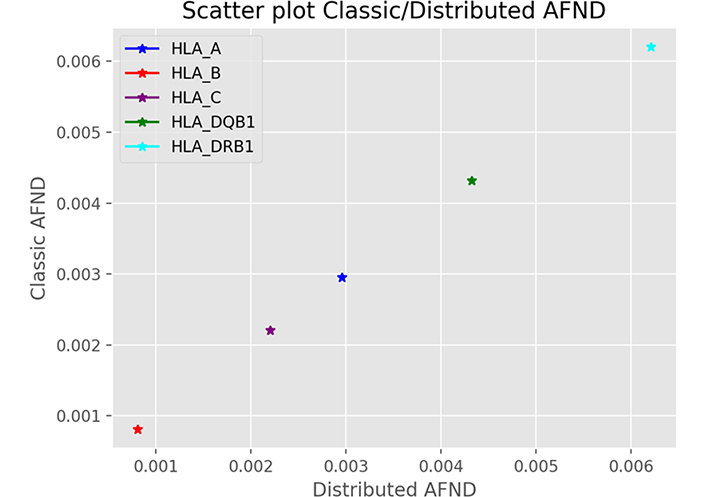
Accuracy of allele frequency estimates. This figure shows the correlation result of an allelic frequency query of 5 alleles taken as example for HLA-A, HLA-B, HLA-C, HLA-DQB1 and HLA-DRB1 genes between the centralized case and the distributed case. This is representative of all alleles with correlation coefficient r2 = 1
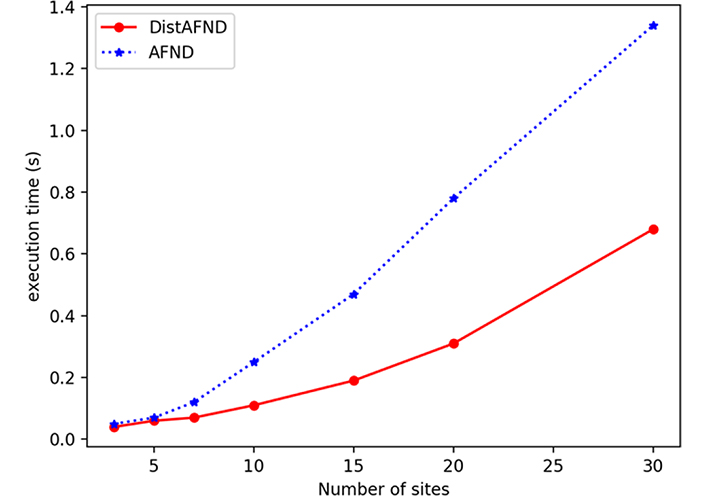
We have then evaluated the execution time per number of sites. The Figure 4 shows the calculation of execution time as a function of the number of sites. Each point presents a test for the same number of sites for both centralized and distributed cases. This figure shows that the more the number of sites is increased, the computation of the allelic frequency is faster in the distributed case. This is due to the fact that we increase the number of sites participating in the calculation for the same size of global data, the calculation will be carried out more quickly. By distributing the calculations on several sites allows to perform calculations on the part of the data on each site separately with an aggregation of the results of the calculations on the server at the end. These speed up the execution time.
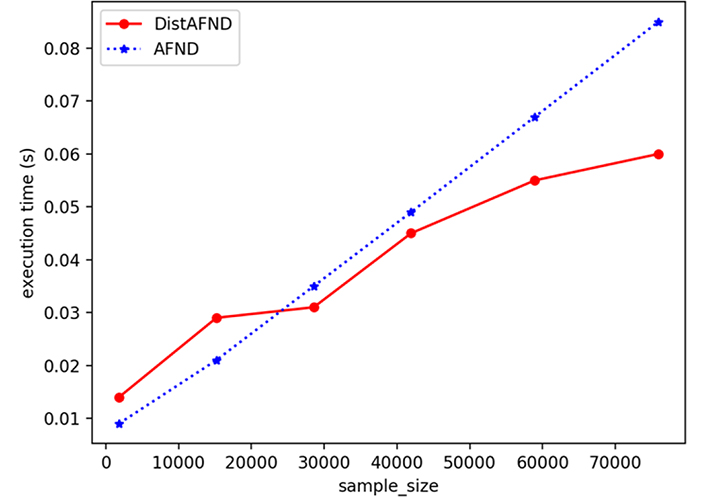
We have also evaluated the execution time according to sample size in the distributed and centralized cases. Each point presents an experiment for both cases for a well-defined sample size. The Figure 5 shows that the estimation of the allelic frequency becomes faster in the distributed case after a certain sample size (30,000 in our case).
Distributed HLA population genetic database, as we proposed it, is an alternative version of the historical centralized version including AFND. It offers distributed analysis among different populations at different levels of analysis of the historical centralized method.
In this article we have proposed this new distribution of HLA database and analysis (1) in order to support stronger security and confidentiality properties of sensitive patient data, (2) to satisfy the various international regulations of data governance [the General Data Protection Regulation (GDPR) and the Health Insurance Portability and Accountability Act (HIPAA)], (3) to enable usage control and (4) to ensure a large-scale distributed analysis without having to move data from their original locations.
Our approach provides a distributed version of the first three levels of HLA analyses such as the distributed calculation of allelic frequencies, the distributed calculation of haplotypic frequencies and the distribution of analyses on individual genotypic data. Our evaluation has shown that our approach yields equivalent accuracy compared to the centralized AFND as well as better performance in terms of execution time relative to increasing sample size and number of sites.
This new secure and distributed model of sensitive data and analyses brings real benefits to the world of precision medicine as it enables precise large-scale calculations without sharing sensitive patient data especially with the emergence of massive data around the world. Distributed calculations have been considered a very important means in IT and this is confirmed by the minimal execution time of our work compared to centralized AFND. Our new model of distribution of HLA analyses and databases offers a great contribution in the biomedical field because of its support of secure and distributed calculations applied to the search for allelic frequencies of HLA data for collaborations between several organizations in the world without having to share HLA data using a single database along with the control of the use of the data it provides (users and data providers know who does what with their data).
Our new distributed HLA population genetics database applies to many populations genetics resource, in particular AFND solution. We used the distributed model Master/Worker and aggregation as a process of synthesizing data in order to facilitate statistical analysis. With this method, we are able to disseminate and exploit aggregate information (information about all of the patients or specific patient groups that we combine so that an individual patient can no longer be identified or mentioned).
The exponential growth of electronic health data has created a huge data governance challenge. Managing an increasing volume of data and merging heterogeneous data sets are complex issues. In Europe, health data protection policies that impose well-founded but strong restrictions on data sharing were defined in 2016 by the GDPR [13]. In the United States, the HIPAA covers the security and privacy of medical information or, in HIPAA’s parlance, protected health information (PHI). By law, “covered entities”, primarily hospitals, policyholders, and organizations that process PHI for them, have a legal responsibility to ensure the protection of PHI [14].
Data governance provides healthcare facilities with a method for sharing medical data that is both standardized and structured, in order to deliver the highest quality care to each patient. Distributed infrastructure, collaborative analysis and data sharing has the potential to revolutionize the healthcare field for the better. However, to achieve this, healthcare establishments (universities, hospitals, research centers and technology companies) must cooperate and adopt a secure distributed analysis where the aggregate data can flow freely and securely throughout the healthcare system. As part of our project, we applied the HIPAA and GDPR regulatory frameworks of data governance for genetic data, and we ensured the flexibility of secure and distributed biomedical analyses on a large scale.
The aggregation method for distributed analysis is a choice to stay within a more standard conceptual and methodological framework better known in biomedical research. Aggregation of calculations is not the only data synthesis solution that can be used for this kind of problem. Synthetic data can be created from different machine learning methods, for instance using the avatarization method [15], which has been validated by the French data protection authority (CNIL), the French data protection agency, as a method of anonymizing a de-identified data set. This method was used as a method of synthesizing HLA genomic data for non-identification of the data in order to go out of the GDPR perimeter. This work has shown its good statistical robustness even with large qualitative data.
We can also use other distributed and secure infrastructures to manage biomedical data on different remote sites in complete security. Blockchain technology, for example, has been used as a solution to the governance challenges associated with sharing genomic data [16].
For distributed analysis, the new federated learning paradigm has also shown its effectiveness in ensuring distributed and secure learning on a large scale. This method consists of sending the model to remote sites, training on datasets that are local to each site, then updating and aggregating the parameters coming from each site and resending the results to the sites.
This algorithm has been used, for example, for the development of a federated learning framework that allows the study of the structural relationships of the brain between diseases and clinical cohorts. This framework allows secure access and meta-analysis of all biomedical data without sharing individual information [17].
Distributed population genetic databases proposed in this paper have been applied as an alternative solution to the historical centralized databases reference such as AFND in HLA Population genetics. Our distributed algorithms have been tested and evaluated on analyses applied to HLA data. Our distributed solution can be used for any type of data like KIR or others.
We have developed Distributed HLA population genetic database. Among others the new distributed version would perfectly apply to the AFND historical database. This repository allows research and analysis on allele frequencies, haplotypic frequencies and individual genotypic data in a distributed and secure manner without requiring institutions to share their data in a common database. We have based our algorithm on the distributed Master Worker model, a realistic distributed experimentation environment (Grid’5000) and realistic data. Experiments have shown good performance results (accuracy and execution time) for our new distributed version. As future work, our distributed approach will extend the distributed version of haplotype frequency estimation, modifying the expectation maximization (EM) method.
AFND: Allele Frequency Net Database
GDPR: General Data Protection Regulation
HIPAA: Health Insurance Portability and Accountability Act
HLA: human leukocyte antigen
IPD: Immuno Polymorphism Database
KIRs: killer immunoglobulin-like receptors
PHI: protected health information
We thank Frédérico Garnier from Agence de la biomédecine for his gracious comments.
SS designed the research study and wrote the manuscript. PAG, MS, VD and NV participated in the study design, corrected and approved the manuscript, and provided intellectual content of critical importance to the work.
Pierre-Antoine Gourraud is the founder of Methodomics (2008, www.methodomics.com) and the co-founder of Wedata (2018, www.wedata.science). He is consulting for major pharmaceuticals companies all dealt with through academic pipelines (AstraZeneca, Biogen, Boston Scientific, Cook, Edimark, Ellipses, Elsevier, Methodomics, Merck, Mérieux, Sanofi-Genzyme, WeData). Pierre-Antoine Gourraud is volunteer board member at AXA Mutual Insurance Company. He has no prescription activity neither drugs nor devices.
Not applicable.
Informed consent to participate in the study was obtained from all participants.
Informed consent to publication was obtained from all participants.
The datasets and the python code for this study can be found in the gitlab site [https://gitlab.inria.fr/ssayadi/distributed_afnd].
This work has been awarded a government grant managed by the National Research Agency under the program “Investissements d’avenir” with the reference KTD-Innov [ANR-17-RHUS-0010]. This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under Grant Agreement No. 754995. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.