 Review
Review

Affiliation:
1Institute for Health and Sport, Victoria University, Footscray Campus, Melbourne VIC. 3011, Australia
2Aegros Therapeutics Pty Ltd, Macquarie Park, Sydney 2019, Australia
Email: nitin.saksena@bigpond.com
ORCID: https://orcid.org/0000-0002-2431-9871
Affiliation:
3Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX 77555, USA
ORCID: https://orcid.org/0000-0002-2888-2418
Affiliation:
4Westmead Institute of Medical Research (WIMR), Herpes Virus Laboratory, Sydney 2145, Australia
ORCID: https://orcid.org/0000-0002-4868-9789
Affiliation:
5OMICS Centre of Excellence, G42 Healthcare, Mazdar City, Abu Dhabi 3079, United Arab Emirates
ORCID: https://orcid.org/0000-0002-4620-6579
Explor Immunol. 2022;2:794–811 DOI: https://doi.org/10.37349/ei.2022.00084
Received: August 01, 2022 Accepted: October 31, 2022 Published: December 29, 2022
Academic Editor: Jorge Kalil, University of São Paulo, Brazil
The article belongs to the special issue Immunology, Immunopathology and Genomics of SARS-COV-2
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection in the human host can lead to various clinical manifestations, from symptomless carriers to mild to moderate to severe/critical illness. Therefore, the clinical classification of SARS-CoV-2 disease, based on severity, is a reliable way to predict disease states in SARS-CoV-2 infection. Recent studies on genomics, transcriptomics, epigenomics, and immunogenomics, along with spatial analysis of immune cells have delineated and defined the categorization of these disease groups using these high throughout technologies. These technologies hold the promise of providing not only a detailed but a holistic view of SARS-CoV-2-led pathogenesis. The main genomic, cellular, and immunologic features of each disease category, and what separates them spatially and molecularly are discussed in this brief review to provide a foundational spatial understanding of SARS-CoV-2 immunopathogenesis.
More recently, on December 31, 2019, we were alerted by the World Health Organization (WHO) and China Health Authority (CHA) [1] that several cases of pneumonia with unknown etiology were affecting Wuhan city in Hubei province in central China. This novel etiology was named severe acute respiratory syndrome (SARS), caused by SARS-CoV-2 [also human coronavirus 2019 (HCoV-19 or hCoV-19)] [2], and the new pandemic called coronavirus disease 2019 (COVID-19) was born. It is characterized by extremely aggressive and rapid dispersal of the SARS-CoV-2 globally, possibly due to certain genetic traits possessed by the newly evolving variants, which epitomizes the ongoing COVID-19 pandemic caused by the virus SARS-CoV-2. The SARS-CoV-2 in critical stages causes acute respiratory distress syndrome (ARDS), leading to subsequent mortality. As of December 3, 2022, the SARS-CoV-2 has caused 649,513,953 infections globally, 6,645,488 deaths, and 626,784,326 recoveries worldwide [2, 3].
The course of COVID-19 disease, caused by SARS-CoV-2, has shown that the disease is typified not only by the evolving variants but also by three disease phases or categories—symptomatic or mild, moderate, severe, and critical, as per the National Institutes of Health (NIH) guidelines. In contrast, according to the WHO, the COVID-19 disease descriptors differ from that put forward by the NIH. For example, rather than mild, moderate, and severe, the WHO descriptors use the terms—non-severe, severe, and critical (Figure 1), where non-severe is typified by the absence of signs usually observed during severe or critical disease; moderate disease invariably shows much reduced respiratory illness, as opposed to acute disease which is characterized by respiratory failure, septic shock with multiple organ dysfunction in infected individuals—a leading cause of mortality in COVID-19 patients seen across the globe. However, despite the disparity in disease descriptor terminology, these descriptors are aligned based on symptoms patients experience during SARS-CoV-2 infection.
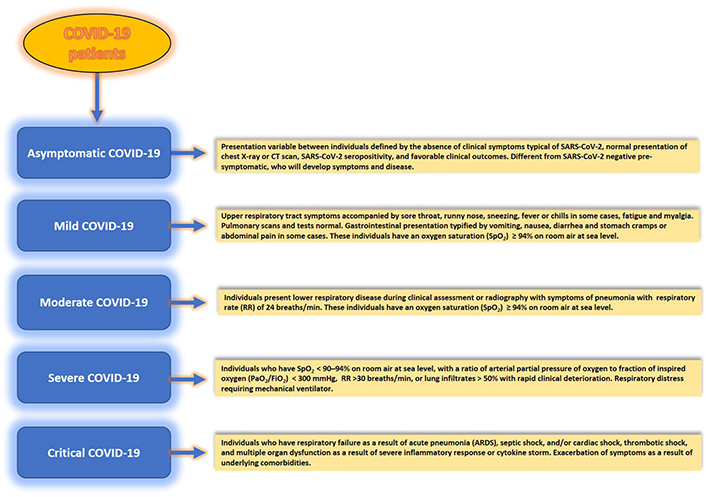
Patients with SARS-CoV-2 infection experience a range of clinical manifestations which define disease severity. Clinical spectrum and disease categories (asymptomatic, mild, moderate, severe, and critical) in SARS-CoV-2 infection of the human host. Considerable inter-host variability in symptomatology exists between infected individuals. Although adults with SARS-CoV-2 infection can be stratified into these disease categories, the criteria for each category could also overlap between different categories. CT: computed tomography
Asymptomatic or pre-symptomatic individuals [4] are the most critical category. Because these individuals can go undetected for SARS-CoV-2 in a virologic test or an antigen test and remain symptomless for COVID-19, it is believed that they have played a significant role in the silent spread of the virus globally. For example, a recent screening of 2,454 published studies included 13 low-risk-of-bias studies from seven countries that tested 21,708 at-risk people, of which 663 were positive, and 111 were asymptomatic [4] confirmed using a real-time reverse transcriptase-polymerase chain reaction test. In this study, the overall asymptomatic proportion ranged from 4% to 41%, while the meta-analysis (fixed effects) found the proportion of asymptomatic to be around 17%. Also, the relative risk (rr) of asymptomatic transmission was 42% lower than that for symptomatic transmission [combined RR 0.58; 95% confidence interval (CI): 0.34–0.99, P = 0.047)]. Thus, these individuals pose considerable concern for public health, its policies, and managing and tracking epidemics and pandemics.
Although we know how these disease descriptors can be classified based on symptomatology, it is beginning to emerge how these disease descriptors also have a biological and molecular basis which can guide us to the development of a new generation of diagnostics, prognostics, and therapeutics for the management of each category promptly. Spatial genomics, transcriptomics, immuno-genomics, and epigenomics are upcoming cutting-edge fields in the field of genomics that hold promise in providing a fine print of a given disease or a disease state (including that of SARS-CoV-2) at the molecular level. As the field develops, it is hoped that we will have deeper insights into the future efficient management of COVID-19 disease both diagnostically and therapeutically. The spatially-defined fine print of host-virus interaction at the single cell or gene level is crucial to understanding any disease.
The questions we are asking for the discussion in this review are—what exactly do mild, moderate, and severe disease category descriptors mean? can they be separated at the biological and molecular levels to make these descriptors diagnostically relevant to SARS-CoV-2 infection, guiding diagnostics, prognostics, and therapeutics? what have we learned from these studies, and what more can be done to improve testing and therapeutic development? This review aims to clarify and cover these aspects to discuss the molecular and biological basis of these descriptors, which other reviews have not covered to date. It remains an important area for diagnostics and prognostics, in addition to guiding the initiation and determining the efficacy of various stage-specific interventions and therapeutics at each stage (Figure 1).
The SARS-CoV-2, like other coronaviruses, is an RNA virus with the largest genome among all RNA viruses comprising 30,000 nucleotides with a single-stranded, positive-sense RNA genome. As shown in Figure 2, the RNA genome encodes 4–5 structural proteins [spike (S) protein, envelope (E) protein, membrane (M) protein, nucleocapsid (N) protein, and in some coronaviruses hemagglutinin (HE) esterase protein], and 16 non-structural proteins [nsps; open reading frames (ORF), ORF1a encoding nsp1–11, ORF1b encoding nsp12–16]. Two polyproteins are directly translated from the positive-sense genome of SARS-CoV-2 from ORF1a and 1b in the cytoplasm immediately upon viral entry into the host cell. These two polyproteins undergo cleavage by two self-activating viral proteases, papain-like protease (PLpro) or nsp3 and 3-chymotrypsin-like protease (3CLpro) or nsp5, resulting in the production of 16 nsps, as mentioned earlier, carrying out diverse functions as indicated in Table 1.
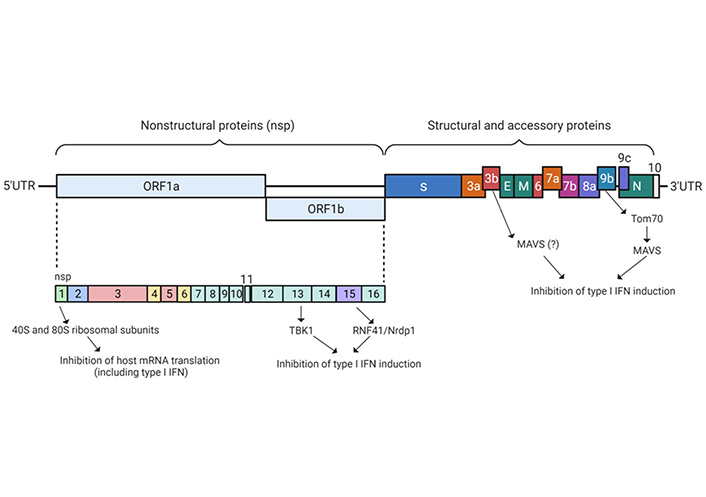
SARS-CoV-2 genomic organization and encoded non-structural and structural proteins. Different structural and nsps and possible mechanistic pathways have been identified that are involved in epigenetic subversion of the host anti-viral machinery through the inhibition of IFN-I induction and inhibition of host mRNA translation. ORF1a/1b encodes a polyprotein, which upon proteolytic cleavage is processed into NSP1–16 (shown in light blue color). Structural proteins, including S (shown in deep blue), E, M, and N proteins (shown in green). Accessory proteins encoded at the 3’ end of the viral genome comprise ORF3a, 3b, 6, 7a, 7b, 8, 9b, 9c, and 10 and are color coded with respective annotations. NSP1, 10, 13, 14, and 15 are involved in the epigenetic muting of the IFN genes that control anti-viral immunity. Moreover, the formation of the NSP14−NSP10 complex enhances translation inhibition, which is executed by NSP14 consequently abolishing the IFN-I-dependent induction of ISGs. Untranslated extremities of the genome are represented by the 5’UTR and 3’UTR. SARS-CoV-2 proteins that interfere with the host IFN induction pathway as well as possible host target proteins such as IFN, MAVS, Nrdp1, RNF41, TANK, TRAF, TBK1, Tom70, and TBK. IFN: interferon; type I IFN: IFN-I; MAVS: mitochondrial antiviral-signaling protein; Nrdp1: neuregulin receptor degradation protein-1; RNF41: ring finger protein 41; TRAF: tumor necrosis factor-receptor associated factor; TANK: TRAF family member-associated nuclear factor kappa B activator; TBK1: TANK-binding kinase 1; Tom70: translocase of outer mitochondrial membrane 70; UTR: untranslated region
Functions of NSPs encoded by ORF1a and 1b of SARS-CoV-2
| NSP proteins | Possible biological functions | References |
|---|---|---|
| Nsp1 | Inhibit translation of host proteins through r-mRNA binding | [5–8] |
| Nsp2 | Disruption of host cell environment by binding with prohibitin-1 and 2 | [6–8] |
| Nsp3 (a PLpro), largest nsp with 1,299 aa) | Viral polyprotein processing | [6–8] |
| Nsp4 | Viral replication and formation of transcription complexes | [6–8] |
| Nsp5 | Viral polyprotein processing | [6–8] |
| Nsp6 (a 3C-like protease) | Viral replication and formation of transcription complexes, biogenesis of replication organelle | [6, 7, 9] |
| Nsp7 | Accessory factors of RdRP, stabilization of the replication/transcription complex, SARS-CoV nsp7 enhances RNA binding by nsp8 | [6–8] |
| Nsp8 | Accessory factors of RdRP, stabilization of the replication/transcription complex | [6–8] |
| Complex formed between Nsp7+8 nsp(7+8) | It is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension activity, and is an accessory factor for RdRP | [6–8] |
| Nsp9 | RNA-binding protein | [6–8, 10, 11] |
| Nsp10 | Cofactor of nsp14 and nsp16, Nsp14−Nsp10 complex enhances translation inhibition, which is executed by NSP14 consequently abolishing the IFN-I-dependent induction of ISGs | [6–8, 12] |
| Nsp11 (smallest nsp with 13 aa) | Intrinsically disordered protein with unknown function | [6–9, 12, 13] |
| Nsp12 | It is an RdRP in addition to being a nucleotidyl-transferase | [6, 7, 13] |
| Nsp13 | It is a helicase with a possible function in nucleic acid metabolism including DNA replication, recombination, transcription, repair, ribosome biogenesis, RNA processing, translation, and decay | [6–8, 13] |
| Nsp14 | It is a proofreading 3’–5’ exoribonuclease and a guanosine-N7 methyltransferase (N7-MTase) for the RNA cap formation, effects of viral infection on the host cell transcriptome, Nsp14−Nsp10 complex enhances translation inhibition, which is executed by NSP14 consequently abolishing the IFN-I-dependent induction of ISGs | [6–8, 12, 13] |
| Nsp15 and 16 | Nsp15 is a uridine-specific endoribonuclease and IFN antagonist, whereas the Nsp16 is ribose 2′-O-methyltransferase for gRNA cap formation | [6–8, 12, 13] |
aa: amino acids; RdRP: RNA-dependent RNA polymerase; ISGs: IFN stimulated genes; gRNA: genomic RNA; mRNA: messenger RNA; r-mRNA: ribosomal-mRNA
The SARS-CoV-2 genome comprises ten ORF [14]. In the first ORF (ORF1a/b) two-thirds of viral RNA is present, which encodes for polyprotein 1a and polyprotein 1b, in addition to 16 nsps (nsp1–16) [14]. The remaining ORFs encode for the SARS-CoV-2 structural proteins like S, M, E, and N, and accessory proteins, which are only expressed from the newly synthesized individual single guide RNAs (sgRNAs), following which the incorporation of S, M, and E (smallest of the structural proteins with 76–109 aa) proteins into viral E occurs leading to the formation of viral particle (virion). Thus, the fully-formed virion of SARS-CoV-2 consists of 4 structural proteins—S, M N, and E, and accessory proteins. All four structural proteins interact with each other for viral morphogenesis and budding [8, 14].
The S protein on the viral E has an affinity to bind in a specific manner to angiotensin-converting enzyme 2 (ACE2) which acts as a viral receptor to latch on to thereby initiating the first step for virus infection [8, 15]. Additionally, the host cell transmembrane protein series 2 (TMPRSS2) specifically serves as the activating protease for the S protein [16], with both ACE2 and TPRSS2 engaged by the virus in facilitating its entry into the host cell and establishing infection. The SARS-CoV-2 infection is initiated with virion attachment to the receptor of host target cells, which involves the interaction of the S protein with the host-cell receptor ACE2 [15]. TMPRSS2 carries out proteolytic cleavage of the S protein culminating in structural changes in S protein thereby initiating the fusion of viral and host cell membrane and subsequent release of viral +gRNA into the host cell cytoplasm—where the SARS-CoV-2 replicates exclusively being an RNA virus. In fact, the viral +gRNA serves as a mRNA for the translation of ORFs 1a and 1b, leading to the production of NSP1–16, in addition to serving as a template for RNA transcription. In the final process a newly formed +gRNA, which is encapsulated in N proteins enclosed by the viral E is eventually released from the infected host cells, thereby completing the viral infection cycle.
The significance of the E protein comes from the vital role it plays in SARS-CoV-2 infection by assisting the release and insertion of SARS-CoV-2 into the host cell. As it is known to alter host cellular processes and modulate viral pathogenicity through the creation of an ion channel in the viral membrane, it is being dubbed as the main virulence element in the novel SARS-CoV-2 [8, 15]. In contrast, the M protein is known to play an important role during viral budding and assembly. In the virion, the M protein has a big carboxyl-terminal region at the interior of the virion, a short aa terminal on the exterior of the virion, in addition to three hydrophobic transmembrane domains. As the M protein assists the S protein, which plays the main role in viral attachment and entry into the host cell, any mutation occurs incurred in the M protein has a significant impact on the interactions with the host cell [17]. The N protein is involved in the binding and packaging of the viral gRNA as a ribonucleoprotein complex in mature virions [18]. In addition to these, there are a number of accessory proteins encoded by the SARS-CoV-2 genome but the functions of these proteins remain obscure, apart from the speculative role they might play in host immunity regulation and viral adaptation in the host [19].
The most common symptoms of SARS-CoV-2 infection include fever and cough (the most common symptoms), myalgia, headache, sore throat, diarrhea, dyspnoea, loss of smell, loss of taste (10% of cases), nausea or vomiting, rhinorrhoea (< 10 % of cases), and the severity of these symptoms vary between infected individuals and also between disease states known for SARS-CoV-2 infection [20].
As mentioned earlier, SARS-CoV-2 can induce three disease states, with asymptomatic or mild disease seen in > 81% of cases; severe disease (typified by dyspnoea, hypoxia, or greater than 50% lung involvement observed on imaging within 48 h) in 14% of cases; and critical illness in 5% of cases characterized by respiratory distress requiring the need for a mechanical ventilator [21]. As discussed in the preceding sections, it is important to iterate that SARS-CoV-2 is a single-stranded RNA beta-coronavirus and the same subgenus as the SARS virus (SARS-CoV-1), which appeared in 2003, and didn’t gain a pandemic potential [22]. Noteworthy is that the ongoing COVID-19 pandemic is being driven by constantly evolving new variants of SARS-CoV-2 and their global spread. Several SARS-CoV-2 variants have been identified and characterized, with some variants carrying a set of mutations that can alter infectivity, antigenicity, and viral interactions with host immunity, thereby leading to higher transmissibility, disease severity, and the duration of disease, which are also guided in response to the changing immune profile of the human population virus encounters [23]. As of December 2021, the variants of SARS-CoV-2 are classified as a variant of concern (VOC) or variant of interest (VOI) and are classified as alpha (also known as B.1.1.7), first identified in the UK; beta (B.1.351), first identified in South Africa; gamma (P.1), first identified in Brazil; delta (B.1.617.2), first identified in India; and Omicron (B1.1.529), first identified in South Africa, and is the most labile variants of all with the rapid and high success of global transmission [24]. Although many sub-lineages for each of these variants have been identified, it is beyond the scope of this review to discuss all of them [25].
Overall, there is a reason for the recognition of these variants, as they are more transmissible than the wild type of SARS-CoV-2 strains. The Omicron variant appears to be more transmissible than other variants, and it has replaced delta as the most dominant variant of SARS-CoV-2 globally, the delta caused a higher rate of severe outcomes than any other SARS-CoV-2 variants. Now the currently circulating variants of Omicron—the BA.4 and BA.5, were able to outcompete and replace the variant one before it, which could be attributed to significant growth advantage that usually leads to increased intrinsic transmissibility, generation of mutants, followed by immune evasion. Consistent with these properties, the Omicron variant shows lower immunogenicity after 2 doses to all the current COVID-19 vaccines in use, and the neutralizing antibody titers appear to be far lower than against other variants, including delta and the ancestral strains. Thus, the reinfections among the vaccinated individuals we are seeing with mild and moderate infections could be attributed to the newly emerging variants [2, 3].
It must be noted that in most cases, the infection caused by SARS-CoV-2 is mild. However, in some patients, severe and critical disease states develop which are characterized by variable degrees of systemic inflammation, ARDS, thromboembolic complications, tissue damage, cardiac injury, and cytokine storm, resulting in mortality [26–28]. This risk for severity or critical disease states is not only exacerbated by specific SARS-CoV-2 variants, as observed throughout the COVID-19 pandemic, but also by the comorbidities, such as age, genetic predisposition, diabetes, obesity, hypertension, ethnicity, and a range of other unknown factors that also play a significant role in the development of these disease states. Although vaccine-experienced status has been thought to prevent severe disease, the surge of breakthrough infections seen among vaccinated individuals across the globe still requires further investigations to make this assertion viable for clinical consideration and risk stratification [28, 29]. It is thus vital to understand the host’s innate immunity during SARS-CoV-2 infection, which will allow a better understanding of the molecular basis of these disease states during SARS-CoV-2 infection.
The host’s innate immunity is the prime target of SARS-CoV-2. For the virus to breach the host immune system and target co-morbidities, it subverts the host antiviral immunity via genomic and epigenomic mechanisms to gain the upper hand in the infection cycle by evading the host antiviral immune attack.
In the infected host, the innate immune cells, which include monocytes, macrophages, neutrophils, dendritic cells (DCs), and innate lymphoid cells (ILCs) such as natural killer (NK) cells, have the arsenal of pathogen recognition receptors [PRRs, such as Toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I), RIG-I-like receptors (RLRs), neutrophil-to-lymphocyte ratios (NLRs), and C-type lectin receptors], sensing through which provide recognition of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) leading to the induction of inflammatory signaling pathways that are vital for mounting sustained immune responses against the pathogen [30–33]. This event is characterized by the production of inflammatory cytokines, chemokines, and induction of apoptosis for clearing infected cells.
The SARS-CoV-2 infection leads to activating these sensors and the associated signaling pathways. As a result, the innate immune system mounts a solid inflammatory immune response that can subdue or reduce the viral replication of SARS-CoV-2 and many other viruses. Intuitively, SARS-CoV-2 has also evolved strategies that allow it to evade the antiviral attack mounted by the host. It achieves this by subduing host IFN-I and IFN-III levels or its production by immune cells fighting the infection. Interestingly, the subversion of IFNs in the host is mainly achieved via the induction of de novo methylation of the IFN-γ promoter leading to epigenetic silencing of the ISGs to block the host’s antiviral arsenal [34]. However, the mechanisms independent of epigenetic silencing directly through viral pathogenic mechanisms have also been described for other DNA and RNA viruses to play a role in the silencing of IFN secreting genes [35]. As this review is focused on disease categories in SARS-CoV-2 infection, it is important to iterate that patients with mild- and moderate-COVID-19 disease consistently display low serum levels of IFN-I and IFN-IIIs, whereas in severe disease, the serum levels and the quality of pro-inflammatory cytokines and chemokines rise considerably above the normal range leading to high inflammation followed by ARDS fatal pneumonia that is linked to severe disease and mortality [36].
Virus limits the production of IFN-I and IFN-III via interference at the post-transcriptional level and by preventing mRNA release from the initial transcription site and degrading it inside the nucleus before the mRNA is transcribed [37]. Several viral proteins are involved in innate immune sensing. These proteins disrupt RLR sensing pathways [RIG-I and melanoma differentiation-associated gene 5 (MDA5) are the critical regulators of IFN pathways] and host antiviral protein induction, in addition to other host signaling and effector functions that counter the virus.
A number of SARS-CoV-2 proteins incur insults on the host anti-viral immunity, a summary of which is shown in Figure 2, and the possible functional interactome of these proteins in Figure 3.
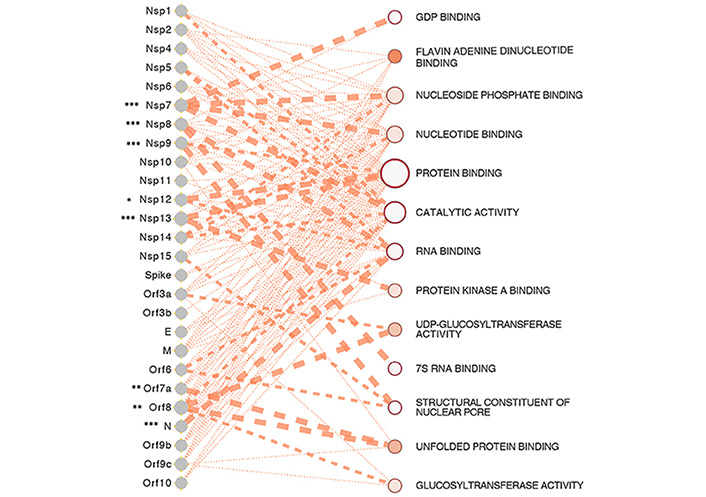
SARS-CoV-2 proteins and gene ontologies of their interacting host genes [31]. The size of the gene ontology is signified by the size of the circle which is proportional to the number of genes in the ontology, such as the genes relating to protein binding occupying the largest space in the interactome. The thickness of the lines linking SARS-CoV-2 proteins and gene ontologies represents the strength of the interaction of gene count between SARS-CoV-2 proteins and host genes in the ontology. SARS-CoV-2 proteins of significance, especially the ones interacting with a significantly higher number of host genes than expected are represented by asterisks, with representation: * P < 0.05, ** P < 0.01, *** P < 0.001
Note. Reprinted from “Epigenetic Lens to Visualize the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) Infection in COVID-19 Pandemic,” by Saksena N, Bonam SR, Miranda-Saksena M. Front Genet. 2021 Mar 22;12:581726 (https://www.frontiersin.org/articles/10.3389/fgene.2021.581726/full). CC BY.
The SARS-CoV-2 uniquely encodes for a shorter protein ORF3b [38] and hampers host innate immune reaction in vitro by limiting the induction of the IFN-I response—an aspect crucial to viral pathogenesis. This protein can block host defenses early in the infection cycle by shutting off the host’s IFN secretion through an epigenetic mechanism before the T and B cells come into play. Further, novel coronaviruses (nCoVs) are also known to encode three ion-channel proteins, also called viroporins, such as protein E, ORF3a, and ORF8a [39]. These are small, hydrophobic proteins with a multifunctional ability that aids in modifying cellular membranes and renders permeability with both features assisting the release of virions from infected cells [40]. ORF3b suppresses the induction of IFN-I more efficiently than its orthologous counterpart SARS-CoV, SARS-CoV-2 ORF6 and ORF8 inhibit expression of IFN-β and lead to activation of ISGs, and the SARS-CoV-2 ORF9b and N and M proteins also cause inhibition of expression of IFN-β and proinflammatory cytokines via interference with RIG-I and MDA5 pathways [41–44]. Further, the SARS-CoV-2 NSP1 and NSP14, in addition to other potential viral proteins, participate in inhibiting translation, thereby preventing the expression of the IFN signaling pathway [45, 46], whereas the SARS-CoV-2 N protein prevents aggregation of viral RNA with MAVS and blocks the induction of host IFN pathway [31]. Another essential protein is NSP5, the central protease of SARS-CoV-2, and has been shown to interact with histone deacetylase 2 (HDAC2)-an epigenetic regulator. This interaction affects inflammatory and IFN responses mediated by HDAC2 [47]. Concurring with these findings, Gordon et al. [38] through a detailed interactome of SARS-CoV-2 and host have also identified high-confidence interaction between Nsp5 and the epigenetic regulator HDAC2 and predicted a cleavage site between the HDAC domain and the nuclear localization sequence, suggesting that Nsp5 may inhibit HDAC2 transport into the nucleus potentially impacting the function of HDAC2 in mediating inflammation and interferon response [38, 48].
Through the involvement of intracellular lysosomal disruption and ion-redistribution, viroporins can lead to the activation of innate immune signaling receptor NOD-, LRR-, and pyrin domain-containing 3 (NLRP3) inflammasome. Following viral binding to the ACE2 receptor for cell entry, the SARS-CoV-2 viral RNA is recognized by TLR3, thereby triggering transcriptional responses and cytoplasmic changes, leading to the activation of the NLRP3 inflammasome. As a consequence, the production of inflammatory cytokines [interleukin 1β (IL-1β), IL-6, and tumor necrosis factor (TNF)] is triggered resulting in tissue inflammation and damage, and severe respiratory illness, as seen in severely and critically ill COVID-19 patients. Several viruses and viral proteins, including that of SARS-CoV-2 are known to be activated by the NLRP3 inflammasome in macrophages [49]. This activation can induce caspase-1-dependent cleavage, thereby forcing the release of crucial proinflammatory cytokines IL-1β and IL-18, which triggers gasdermin D-mediated pyroptotic cell death. The severity of COVID-19 disease corresponds to the extent of NLRP3 activation [50]. As the inflammasome is a vital arm of innate immunity that detects and senses various endogenous or exogenous, sterile, or infectious stimuli encountered within the cell, it induces cellular responses and effector mechanisms to ward off unwanted pathogens or threats. New evidence suggests epigenetic factors, including DNA methylation and histone acetylation, regulating NLRP3 mRNA expression [51], which makes NLRP3 inflammasome an attractive drug target for the treatment of SARS-CoV-2 [52].
From the preceding sections, several studies discussed already show a good body of evidence that SARS-CoV-2 subverts the components of host antiviral immunity, therefore, this section discusses the recent developments in multi- and single-cell-OMICS in delineating the role of various immune cells that play a crucial role in host immunity during SARS-CoV-2 infection, and the relevance of genomic/molecular changes associating with mild, moderate and severe/critical SARS-CoV-2 disease. Furthermore, several studies have emphasized the utility of OMICS in creating a fine print of SARS-CoV-2 pathogenesis, in addition to genomically visualizing single cells and shifts in SARS-CoV-2-mediated disease states. This is promising for future therapeutic strategies to attack SARS-CoV-2 and stop it while on track.
A study by Gordon et al. [38] is the most comprehensive study demonstrating the use of multi-OMICS in segregating disease states from mild to moderate and severe disease during SARS-CoV-2 infection [53]. Using serial blood draws from 139 COVID-19 patients, they performed an extensive multi-omics analysis on single peripheral blood mononuclear cells, analyzed and collected data on plasma proteome metabolites, secreted proteins, cellular transcriptome, surface protein data, immune receptor sequences in alignment with electronic health records. This integrative analysis identified immune coordination during infection accompanied by a significant immunological shift between mild and moderate infection, with moderate and severe infection showing an increase in inflammation, drop in blood nutrients, and the emergence of novel immune cell subpopulations intensifying with disease severity during SARS-CoV-2 infection.
Previously the evidence of dysregulated plasma metabolites has already been shown in severe SARS-CoV-2 infection [54]. However, a comparison of different disease states in COVID-19 at a more refined level using multi-omics and a clear understanding of the coupling between elevated inflammation signals, plasma metabolites, proteins, and dysfunction in immune cells, is lacking at the patient level, which Su et al. [53] clarified demonstrating that the host metabolic and plasma compositions correlate with disease severity. Moreover, they also determined the power of plasma analytes as clinical measures in predicting disease severity in clinically-stratified COVID-19 patients.
Principal component analysis (PCA) of plasma proteins and metabolites clearly demonstrated shifts in plasma profiles between healthy donors, mild versus moderate, and moderate versus severe COVID-19 patients. In particular, the pro-inflammatory cytokines showed proteomic separation of healthy donors from COVID-19 patients [e.g., chemokine (C-X-C motif) ligand 6 (CXCL6)], along with the proteins associated with immune cell activation (e.g., CD244 and CD40), implying immune activation in even mild COVID-19 cases. Overall, the most significant proteomic shift was observed in the transition from healthy donors to mild infections with 307 differentially-expressed (DE) proteins and only 27 downregulated. Whereas the mild to moderate transition revealed relatively fewer changes with 132 DE proteins and 91 downregulated. In contrast, only a few proteins were identified for the transition of moderate to severe disease with 5 up-regulated DE proteins and no down-regulated proteins. Consistent with previous literature [55], there was also the upregulation of C-C motif chemokine ligand 7 (CCL7), IL-10, and IL-6 (highly significant disease correlation between moderate and severe COVID-19 disease). Keratin-19 (KRT19, marker of tissue damage), mainly involved in the organization of muscle fibers [56], was significantly upregulated across comparisons. In contrast, the global metabolic profile alterations caused a significant shift between mild and moderate disease states, which was evident from the excessive loss of several classes of circulating metabolites during COVID-19 disease. Overall, these proteomic comparisons displayed a striking overlap between moderate and severe COVID-19 disease states, along with the augmentation of inflammatory signals, coupled with the drop in metabolic resources and xenobiotic metabolism, implying that a stressed environment may influence the immune response correlating with disease states in COVID-19 patients [53, 54].
The CD8+ T cell single-cell transcriptome data analysis resolved nine sub-populations characterized by both protein and transcriptional signatures. Su et al. [53] identified naive, memory, effector, exhausted-like, and proliferative phenotypes. Exhausted CD8+ T cells have been documented in COVID-19 patients [57] consistent with lymphocyte activation gene-3 (LAG3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT) markers that were upregulated. Interestingly specific CD8+ sub-populations also correlated with disease severity. For instance, the cellular cluster with naive-like markers exhibited the highest density in healthy donors, whereas they showed a decreased subset in COVID-19 patients. The effector like cells, as expected, were found to be enriched in COVID-19 samples [58]. Similarly, CD8+ T cells displayed higher levels of effector associated transcripts [granzyme H (GZMH), killer cell lectin like receptor D1 (KLRD1), SLC9A3 regulator 1 (SLC9A3R1)] in COVID-19 patients who improved with time from SARS-CoV-2 infection. Thus, mainly the severe patients were typified by an increase of naive clusters and a decrease in activated effector T cells relative to moderate disease [53].
Further, the integration of single-cell omics with T-cell receptor (TCR) datasets resolved two novel and distinct phenotypically-specific groups of TCRs (the cytotoxic effector phenotype and the memory-like phenotype) in CD8+ T cells. The TCRs from the cytotoxic effector phenotype were mainly observed as unique to patients with moderate or severe COVID-19, whereas the memory-like phenotype was predominant in mild COVID-19 patients. While an enhanced CD8+ T cell clonal expansion was a feature of mild and moderate disease states as opposed to healthy donors, noteworthy is that the expansion of the CD8+ clones serves as predictors of a sharp transition between mild and moderate COVID-19 disease, implying the value of these cells in disease staging of COVID-19 [43, 53].
Polyfunctional T cells are an adequate and robust measure of immunity, and this very function robustly guides the production of several different cytokines, which are considerably higher than their T cell counterparts, thereby dominating an immune response [59, 60]. The measure of polyfunctional strength and the secreted proteins has shown that it was similar between healthy and mild disease states, whereas there was an increased frequency of cells secreting granzyme B (GZMB) and perforin (PRF1) during severe disease, implying that polyfunctionality is reduced in severe disease states of COVID-19 patients. The highest polyfunctionality and the highest percentages of effector phenotypes were observed in moderate COVID-19 disease.
Monocytes play a vital role in innate and adaptive immunity, with their primary function in immune defense, inflammation, and tissue remodeling. The dichotomy of monocyte subsets in humans and its classification is based on the expression levels of CD14, a component of the lipopolysaccharide receptor complex, and CD16, the FCγRIII immunoglobulin receptor [61]. Three subsets of monocytes have been recognized in humans: classical (CD14++CD16−), intermediate (CD14+CD16+), and non-classical (CD14+CD16++) monocytes [62, 63]. Previously, the value of these subsets in human immunodeficiency virus (HIV) disease has been shown. By categorizing the monocyte population into CD14+, CD16high, CD16medium, and CD16low, clear discrimination between viremic and aviremic HIV patients was seen, with a significant elevation of CD16low population (80%) of HIV-negative individuals and non-progressors (57%), thereby underpinning its importance in non-progressive HIV disease [64]. Overall, our understanding of the fate of these monocyte subsets is constantly evolving, along with their role in inflammation, infection, and injury in the context of other viruses, including SARS-CoV-2.
Recently, the S100highHLA-DRlow dysfunctional monocyte subpopulation was shown in COVID-19 severity. There was a clear separation of non-classical monocytes (CD14lowCD16high), typified by the upregulation of transcripts Fc gamma receptor IIIa (FCGR3A), CX3C motif chemokine receptor 1 (CX3CR1), and surface protein CD16. Thus, there was a decrease in non-classical monocyte fraction in moderate and severe infections [53, 65, 66]. In contrast, the classical monocytes (CD14highCD16low) percentage increased for moderate and severe disease cases accompanied by the expression of inflammation-related transcripts such as S100A4, S100A9, S100A12, and decreased HLA-DR surface protein level. Interestingly, SARS-CoV-2 patients who improved their monocytes showed higher HLA-DRB5 and S100A8.
In relation to the role of these monocytic subsets in cellular dysfunction during SARS-CoV-2 infection, it was thought that the monocyte responses to COVID-19 could also correlate with the metabolic and proteomic plasma compositions. Pathway analysis of the top 50 proteins [3] showed a possible association with cytokine signaling, inflammation response, monocyte chemotaxis, IFN-γ, and mitogen-activated protein kinase (MAPK) signaling. These plasma protein signatures, most of which were reflected in the cross-OMICS interaction network, could be compared against monocyte gene expression signatures that increased with disease severity. Additionally, there was a positive correlation of metabolites N1-methyladenosine, two lipids, mannose, and kynurenine, of which kynurenine has been previously associated with COVID-19 severity [54]. Overall, these associations of transcripts, proteins, and metabolites, which associate with monocytic function, clearly suggest that the monocytes may play a potential role in the coordinated immune response during severe COVID-19 infection, and plasma proteins (IL-6 and CCL23) could potentially serve as clinical biomarkers for assessing disease severity and dysfunctional state of monocytes.
NK cells or neutrophils are white blood cells that form an integral part of the innate branch of the immune system and are vital in tackling and controlling viral infections, which they control by rapid activation [67], and efficiently eliminating virus-infected cells by releasing cytotoxic granules that directly kill the virus-infected cells, and also by producing cytokines and chemokines by modulating the behavior of different immune cells in the infected host. It is important to note that the NLR bears a correlation with disease severity and is an excellent surrogate marker for predicting systemic inflammation. As the disease severity increases with comorbidities, such as age, obesity, and diabetes, the NLR ratio also shows a positive correlation with these comorbidities [67].
Monitoring of the immune response of NK cells in individuals with COVID-19 showed lower than normal levels of NK cells in people with the disease, but the exact role of these cells in severe COVID-19 disease remains obscure [66, 68, 69]. Through the analysis of clinical samples, it has been reported that NK cells from healthy individuals can directly kill SARS-CoV-2-infected cells in vitro [70, 71], but that NK cells from people with moderate or severe COVID-19 had impaired cell killing activity. According to this study, these defective NK cells display higher than normal levels of expression of cytotoxic molecules that kill cells. However, they are impaired in their ability to bind to target cells and to export their cytotoxic granules at the cell surface. In addition, NK cells from people with severe COVID-19 were less efficient in producing cytokines and chemokines than the cells from healthy donors or infected individuals with no or minimal symptoms. Concurring with these data, there was a direct correlation between a rapid decline in the level of the virus (the viral load) and high levels of NK cells at the beginning of the infection. However, the exact correlation was lacking for the T or B cells. Thus, the rapid decline in the blood NK cells throughout SARS-CoV-2 infection could be attributed to apoptosis [72], or possibly due to their recruitment from the bloodstream to the lungs.
Unsupervised clustering and uniform manifold approximation and projection (UMAP) visualization of NK cell transcriptomes, which resolved CD56bright and CD56dimCD16bright subsets of NK cells, suggest the association of proliferative NK cell sub-population with increased severity of SARS-CoV-2 infection [73]. The CD16bright subpopulation expresses markers associated with less differentiation, including IL-7 receptor (IL-7R), CD27, and L-selectin (CD62L), display high levels of cytotoxic transcripts [PRF1 and granzyme B (GZMB)], and elevated levels of CD57 (the terminal-differentiation marker) and the cell exhaustion marker LAG3. This population of NK cells is significantly repressed in patients with moderate and severe disease patients, implying the differentiation of NK cells toward more cytotoxic phenotypes pointing at the sharp transition from mild to moderate disease. This also concurred with the decrease of CD56, CD127, and IL-7R and the increase of PRF1, GZMB, and CD69 in patients with moderate and severe disease.
Further, the evaluation of NK cell transcriptomic signatures also showed that these signatures varied with the disease trajectory [52], and there was an association between expression of the cytotoxic NK cell marker PRF1 and DNA damage inducible transcript 4 (DDIT4-a) DNA repair that is seen during the improvement in SARS-CoV-2 infection state. In contrast, in the patients who did not show improvement, the nuclear factor kappa B (NF-κB)-inhibiting protein, COMM domain containing 6 (COMMD6), concomitantly increased suggesting the role of decreased inhibition of NF-κB signaling in NK cells, correlating with COVID-19 patient recovery.
In the preceding sections, a good body of evidence shows that proteomic, metabolite and transcriptomic changes at the level of plasma and single cells do bear a significant relationship with the severity of SARS-CoV-2 infection, but how they coordinate immune responses between them is an essential aspect of pathogenesis. Furthermore, multi-omics data integration sheds light on the cumulative visualization of gene expression and its correlation with disease trajectories [4]. Previously the value of coordinated cellular immune responses has been shown in a parallel retrospective analysis of CD4+ T cells, CD8+ T cells, and CD14+ monocytes from viremic and aviremic phases of HIV disease using protein microarray [74]. Both these analyses, based on different viruses (SARS-CoV-2 and HIV), demonstrate that simultaneous visualization of gene and protein expression in diverse immune cell types shows covariation of cellular immune responses, gene and protein expression in parallel, which can be a better guide to understand a disease, its states and also targeting it therapeutically.
In SARS-CoV-2 infection the innate immune cells also orchestrated transcriptional alterations, which includes increased IFN-α response for all cell classes, implying an integral role of IFN-I response to infection, and also agrees with the first-line strategies virus deploys to target the innate immunity to suppress this coordinated immune response in the host [53, 75]. An increased antigen presentation in both CD4+ and CD8+ T cells and decreased antigen presentation expression in monocytes with a unique S100highHLA-DRlow monocyte and the presence of the proliferative NK cells mainly emerged at the transition from mild to moderate disease. In addition, the CD4+ T cells exhibited proliferative exhaustion and a clonally-expanded CD4+ cytotoxic phenotype with distinct signatures and TCR-sharing patterns. The relevance of these two phenotypes remains unclear, but their relative increase with infection severity, especially at the transition between mild and moderate disease, is of biological significance. Cumulatively, these results show that immune cell coordination correlates with pro-inflammatory signals, profound loss of circulating lipids, and other biological processes associated with COVID-19. Furthermore, integrating the transcriptomic changes defining the immune responses from diverse immune cell types points to significant physiological changes distinguishing mild from moderate and severe COVID-19 disease states.
Lastly, it is worthy of discussion that the immune cell numbers, like in any other virus infection, also play a crucial role in defining disease stages, including in COVID-19. Using single-cell sequencing analysis to demonstrate how the dynamic changes in immune cell numbers in the blood of COVID-19 patients has shown a clear stratification of disease categories [76]. Interestingly, the immune response in the T cell subgroup appears to be mainly concentrated in the high to the low-cell group. In addition, the disease-related complications were mainly in the low to the high-cell group. Thus, how the modulation in immune cell numbers causes dynamic shifts in cellular biomarkers that can be used in defining and diagnosing disease states is an important aspect of future research.
An infection with SARS-CoV-2 can lead to life-threatening hyperinflammatory disease in some cases. Each of the disease states in COVID-19 can lead to considerable immunological damage, in addition to long COVID in some individuals. Therefore, global efforts are needed to understand better SARS-CoV-2-specific immune responses along with the immunopathological mechanisms underlying mild, moderate, and severe disease. The power of genomics in infectious disease research has been highlighted during the COVID-19 pandemic. Using high-throughput genomic tools, there is a global effort in engaging with the most critical aspects of SARS-CoV-2 pathogenesis and epidemiology. With more focus than ever on full-genome sequencing, researchers are exploring ways to apply next-generation sequencing (NGS) to SARS-CoV-2, in addition to other recently emerging pathogens. The high throughput technologies have the potential to unveil disease mechanisms and stratify disease states of SARS-CoV-2, and other viruses, as evident from this review. The take-home message from the discussion in this review is to develop artificial intelligence-based immune surveillance at the genomic, proteomic, and metabolite levels, in addition to using integrative systems immunology approach in understanding immune responses at the individual and cumulative levels for a holistic nature of the coordinated response to pathogens that can guide us in visualizing the disease stages accurately in SARS-CoV-2 infection and other viruses, and developing effective therapeutics to target viruses.
Lastly, although not discussed due to lack of data, there are many unanswered questions about asymptomatic cases with SARS-CoV-2 infection, as these cases significantly contribute to viral spread globally. Therefore, in these individuals, there is a need to delineate the role of viral load and virus-shedding dynamics as opposed to symptomatic cases, which will facilitate the understanding of onward transmission, disease length, and severity, and the possible biomarkers [77] that could segregate asymptomatic from mild, moderate, and severe categories.
aa: amino acids
ACE2: angiotensin-converting enzyme 2
ARDS: acute respiratory distress syndrome
COVID-19: coronavirus disease 2019
DE: differentially-expressed
E: envelope
gRNA: genomic RNA
HDAC2: histone deacetylase 2
HIV: human immunodeficiency virus
IFN: interferon
IFN-1: type I interferon
IL-1β: interleukin 1β
ISGs: interferon stimulated genes
M: membrane
MAVS: mitochondrial antiviral-signaling protein
mRNA: messenger RNA
N: nucleocapsid
NK: natural killer
NLRP3: NOD-, LRR-, and pyrin domain-containing 3
NLRs: neutrophil-to-lymphocyte ratios
NSPs: non-structural proteins
PRF1: perforin
RIG-I: retinoic acid-inducible gene I
S: spike
SARS-CoV-2: severe acute respiratory syndrome coronavirus-2
TCR: T-cell receptor
WHO: World Health Organization
NS, SRB: Conceptualization, Writing—original draft, Visualization. THC, MMS: Supervision. NS: Supervision, Project administration. NS, SRB, MMS, THC: Validation.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Naima G. Cortes-Perez
Sneha Das ... Rupesh K. Srivastava
Junming Chen ... Jianshe Yang
Hana Ratnawati ... Steven Felim
