
Abstract
Human cytomegalovirus (HCMV), whose genome is around 235 kb, is a ubiquitous human herpesvirus that infects between 40% and 95% of the population. Though HCMV infection is commonly asymptomatic and leads to subtle clinical symptoms, it can promote robust immune responses and establish lifelong latency. In addition, in immunocompromised hosts, including individuals with acquired immunodeficiency syndrome (AIDS), transplant recipients, and developing fetuses it can lead to severe diseases. Immunosenescence, well-defined as the alterations in the immune system, is linked mainly to aging and has been recently gathering considerable attention. Senescence was characterized by an elevated inflammation and hence considered a powerful contributor to “inflammaging” that is measured mainly by tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and C-reactive protein (CRP) levels as well as latent viral infections, for instance, cytomegalovirus (CMV). Inflammaging resulted in a senescence-associated secretory phenotype (SASP). HCMV is markedly associated with accelerated aging of the immune system as well as several age-associated diseases that accumulate and subsequently deteriorate the immune responses, thus have been linked to mortality, declined vaccine efficacy, serious diseases, and tumors in the elderly. HCMV triggers or exacerbates immunosenescence; on the other hand, the weakened immune responses and inflammaging favor viral reactivation and highlight the role of HCMV in aging as well as viral-associated tumors. HCMV reactivation resulting in sequential lytic and latent viral cycles could contribute to HCMV genomic variability. Besides the oncomodulatory role and transforming capacities of HCMV, the immune-privileged tumor microenvironment has been considered the main element in tumor progression and aggressiveness. Therefore, the interplay between HCMV, immunosenescence, and cancer will aid in discovering new therapeutic approaches that target HCMV and act as immune response boosters mainly to fight cancers of poor prognosis, particularly in the elderly population.
Keywords
Cytomegalovirus, immunosenescence, oncomodulation, oncogenesis, inflammaging, tumor microenvironmentIntroduction
Human cytomegalovirus (HCMV) belongs to the β-herpesvirus family and is a prevalent human pathogen infecting 40% to 95% of the world’s population [1]. Following primary infection, cytomegalovirus (CMV) possesses the capacity to induce both lytic and latent infections to establish lifelong persistence in human hosts [2]. The clinical course of HCMV infection is widely variable, it depends on the age and is highly influenced by the immune fitness of the host. While the immunocompetent healthy individuals are asymptomatic, the congenitally infected infants could be symptomatic at birth and suffer long-term neurologic sequelae [3]. However, over time pronounced changes occur in the human immune system, known as immunosenescence. These age-related changes decrease immune protection and have been linked to mortality, decreased vaccine responsiveness, cardiovascular diseases, and cancer in the elderly [4, 5]. Importantly, while some studies implicate HCMV in driving immunosenescence and disease risk, others suggest it enhances immune function [6, 7]. Recently, HCMV has been reported to induce oncomodulation and even oncogenesis in some cases [2]. In this review, the role of HCMV in driving immunosenescence and its impact on oncogenesis were highlighted.
Concepts and hallmarks of immunosenescence
Immunosenescence is a normal physiologic process in which immune system function slowly changes with age. In fact, immunosenescence is regulated by many factors including aging, particularly the degeneration of the thymus resulting in a decrease of the T cell population and decrease of CD8+ naive T cells, one of the main manifestations of immunosenescence [8]; as well the inflammation which produces senescence-associated secretory phenotype (SASP) wherein aging cells secrete soluble factors such as growth factors, cytokines, chemokines, and extracellular matrix inducing several senescence-related diseases, including various malignancies [9] in addition to the overlooking of intrinsic and extrinsic factors of the immune system [10]. Moreover, the characterization of the hallmarks of immunosenescence is essential, especially for understanding its impact on the disease risk and tumor progression. With aging, the cytotoxic effect of immune cells as well as the expression of interferon γ (IFN-γ) and cytotoxic molecules such as granzyme B and perforin decrease [11, 12]. In addition, memory T cells which differentiate from naive T cells upon primary antigenic stimulation play a crucial role in the adaptive immune system and enable a robust immune response over the human lifespan. Nevertheless, pronounced age-associated changes occur in the composition of T cell populations (naive versus memory cells). Despite the increased number of memory cells during early life, it shows senescent changes after 65 years [13]. One of the most prominent markers of T cell senescence is the loss of the costimulatory molecule CD28 and the accumulation of highly differentiated effector memory T cell [CD27−CD28−CD57+ killer cell lectin-like receptor G1 (KLRG1)+], which are hallmarks of immunosenescence [14]. Another hallmark of senescent T cells is telomerase shortening [15]. It should be noted that, in the process of immunosenescence, there is remodeling of mature natural killer (NK) cells and reduced expression of the activated receptors which may affect the immune monitoring effect of NK cells in the elderly [16]. Age-related modifications also occur in naive/memory B cell subsets. Indeed, in the elderly, there is a reduction of naive B cells, accompanied by the expansion of memory B cells that show a senescence-associated phenotype [17]. While the functions of dendritic cells (DCs) such as antigen presentation, endocytosis, and IFN production are reduced in elderly individuals [18], the phagocytic ability of neutrophils decreases [19]. Aged macrophages reduce their functional activity leading to the accumulation of unphagocytosed debris, increased senescent-associated markers, increased inflammatory cytokine production, reduced autophagy, and a decrease in Toll-like receptor (TLR) expression [20].
HCMV infection driving immunosenescence
Following primary infection, HCMV has the capacity to induce both lytic and latent infection to establish lifelong persistence in human hosts. Despite the extensive innate and adaptive response elicited by HCMV infection during both lytic and latent infection, the virus develops diverse immune evasion strategies to alter the host immune recognition [21]. The clinical presentation of HCMV infection is highly influenced by the immune response of the host during distinct life stages. In healthy immunocompetent hosts, viral reactivation which occurs throughout life induces the establishment of immune memory leading to the control of viral replication. However, in immunocompromised hosts, the loss of CMV-specific CD4+CD8+ T cells favors uncontrolled viral replication and dissemination leading to serious clinical diseases and even death [22]. In addition, long-term HCMV persistence will modulate the immune system composition and function even in healthy HCMV-infected individuals (Figure 1). Many epidemiologic studies on aging indicate that HCMV seropositivity is associated with immunosenescence and increased mortality in the elderly [23, 24]. Moreover, HCMV seropositive older individuals have a reduced response to vaccination [25]. Reactivation of HCMV triggered in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infected patients exacerbates the risk of coronavirus disease [26]. Although, the link between HCMV and immunosenescence request more investigation, HCMV persistence is thought to be a driver of immunosenescence in humans [7]. It was postulated that inflation of HCMV-specific T cells during viral persistence compromised host immunity. In HCMV-infected elderly individuals, the CD8+ T cells response to HCMV antigens constitutes 50% of the entire memory CD8+ T cells compartment in peripheral blood, while around 30% of total circulating CD4+ T cells can be HCMV responsive [27]. The majority of this terminally differentiated inflationary HCMV-specific CD8+ T cell subset has a typical age-related senescent T cell phenotype [CD57+CD28− C-C motif chemokine receptor 7 (CCR7)−], lacking CD28 expression which is a major characteristic of T cell aging [28]. This finding is supported by the fact that this large population of HCMV-specific CD8+CD28– T cells is absent in seronegative elderly individuals [29]. Moreover, in elderly hosts, impairment of T cell immunity is also linked to memory inflation in HCMV infection. Despite the diverse CD8+ T cells repertoire to recognize different viral epitopes soon after HCMV primary infection, this diversity starts to decrease with age often manifested as large clonal expansions of cells of limited antigen specificity together with a marked shrinkage of the T cell antigen receptor repertoire [7]. In elderly hosts, more than 25% of total CD8+ T cells show a specific response to an individual immunodominant HCMV epitope such as p65 tumor suppressor protein (p65) and immediate early 1 (IE1) [30]. This limited diversity of HCMV-specific T cell clones during memory inflation may affect the immune protection to novel and vaccine antigens through decreased T cell receptors (TCRs) diversity in the elderly and thereby exposing them to the risk of life-threatening diseases [31]. Additionally, in older adults, most inflationary CD4+ T cells induce T-helper 1 (Th1) responses by producing IFN-γ which explains the poor humoral responses seen in the elderly [32]. Although HCMV has a more immunologic impact on memory T cells than naive T cells, it also alters naive T cells. Thus, mainly and exclusively in older subjects with elevated anti-CMV antibody titers, there is a significant decrease in CD4+ naive T cells parallel to an absolute increase in effector/effector memory CD4+CD8+ T cells [33].
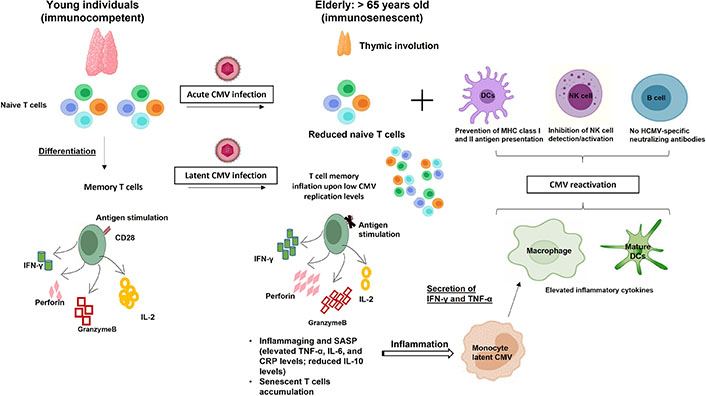
HCMV-induced immunosenescence. Acute and chronic HCMV infections modulate the host immune system resulting in naive T cell pool depletion and memory inflation, respectively which further drive/accelerate immunosenescence. The weakened immune responses and inflammaging favor HCMV reactivation which is the key to the accumulated HCMV-specific immune responses. IL-2: interleukin-2; TNF-α: tumor necrosis factor-α; CRP: C-reactive protein; MHC: major histocompatibility complex
Moreover, cell exhaustion, another form of T cell dysfunction, can arise during chronic infection which is often associated with inefficient control of persisting infections due to the loss of proliferative potential, decreased cytotoxicity, impaired cytokines secretions, and high expression of several inhibitory receptors [programmed death 1 (PD1), KLRG1, and CD57]. HCMV-specific CD8+ T cells are characterized by a low proliferative capacity and expression of senescent markers such as KLRG1 and CD57. These cells are not totally exhausted since they are still highly cytotoxic and produce Th1 cytokines in response to viral replication [31]. It’s worth mentioning that HCMV infection leads to the accumulation of functional exhausted cells that could accelerate immunosenescence in immunocompromised and immunosuppressed individuals [34].
Immunosenescence—a key player in cancer
Due to the increased rates of mortality and morbidity of various tumors with age, cancers are generally defined as aging diseases. The immunosenescence features and mechanisms were described as an important player in the tumoral process resulting in a high-risk of tumors in elderly groups [35]. Immunosenescence in the tumorogenesis process reflects the senescence of both innate (NK cells and macrophages) and acquired immune cells (B cells and T cells) affected on one side by the age-related change and the other side by the factors of the tumor microenvironment (TME). In acute myeloid leukaemia (AML) in elderly patients, NK cells harbor diminished levels of several activating receptors that contribute to impairing NK function and thereby favoring disease progression and decreased survival [36]. Age-modified changes in tissue-specific macrophages and neutrophils cause chronic low inflammation that is associated with a macrophage pro-tumorigenic phenotype [19]. Cytotoxic T lymphocytes (CTLs) are critical in eliminating tumor cells as well as virally infected cells. Thus, alterations of CTL function observed during aging could favor both viral infection and cancer. In patients with HCMV-positive glioblastoma multiforme (GBM), the signs of immunosenescence in the CD4+ T cells compartment are associated with poor prognosis which may reflect the activity of HCMV [37]. Aging can also alter the TME. Thus, the increased presence of tumor-associated macrophages (TAMs) and tumor-derived γδ regulatory T cells (Tregs) in TME has been reported to decrease both innate and adaptive immunity; TAMs produce cytokines that promote T cell inactivation and inhibition of DCs activity thus increasing cancer cell proliferation [38, 39]. In hypoxic TME, tumor-derived cyclic adenosine monophosphate (cAMP) activates DNA damage and induces T cell senescence [40]. Similarly, glucose deprivation triggers DNA damage and activates the p38 pathway leading to cell cycle arrest and inhibition of T cell proliferation [41]. Moreover, the TME oncogenic stress activates signaling pathways including nuclear factor-kB (NF-kB), p38, CCAAT/enhancer binding protein β (C/EBPβ), and mechanistic target of rapamycin complex 1 (mTORC1) which play a role in regulating T cell SASP [42]. Furthermore, in aging, secretion of SASP molecules, such as IL-6, IL-8, and IL-10 in TME favors tumor progression through an inflammaging mechanism [43, 44]. Finally, HCMV favors immunosenescence with decreased immune defenses and inflammaging that could trigger viral reactivation from latency and further supports its role in aging as well as viral-driven malignancies.
Oncomodulatory and oncogenic properties of HCMV
Besides HCMV-induced immunosenescence, presented evidence reveals HCMV presence in numerous solid tumors [45, 46]. HCMV proteins have been found in 90–100% of breast, ovarian, colon, and prostate tumors, in sarcomas as well as in neural-derived tumors such as neuroblastoma, glioblastoma, and medulloblastoma [47]. Taking into account the broader concept of cancer hallmarks, the TME and cancer progression are considered essential oncomodulatory mechanisms relating tumor initiation to viral infections caused by oncoviruses [48]. HCMV mediates both oncomodulation and oncogenesis (Figure 2) [49]. During HCMV infection, the virus expresses viral gene products possessing potential transforming capacities and activating specific molecular pro-oncogenic pathways. HCMV key products involves IE1, IE2, unique short 28 protein (pUS28), viral IL-10 (vIL-10), unique long 76 protein (pUL76), pUL97, pUL82, pUS2, pUL16, 65-kDa tegument protein (pp65), pUL36, pUL37x1, and long non-coding RNA4.9 (lncRNA4.9) [49]. Early HCMV proteins regulate main cellular factors, for instance, retinoblastoma protein family, p53, cyclins, Wnt, phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt), and NF-kB, hence affecting the cell cycle, differentiation, cellular proliferation, apoptosis, and metabolism [50–52]. HCMV-unique short 28 (US28) stimulates signaling pathways that are well-known to interfere with proliferation as well as survival, migration, angiogenesis, and inflammation [53–56]. pp65 tends to incapacitate the intrinsic cellular immune responses [57]. By expressing CMV IL-10 (cmvIL-10), HCMV displays a potent immunosuppressive effect thereby promoting the maturation of protumoral M2 macrophages TAM [58]. Thus, HCMV can entirely promote the steps of classical hallmarks of cancer through the expression of many gene products [49]. Furthermore, a direct oncogenic outcome was revealed by a few HCMV strains, namely, high-risk strains [59], that could promote cellular stress, polyploid giant cancer cells (PGCCs) generation, stemness, and epithelial- to-mesenchymal transition (EMT) plasticity explaining the appearance of aggressive tumor phenotypes, particularly adenocarcinomas, having poor prognosis, metastasis, therapy resistance, and relapse [46, 60]. HCMV-DB and BL, isolated high-risk clinical strains, displayed oncogenic potential in human mammary epithelial cells (HMECs) and replicated in epithelial cells with the interchange of lytic and latent viral cycles promoting the appearance of CMV-transformed HMECs (CTH cells) [60–62]. Two additional high-risk HCMV strains, namely biopsy 544 (B544) and B693, recently isolated from enhancer of zeste homolog 2 (EZH2)High MycHigh triple-negative breast cancer (TNBC) biopsies revealed oncogenic and stemness potential [63, 64]. EZH2 was identified as a downstream target for HCMV-induced Myc upregulation upon HMECs infection with high-risk HCMV strains [64]. In GBM tissues harboring HCMV, EZH2 overexpression was detected [65]. EZH2 is overexpressed in PGCCs highlighting the presence of a potential link between HCMV infection, Myc/EZH2 upregulation, and PGCC generation [64]. Hence, this evidence suggests that some HCMV strains may not only possess an oncomodulatory role but in a certain cellular context, it unveils direct tumor-promoting strategies. Besides the transforming capacities of HCMV, the TME has increasingly been recognized as a key element in tumor progression and metastasis [2]. Restricted immune control against HCMV will favor the productive viral infection in the immune-privileged TME thereby supporting a stem-cell-like state of cancer cells and promoting cancer aggressiveness [66, 67]. HCMV reactivation in M1 macrophages promotes an M2/TAM shift, thus driving the neoplastic progression [21, 68]. Tumor cells evade immune responses promoting EMT, metastasis, and relapse. Therefore, in tumor cells, the association of cellular machinery and viral immune evasion mechanisms may give rise to an environment that enhances limited HCMV replication and triggers cancer cells to evade immune surveillance revealing the bidirectional association between cancer cells and HCMV [21, 69]. Persistent HCMV infection markedly alters the host immune system and has been suggested to trigger or exacerbate age-associated diseases as well as immunosenescence (Figure 3) [70, 71].
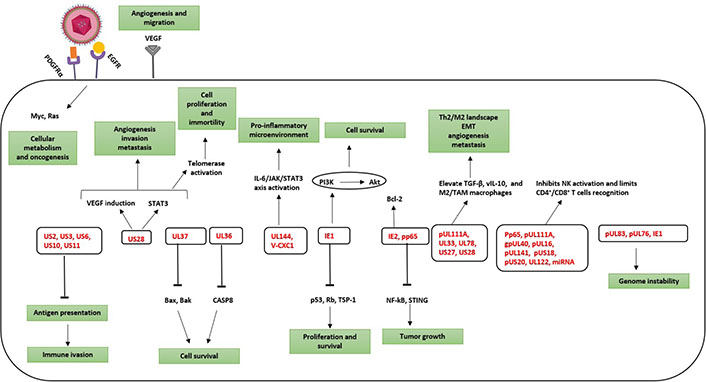
HCMV from oncomodulation to oncogenesis. Activation of specific molecular pathways that are implicated in oncomodulation and cellular transformation. Oncomodulation, which favors the growth and spread of tumor cells, as well as oncogenesis are mainly targeted by HCMV gene products (in red). PDGFRα: platelet-derived growth factor receptor α; EGFR: epidermal growth factor receptor; VEGF: vascular endothelial growth factor; STAT3: signal transducer and activator of transcription 3; JAK: Janus kinase; TGF-β: transforming growth factor β; UL37: unique long 37; V-CXC1: viral chemokine (C-X-C motif) ligand 1; gpUL40: glycoprotein unique long 40; miRNA: microRNA; STING: stimulator of interferon genes; TSP-1: thrombospondin 1; CASP8: caspase 8; Bcl-2: B-cell lymphoma 2; Bax: Bcl-2-associated X protein; Bak: Bcl-2 homologous antagonist/killer; Rb: retinoblastoma protein
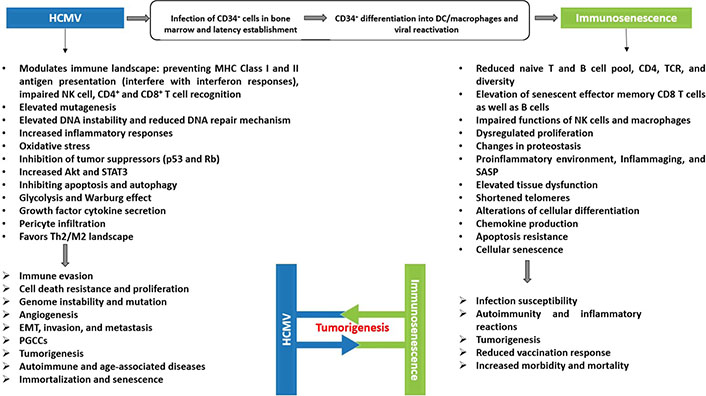
The interplay between HCMV-induced immunosenescence and oncogenesis. A scheme showing the potential association between HCMV, immunosenescence, and tumorigenesis. Persistent HCMV infection fulfills all the cancer hallmarks and is associated with enhanced aging of the immune system. Immunosenescence and inflammaging play a substantial role in the pathogenesis of several serious diseases in the elderly. Both players, HCMV and immunosenescence, have been associated with high mortality rates, chronic diseases, and tumors
Conclusions
HCMV infection and immunosenescence in clinical conditions such as organ transplantation, cancer, immunodeficiency, as well as autoimmune and inflammatory illnesses, support the concept that HCMV can affect their progression by inducing immunosenescence. In return, immunosenescence favors HCMV reactivation from latency in an inflammatory microenvironment (inflammaging). Viral reactivation will trigger HCMV-driven oncomodulation by low- and high-risk HCMV strains, and could also promote the initiation of tumorigenesis, particularly by high-risk HCMV strains, thus directly favoring the appearance of tumors especially adenocarcinoma and glioblastoma in an immunocompromised TME with immunosenescence and inflammaging traits. A better understanding of the complex interaction between HCMV, immunosenescence, and tumors will open new perspectives to explore novel therapeutic approaches that will reverse immunosenescence and boost the immune system to fight viral infections, especially HCMV, and tumor development in the elderly.
Abbreviations
| Akt: | protein kinase B |
| CMV: | cytomegalovirus |
| DCs: | dendritic cells |
| EMT: | epithelial-to-mesenchymal transition |
| EZH2: | enhancer of zeste homolog 2 |
| HCMV: | human cytomegalovirus |
| HMECs: | human mammary epithelial cells |
| IE1: | immediate early 1 |
| IFN-γ: | interferon γ |
| IL-6: | interleukin-6 |
| KLRG1: | killer cell lectin-like receptor G1 |
| NF-kB: | nuclear factor-kB |
| NK: | natural killer |
| p65: | p65 tumor suppressor protein |
| PGCCs: | polyploid giant cancer cells |
| pp65: | 65-kDa tegument protein |
| pUL76: | unique long 76 protein |
| pUS28: | unique short 28 protein |
| SASP: | senescence-associated secretory phenotype |
| TAMs: | tumor-associated macrophages |
| Th1: | T-helper 1 |
| TME: | tumor microenvironment |
| UL37: | unique long 37 |
| US28: | unique short 28 |
Declarations
Author contributions
FB, REB, SMB, MDA, and GH: Writing—original draft, Writing—review & editing. GH: Conceptualization, Supervision.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
GH received grants from the University of Franche-Comté [3300]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2023.