
Abstract
Tumor necrosis factor receptor (TNFR)-associated factors (TRAFs) are a family of intracellular signaling adaptors that associate with the cytoplasmic tails of a diverse range of lymphocyte receptors, including members of the TNFR superfamily, the Toll-like receptor (TLR)/interleukin-1 (IL-1) receptor superfamily, and the IL-6 receptor family that are major targets for therapeutic intervention for inflammatory diseases. TRAF5 is one of the seven family members of the TRAF family and is highly expressed by B- and T-lymphocytes. As compared to other family members, the biological and pathophysiological functions of TRAF5 have remained ambiguous since its discovery. TRAF5 promotes lymphocyte signaling for the TNFR family molecules such as glucocorticoid-induced TNFR family-related protein (GITR), CD27, and CD40. In contrast, TRAF5 limits the activity of the common signaling receptor subunit glycoprotein 130 kDa (gp130) in CD4+ T cells that requires signaling by IL-6 and IL-27. TRAF5 also restrains TLR signaling in B cells. Thus, TRAF5 regulates lymphocyte signaling in both positive and negative ways. This review will summarize the findings of recent studies of TRAF5 in terms of how TRAF5 regulates signaling in lymphocytes and other cell types and how TRAF5 expression contributes to inflammatory and autoimmune diseases in mice and humans.
Keywords
Tumor necrosis factor receptor-associated factor 5, interleukin-6, Janus kinase, signal transducer and activator of transcription, Toll-like receptor, tumor necrosis factor receptor superfamily, inflammation, autoimmunityIntroduction
Two proteins that were associated with tumor necrosis factor receptor 2 (TNFR2) [also known as TNFR superfamily member 1B (TNFRSF1B)] were firstly identified and designated TNFR-associated factor 1 (TRAF1) and TRAF2 [1]. To date, seven members numbered sequentially TRAF1 through TRAF7, have been identified in the TRAF family. TRAF molecules are a family of signaling adapters that play vital roles in regulating cellular signaling not only by members of the TNFRSF and the Toll-like receptor (TLR)/interleukin-1 (IL-1) receptor superfamily but also by unconventional receptors for cytokines including IL-6 [2–8]. Unlike the non-conventional TRAF7, TRAF1 to TRAF6 share the conserved C-terminal TRAF domain, which binds to the cytoplasmic domains of a range of cell surface receptors (Figure 1). The TRAF domains of TRAF1, 2, 3 and 5 share overlapping binding sites on those receptors [9, 10]. Signaling mediated by TRAFs greatly impacts the activation, differentiation, and survival of cells of innate and adaptive immunity [2].
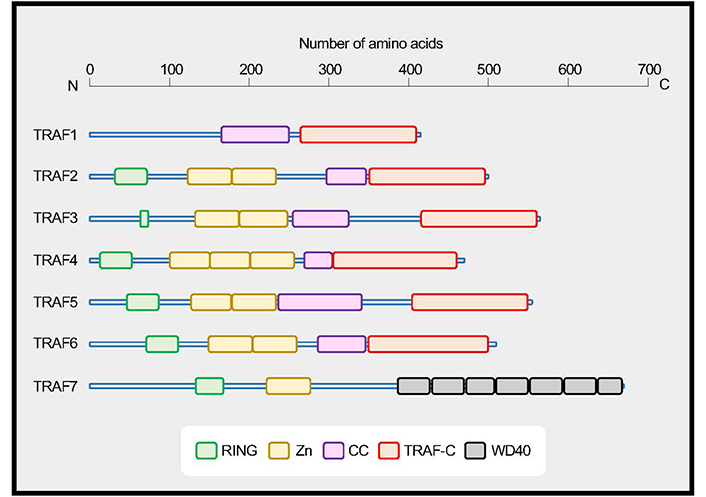
TRAF family proteins. TRAF1 to TRAF6 share a conserved coiled-coil (CC)/TRAF-C domain, and TRAF7 has a series of 40 amino acid peptide repeats terminating in tryptophan (W) and aspartic acid (D) residues (WD40) domains. TRAFs, with the exception of TRAF1, have an N-terminal really interesting new gene (RING) domain and zinc finger motifs (Zn). Data are based on UniProt (https://www.uniprot.org). TRAF1, Q13077; TRAF2, Q12933; TRAF3, Q13114; TRAF4, Q9BUZ4; TRAF5, O00463; TRAF6, Q9Y4K3; TRAF7, Q6Q0C0
The function of TRAF5 in vivo has remained ambiguous since its discovery. TRAF5 was discovered as a potential signaling regulator for the lymphotoxin-β receptor (LT-βR) [11] and CD40 [12]. TRAF5 is highly expressed in the lung, spleen, and thymus [11, 12] and abundantly expressed in resting B- and T-lymphocytes [13]. Other members of the TRAF family that are structurally similar to TRAF5 are TRAF2 and TRAF3 (Figure 1). Whereas germline deletion of Traf2 or Traf3 in mice leads to premature death with severe developmental abnormalities [14–16], that of Traf5 displays no obvious abnormalities [17], suggesting that TRAF5 likely plays minor roles at a steady state. However, Traf5 deficient mice showed exacerbated disease phenotypes in the context of a proinflammatory milieu [13, 18–23]. This leads to the question of how TRAF5 regulates cellular signaling in the context of inflammatory diseases. This review discusses the current understanding of the significance of TRAF5 in regulating signaling in B- and T-lymphocytes and also other cell types in both positive and negative ways that would be critical for the development of inflammatory and autoimmune diseases.
Traf5-knockout mouse
Knockout mouse studies have greatly helped to understand the biological roles of TRAF molecules in the mammalian immune system. TRAF5 and TRAF3 share a high degree of sequence homology in the TRAF domain, however, the phenotype of Traf3−/− mice is totally different from that of Traf5−/− mice. Traf3−/− mice display decreased spleen size and lymphocyte count, stunted growth and progressive hypoglycemia, hypercortisolemia, and leucopenia, resulting in premature death within 2 weeks of age [16]. In contrast, Traf5−/− mice are born healthy and show no obvious abnormalities through 24 weeks of age. Furthermore, there is no developmental defect of lymph nodes (LNs) in Traf5−/− mice. The expression of CD3, CD4, CD8, and B220 on thymocytes, splenocytes, or LN cells is comparable between Traf5+/+ and Traf5−/− mice [17]. The development of secondary lymphoid organs including LNs and Peyer’s patches (PPs) requires the interaction between LT-α1β2 (LT-β) and LT-βR. TRAF2 and TRAF5 work as signal transducers for the LT-βR [11, 24]. The development of PPs, but not LNs, was severely impaired in Traf2 and Traf5 double knockout mice, and a mild defect in PPs was observed in Traf5 single knockout mice [25]. Thus, TRAF5 may play a minor role in the development of lymphoid tissues. Expression of Traf5 messenger RNA (mRNA) was significantly higher in B cells than in T cells, natural killer (NK) cells, plasmacytoid dendritic cells (pDCs), or macrophages [3, 13]. Traf5−/− B cells showed defects in proliferation and up-regulation of various surface molecules, including CD23, CD54, CD80, CD86, and CD95 in response to CD40 stimulation in vitro. Traf5−/− B cells produced decreased amounts of immunoglobulin M (IgM) and IgG after stimulation with CD40L and IL-4 in vitro [17], indicating that TRAF5 promotes CD40 signaling in B cells.
Traf5−/− mice have decreased pDCs in bone marrows and spleens, and the levels of the transcription factor 4 (Tcf4), Spi-B transcription factor (Spib), and B cell CLL/lymphoma 11A (Bcl11a), which are critical for pDC commitment, were significantly reduced in bone marrow pDCs in Traf5−/− mice [26]. This study suggests that TRAF5 promotes the development of pDCs from bone marrow progenitors, although the detail is unknown.
Traf5 displays both anti-inflammatory and pro-inflammatory functions in various murine models of autoimmune and inflammatory diseases.
Anti-inflammatory function
TRAF5 limits the induction of T helper 2 (Th2) immunity. In a mouse model of asthma, after intraperitoneal immunization of ovalbumin (OVA) in alum adjuvant, Traf5−/− mice displayed enhanced susceptibility to the development of allergic lung inflammation when challenged with aerosolized OVA via the airway, which was characterized by eosinophilia, airway hyperreactivity, higher levels of IL-5 and IL-13 in bronchoalveolar lavage fluids, and OVA-specific IgE in plasma [18].
TRAF5 inhibits the differentiation of IL-17-producing CD4+ T cells. Traf5−/− B6 mice immunized with amino acids 35–55 myelin oligodendrocyte glycoprotein (MOG35–55) peptide emulsified in complete Freund’s adjuvant (CFA) displayed exacerbated Th17 cell-dependent neuroinflammation in a model of experimental autoimmune encephalomyelitis (EAE). Draining LN cells from Traf5−/− mice produced a higher amount of IL-17 in response to the MOG peptide. Accumulation of IL-17+CD4+ T cells in the central nervous system was enhanced in Traf5−/− mice. Sublethally irradiated wild-type recipient mice with Traf5−/−CD4+ T cells induced significantly higher EAE scores than those of recipients with Traf5+/+CD4+ T cells. This study demonstrates that TRAF5 expressed by CD4+ T cells limits the generation of pathogenic Th17 cells that are responsible for the induction of EAE disease [13].
The expression of TRAF5 in mature pDCs in the periphery negatively regulates the wound healing process driven by pDCs in the skin. Traf5−/− mice displayed increased wound closure after skin injury. Traf5-deficiency caused enhanced accumulation of pDCs in skin wounds and promoted wound-repair responses. Traf5−/− pDCs produced higher amounts of pro-inflammatory cytokines including IL-6, TNF-α, and interferon α/β (IFN-α/β) in response to TLR7 and TLR9 stimulation. In the injured skin of Traf5−/− mice, pro-inflammatory cytokines and chemokines including IL-6, TNF-α, IFN-α/β, macrophage inflammatory protein-1α (MIP-1α), and transforming growth factor-beta (TGF-β) required for wound repair were significantly elevated. Traf5−/− pDCs had higher expression of C-X-C motif chemokine receptor 3 (CXCR3) and IFN regulatory factor 5 (IRF5), and the increased IRF5 in Traf5−/− pDCs supported pro-inflammatory and skin-tropic characteristics of pDCs that play important roles for wound healing in the skin [27].
TRAF5 exhibits anti-inflammatory function in atherogenesis. Traf5 and low-density lipoprotein receptor (Ldlr) double knockout mice consuming a high-fat diet (HFD) for 18 weeks developed enhanced atherosclerosis lesions as compared with Ldlr single knockout mice. Atherosclerotic plaques of the double knockout mice contained more macrophages. Traf5-deficiency increased the expression of adhesion molecule vascular cellular adhesion molecule-1 (VCAM-1) and chemokine monocyte chemoattractant protein-1 (MCP-1). Bone marrow derived macrophages (BMMs) from the double knockout mice expressed increased scavenger receptor CD36 and internalized more LDL. BMMs and endothelial cells from the double knockout mice showed increased c-Jun N-terminal kinase (JNK) activation in response to TNF-α. In accordance with the murine study, total blood from patients with stable or acute coronary heart disease contained decreased amounts of TRAF5 mRNA as compared with healthy controls [21].
Similarly, Traf5−/− mice consuming HFD for 18 weeks showed increased body weight with fat depots, insulin resistance, hyperinsulinemia, and increased fasting glucose plasma levels. Livers of Traf5−/− mice with HFD displayed enhanced weight, fat accumulation, and signs of steatosis. Visceral adipose tissues of Traf5−/− mice with HFD contained higher numbers of pro-inflammatory CD8+ T cells and CD11c+ macrophages and expressed enhanced levels of pro-inflammatory cytokines TNF-α and IL-12. Traf5-deficiency in adipocytes promoted the induction of pro-inflammatory chemokines, such as MCP-1 and MIP-1α, and regulated upon activation, normally T-expressed, and presumably secreted (RANTES). Blood cells and subcutaneous adipocytes from patients with a high cardiovascular risk (a body mass index > 25 kg/m2) contained decreased TRAF5 mRNA. This study suggests that decreasing expression of TRAF5 in adipose tissue causes increased production of chemokines and subsequent homing of inflammatory cells into fat pads [20].
TRAF5 limits the development of hepatic steatosis. Traf5−/− mice fed with HFD exhibited enhanced hepatic steatosis. After 24 weeks of HFD feeding, Traf5−/− mice displayed higher liver weights, enhanced lipid accumulation in the liver, and increased serum levels of alanine aminotransferase (ALT) and aspartate amino transferase (AST), indicating compromised hepatic functions. Traf5−/− mice livers and sera contained increased levels of pro-inflammatory cytokines IL-1β, IL-6, TNF-α, and MCP-1 and decreased levels of anti-inflammatory cytokine IL-10. Traf5−/− mice showed enhanced HFD-induced hepatic fibrosis, as evidenced by the higher expression of fibrogenic genes including collagen type 1 alpha 1 chain (Col1a1) and Tgfβ1. Traf5−/− mice livers contained enhanced JNK1 which was responsible for the obesity-related pathology. Liver-specific overexpression of TRAF5 restored the inflammatory phenotype in the liver. In accordance with this, the level of TRAF5 protein was decreased in the liver of nonalcoholic fatty liver disease (NAFLD) patients. This study suggests that TRAF5 ameliorates nonalcoholic steatohepatitis (NASH)-like phenotypes [23].
TRAF5 expression protects against cardiac damage in response to myocardial ischemia reperfusion injury. Cardiomyocytes from wild-type mice expressed higher levels of Traf5 mRNA and Traf5 protein after ischemia reperfusion injury. Cardiac tissues from Traf5−/− mice displayed exacerbated inflammation and cell death after myocardial ischemia reperfusion injury, which led to severe heart damage. Cardiac tissues from Traf5−/− mice following myocardial ischemia reperfusion injury contained higher levels of pro-inflammatory mediators TNF-α, IL-6, IL-1β, and MCP-1 and lower levels of AKT and its downstream regulators’ forkhead box O1 (FOXO1), mechanistic target of rapamycin (mTOR) and protein 70 S6 kinase (p70S6K). This study suggests that increased expression of TRAF5 is protective against myocardial ischemia reperfusion injury [22].
Pro-inflammatory function
TRAF5 augments lung inflammation mediated by oncostatin M (OSM). Traf5−/− mice intranasally treated with OSM, which is highly upregulated in human inflammatory diseases such as lung fibrosis and inflammatory bowel diseases, displayed reduced infiltration of eosinophils in the lung. Lung stromal/fibroblast cells and endothelial cells in Traf5−/− mice administered with OSM expressed reduced levels of VCAM-1, which is a ligand for very late activation antigen-4 (VLA-4) and plays an important role in extravasation and accumulation of eosinophils in the lung. Upon stimulation with OSM in vitro, lung fibroblasts purified from Traf5−/− mice expressed reduced levels of VCAM-1, as compared with those from Traf5+/+ mice. In contrast, upon stimulation with TNF-α or IL-6, Traf5−/− lung fibroblasts expressed a similar level of VCAM-1 as Traf5+/+ lung fibroblasts, suggesting that the effect of OSM on VCAM-1 induction is independent of TNF-α or IL-6 [28]. This study suggests that TRAF5 works as a pro-inflammatory mediator for OSM-mediated lung inflammation, although the detail remains unclear.
TRAF5 promotes acute chemical-induced colitis. Traf5−/− mice showed attenuated inflammation in dextran sulfate sodium (DSS)-induced colitis. Intestinal epithelial cells from Traf5−/− mice had decreased levels of nuclear factor kappa B (NF-κB)-dependent genes as early as day 3 after DSS treatment. Traf5-deficiency maintained the barrier integrity of the intestine and suppressed the infiltration of neutrophils. Experiments with bone marrow chimeras indicated that Traf5-deficiency in nonhematopoietic cells ameliorated colitis. The levels of Traf2 protein in the colons and intestinal epithelial cells from Traf5−/− mice were significantly reduced after DSS treatment. Proinflammatory cytokines TNF-α and IFN-γ increased the degradation of Traf2 protein in a proteasome-dependent manner in Traf5−/− nonhematopoietic cells. This study suggests that Traf5 expressed in nonhematopoietic cells in the colon supports inflammatory responses mediated by TNFR-TRAF2/5-NF-κB signaling and is responsible for acute inflammation in the intestine [29]. However, the above results from our group are inconsistent with the former study that showed that Traf5−/− mice displayed enhanced DSS colitis with enhanced CD4+ T cell-mediated inflammation in the colon [30]. The discrepancy may be due to differences in the intestinal microbiota and the gender and age of mice.
Human diseases
The association of TRAF5 genetic variants with several human autoimmune diseases has been reported.
Ankylosing spondylitis (AS) is a type of arthritis that causes long-term inflammation in the joints and ligaments of the spine. Compared with healthy controls, the cytosine-phosphate-guanine (CpG) island of the TRAF5 gene promoter was highly methylated in AS patients. In addition, the relative mRNA expression level of TRAF5 was significantly reduced in AS patients. This study suggests that decreased expression of TRAF5 contributes to the pathogenesis of AS [31].
Rheumatoid arthritis (RA) is a multifactorial disease that causes persistent synovial inflammation in the joints, deforming them and affecting their functionality. A single nucleotide polymorphism (SNP), located upstream of the TRAF5 gene, has demonstrated a significant association with RA in a large cohort of RA patients [32].
Uveitis is a type of intraocular inflammation that causes visual impairment and loss. Two SNPs of the TRAF5 gene were associated with acute anterior uveitis, and one of the SNPs was also associated with pediatric uveitis [33]. Vogt-Koyanagi-Harada (VKH) syndrome and Behçet’s disease (BD) are two important uveitis entities. VKH syndrome is a granulomatous inflammatory disease directed against pigmented structures, such as the eye, inner ear, meninges, skin, and hair. BD is characterized by bilateral granulomatous uveitis and a type of inflammatory disorder that causes recurrent oral aphthae, uveitis, multiform skin lesions, and genital ulceration. Three SNPs of TRAF5 were significantly associated with VKH syndrome and BD [34]. These results suggest that gene polymorphisms of TRAF5 increase the risk for uveitis.
The reduced level of TRAF5 expression may cause inflammatory diseases.
Sjögren syndrome (SS) is an autoimmune disorder characterized by lymphocytic infiltrates in exocrine organs, the tear glands, and salivary glands. Patients with SS displayed cardinal clinical signs of dry eye keratoconjunctivitis sicca (KCS). Decreased TRAF5 gene expression was observed in the conjunctiva of SS KCS patients [35].
Systemic lupus erythematosus (SLE) is an autoimmune disease with multiple associated autoantibodies, which affects many organs and systems in the body. In SLE patients, a subset of CD27−IgD−CXCR5−CD11c+ (DN2) B cells, representing pre-plasma cells, was elevated, and DN2 B cells expressed higher and lower amounts of TLR7 and TRAF5, respectively. DN2 B cells were hyper-responsive to TLR7 and hypo-responsive to CD40 stimulation, suggesting that downregulation of TRAF5 in B cells augments TLR7 signaling and promotes the development of autoreactive B cells that are responsible for SLE [36].
Human TRAF5 was included in a gene cluster that was downregulated with aging [37], implying that the lower expression of TRAF5 may cause age-associated chronic inflammation.
The role of TRAF5 in lymphocyte signaling
TNFR superfamily
Glucocorticoid-induced TNFR family-related protein
TRAF5 promotes glucocorticoid-induced TNFR family-related protein (GITR) signaling. GITR is expressed at low levels on naive or resting CD4+ and CD8+ T cells and is upregulated after T cell activation. GITR can interact with TRAF2, 3, and 5. Ligation of GITR on T cells activates NF-κB1/2 as well as the extracellular-signal-regulated kinases (ERK), JNK, and p38 mitogen-activated protein kinase (MAPK) pathways that enhance T cell proliferation, survival, and cytokine production [38–42]. GITR cross-linking on antigen-activated GITR+CD4+ T cells from Traf5−/− DO11.10 mice displayed diminished activation of NF-κB1, p38, and ERK pathways. Consistently, Traf5-deficiency in CD4+ T cells resulted in diminished T cell proliferation and IL-2 production mediated by T cell receptor (TCR)/CD3 and GITR [19, 43]. Small interfering RNA-mediated Traf5 knockdown in primary CD8+ T cells caused impaired NF-κB1 activation mediated by GITR [44]. These results show that TRAF5 positively regulates co-signaling mediated by GITR expressed by CD4+ and CD8+ T cells.
CD27
TRAF5 increases CD27 signaling. CD27 is constitutively expressed on naive CD4+ and CD8+ T cells. CD27 can interact with TRAF2, 3, and 5. As with GITR, engagement of CD27 on T cells activates NF-κB1/2 as well as MAPK pathways that enhance T cell proliferation, survival, and cytokine production [38, 45–47]. The initial study demonstrated that Traf5−/− thymocytes showed attenuated proliferation mediated by anti-CD3 monoclonal antibody and membrane-bound CD27 ligand (CD70). However, this study demonstrated that CD27-mediated activation of NF-κB and JNK pathways was intact in Traf5−/− thymocytes [17].
Traf5−/− mice showed decreased CD8+ T cell responses against infection with Listeria monocytogenes, which resulted in lower production of memory CD8+ T cells and delayed bacterial clearance from the liver. Traf5−/− CD8+ T cells displayed the increased activity of caspase-3 and increased susceptibility to apoptosis following stimulation with TCR/CD27. However, Traf5−/− CD8+ T cells did not show defects in NF-κB1 as well as the ERK, JNK, and p38 MAPK pathways in response to membrane-bound CD70 in vitro. This study suggests that TRAF5 works as a positive regulator of CD8+ T cell survival via TNFR family molecules including CD27 [48].
CD40
TRAF5 augments CD40 signaling. Upon interaction with the CD40 ligand (CD40L) expressed by activated CD4+ T cells, CD40 expressed by B cells delivers activating signals, which are critical for germinal center formation and class switch recombination. CD40 interacts with TRAF1, 2, 3, 5, and 6. Engagement of CD40 on B cells activates NF-κB1/2 as well as MAPK pathways [49, 50]. TRAF5 has been suggested to bind to CD40 via TRAF3 and indirectly regulate CD40 signaling [51, 52]. Traf5−/− B cells displayed impaired proliferation mediated by CD40 signaling and decreased up-regulation of various surface molecules, including CD23, CD54, CD80, CD86, CD95, intercellular adhesion molecule 1 (ICAM-1), LT-α in response to CD40 stimulation in vitro. CD40-mediated activation of NF-κB and JNK pathways was intact in Traf5−/− B cells. CD40-mediated Ig production and class switching in vitro were significantly reduced in Traf5−/− B cells and mild defects in affinity maturation of IgG to a T-dependent antigen in vivo were observed in Traf5−/− mice [12, 17, 19]. These studies indicate that Traf5-deficiency leads to partial impairments of CD40 signaling in B cells.
The above findings argue for positive regulatory or pro-inflammatory roles of TRAF5 in TNFR family signaling. Additionally, TRAF5 works as a negative regulator or an anti-inflammatory mediator in TLR and IL-6 receptor family signaling, which might be critical for the regulation of inflammatory and autoimmune diseases.
TLR family
TLRs are pattern recognition receptors, providing a first-line defense against pathogens by recognizing pathogen-associated molecular patterns. Although cells of innate immunity such as dendritic cells (DCs) and macrophages highly express TLRs, B- and T-lymphocytes also have sufficient levels of some TLRs that can recognize and respond to TLR ligands. TRAF adaptor proteins are involved in two major TLR signaling pathways, myeloid differentiation primary response gene 88 (MyD88)-TRAF6 and Toll-interleukin-1 receptor (TIR) domain-containing adapter protein inducing IFN-β (TRIF)-TRAF3/6 pathways [53–55].
Traf5−/− B cells produced significantly higher levels of IL-6, IL-10, IL-12p40, and TNF-α in response to lipopolysaccharide (LPS)(TLR4), R848 (TLR7), or CpG DNA (TLR9) in vitro, although B cell survival and proliferation were unaltered by the deficiency of Traf5. In line with this observation, bone marrow-derived DCs and macrophages generated from Traf5−/− mice also produced increased amounts of cytokines, albeit less pronounced, as compared to B cells. Traf5−/− B cells produced enhanced levels of IgM in response to TLR4 or TLR7 stimulation in vitro. Although NF-κB activation was unaltered, Traf5−/− B cells displayed enhanced activation of the ERK and JNK MAPK pathways in response to TLR7 stimulation in vitro. After ligation of TLR7 in B cells, TRAF5 interacted with MyD88 and TGF-β activated kinase 1 binding protein 2 (TAB2) in the TLR signaling complex and inhibited the interaction between TAB2 and TRAF6. Traf5−/− mice have increased marginal zone B cells (IgMhiCD21hi) in the spleen. This study indicates that TRAF5 incorporated into the TLR signaling complex disrupts the interaction between TAB2 and TRAF6 that requires for the activation of the TLR signaling pathway. Thus, TRAF5 in B cells negatively regulates TLR signaling [56].
In accordance with the above study, a subset of B cells, called DN2 cells, from patients with SLE had decreased amounts of TRAF5 and displayed hyper-responsive to TLR7 stimulation [36].
IL-6R family
IL-6
Pro-inflammatory cytokine IL-6 interacts with the ligand-binding subunit IL-6R, and the IL-6-IL-6R complex binds to the signaling transducing common chain glycoprotein 130 kDa (gp130), which leads to dimerization of gp130 and activates the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway [57, 58].
TRAF5 negatively regulates the IL-6R signaling in CD4+ T cells. In the presence of exogenous IL-6 in the differentiating culture in vitro, naive CD4+ T cells from Traf5−/− mice produced higher amounts of IL-17 and IL-21, whose production is controlled by IL-6. However, endogenous production of IL-6 in the T cell culture was comparable between Traf5+/+ and Traf5−/− CD4+ T cells, showing that TRAF5 limits the activity of IL-6R signaling but not the production of IL-6 from T cells [13].
Th17-associated genes including Il17a, Il17f, retinoid-related orphan receptor C (Rorc), and IL-23 receptor (IL-23r) were significantly increased in Traf5−/−CD4+ T cells in Th17-skewing cultures containing IL-6 and TGF-β. Adoptively transferred Traf5−/− naive CD4+ T cells from OT-II mice, which have a transgenic expression of OVA-specific TCR, exhibited enhanced Th17 differentiation in recipient wild-type mice that had been immunized with OVA in CFA. Thus, TRAF5 expressed in CD4+ T cells antagonizes the differentiation of Th17 cells both in vitro and in vivo [13].
Traf5−/− naive CD4+ T cells stimulated with the IL-6-IL-6R complex without TCR or CD28 triggering displayed enhanced phosphorylation of JAK1 and STAT3, although both Traf5+/+ and Traf5−/− CD4+ T cells expressed comparable levels of IL-6R and gp130 on the surface. Phosphorylations of STAT3 mediated by IL-10 and IL-21 in CD4+ T cells were comparable between Traf5+/+ and Traf5−/− CD4+ T cells, indicating the specific activity of TRAF5 for IL-6R signaling. Retrovirally transduced Traf5 in CD4+ T cells inhibited the phosphorylation of STAT3 mediated by IL-6-IL-6R, and the exogenous Traf5 significantly suppressed the development of IL-17-producing Th17 cells in vitro. These results demonstrate that TRAF5 expressed by CD4+ T cells negatively regulates IL-6R signaling that requires the differentiation of IL-17-producing pro-inflammatory CD4+ T cells [13, 59, 60].
Traf5-deficiency exacerbates neuroinflammation in EAE, which is an IL-6-dependent disease mediated by pathogenic Th17 cells [61]. Traf5−/− B6 mice immunized with MOG35–55 emulsified in CFA exhibited significantly higher clinical scores of EAE and body-weight loss when compared with Traf5+/+ B6 mice. The draining LNs and central nervous system of Traf5−/− mice immunized with MOG35–55 contained higher numbers of IL-17+CD4+ T cells. Recipient mice with Traf5−/− donor CD4+ T cells showed significantly higher EAE scores than those of recipients with Traf5+/+ donor CD4+ T cells, indicating that Traf5 expressed in CD4+ T cells limits the generation of pathogenic Th17 cells responsible for the disease [13].
TRAF5 interacted with gp130 via its C-terminal TRAF-C domain, and a cytoplasmic region of gp130 was responsible for the interaction (TRAF-binding region in Figure 2). The C-terminal domain TRAF2 is also bound to the TRAF-binding region in gp130. Without this TRAF-binding region in gp130, TRAF5 or TRAF2 could not inhibit the signaling activity of the IL-6R complex. Although TRAF5 did not inhibit the binding between gp130 and JAK1, TRAF5 decreased IL-6-IL-6R-driven phosphorylations of JAK1, gp130, and STAT3, suggesting that TRAF5 associated with gp130 regulates the dimer formation of gp130 and the transphosphorylation of JAK proteins in the receptor complex [13, 59, 60].
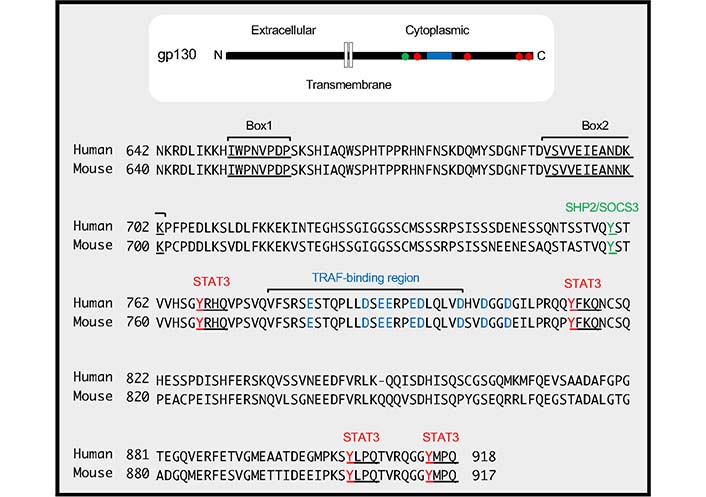
TRAF-binding region in gp130. The cytoplasmic amino acid sequences of human gp130 (residues from 642 to 918) and mouse gp130 (residues from 640 to 917) are shown. The positions of Box1 and Box2 in gp130, which are responsible for JAK binding, are indicated by underlines. The TRAF-binding region, which includes an amino acid sequence VFSRSESTQPLLDSEERPEDLQLVD (residues from 776 to 800 in humans and from 774 to 798 in mice), is shown. Acidic amino acid residues (D and E) in and around the TRAF-binding region, which are critical for the binding between TRAF2/5 and gp130, are indicated in blue. The phosphorylation of tyrosine residue, 759 in humans and 757 in mice indicated by green is involved in binding to Src homology 2 (SH2)-containing protein tyrosine phosphatase-2 (SHP2) and suppressor of cytokine signaling 3 (SOCS3). The distal four tyrosine residues, 767, 814, 905, 915 in humans and 765, 812, 904, 914 in mice indicated by red and underlines, form YxxQ motifs (where ‘x’ indicates any amino acid), which are needed for STAT3 binding
Unstimulated naive CD4+ T cells express relatively higher levels of TRAF5, gp130, and IL-6R, and those expressions decreased after TCR/CD28 triggering. For this reason, TRAF5 could not inhibit IL-6R signaling in the later phase of T cell activation [59]. Unlike CD4+ T cells, Traf5+/+ macrophages had lower amounts of Traf5 as compared to Traf5+/+CD4+ T cells, and phosphorylation of STAT3 mediated by IL-6-IL-6R was comparable between Traf5+/+ and Traf5−/− macrophages [13]. Thus, substantial amounts of cellular expression of TRAF5 would be critical for the inhibitory activity of TRAF5 for IL-6R signaling.
Since OX40 (TNFRSF4 or CD134) can recruit TRAF5 to its TRAF-binding motif [62, 63], initial experiments performed by our group [18] suggested that TRAF5 directly regulated OX40 signaling. Our group observed that an agonistic monoclonal antibody to OX40 (OX86) significantly increased the production of IL-17 and IL-21 from activated Traf5−/− CD4+ T cells in vitro when compared with Traf5+/+CD4+ T cells and that this response was mediated by IL-6 produced from anti-OX40 stimulated cells (blocking IL-6 activity canceled the overproduction of IL-17 and IL-21) [13]. IL-6 stimulates CD4+ T cells to produce IL-17 and IL-21 and also supports the differentiation of Th2 and Th17 cells [64, 65]. From these observations, our group thought that IL-6R signaling might be negatively controlled by TRAF5. Indeed, exogenous IL-6 added into T cell cultures induced more production of IL-17 and IL-21 by Traf5−/− CD4+ T cells and more phosphorylation of STAT3 in Traf5−/− CD4+ T cells [13]. The inhibitory activity of TRAF5 for IL-6R signaling does not rule out the authentic activity of TRAF5 for the TNFR family molecules, but the inhibitory role of TRAF5 for IL-6R signaling would provide a logical explanation for the increased development of Traf5−/−CD4+ T cells into the Th2 and Th17 lineages [13, 18] and for the anti-inflammatory activity of TRAF5 observed in several autoimmune and inflammatory disease models using Traf5−/− mice [13, 18, 20, 22, 23].
The phenotype of Traf5-deficiency in the JAK-STAT3 signaling was more obvious when CD4+ T cells were stimulated with IL-6-IL-6R instead of IL-6 alone, suggesting that TRAF5 preferentially inhibits IL-6 trans-signaling. The IL-6 trans-signaling plays dominant roles in inflammatory and autoimmune diseases including colitis, tissue fibrosis, arthritis, allergy, cardiovascular diseases, and cancers [66]. Thus, it will be important to understand the molecular mechanisms by which TRAF5 controls the JAK-STAT3 pathway mediated by the IL-6 trans-signaling in CD4+ T cells in the context of chronic inflammatory disease conditions.
IL-27
Given the negative regulatory function of TRAF5 for IL-6R signaling, our group decided to examine whether TRAF5 also displays a similar function for gp130 utilized by other IL-6 family cytokines. IL-27, a member of the IL-6 family, which is composed of IL-27p28 and Epstein-Barr virus (EBV)-induced gene 3 (EBI3), binds to a heterodimer receptor composed of WSX-1 [IL-27Rα or T-cell cytokine receptor (TCCR)] and gp130. CD4+ T cells express both WSX-1 and gp130, and IL-27 binding to these receptors activates the JAK-STAT pathway [67, 68]. The IL-27-WSX-1-gp130 predominantly activates STAT1 as compared to STAT3, whereas the IL-6-IL-6R-gp130 mainly activates STAT3 as compared to STAT1 [69, 70].
Indeed, TRAF5 acts as a negative regulator in IL-27R signaling in CD4+ T cells. Upon short-term stimulation with IL-27, Traf5−/− naive CD4+ T cells displayed rapidly upregulated phosphorylated STAT1 and STAT-regulated genes, Soxs3 and T-box transcription factor 21 (Tbx21) as compared to Traf5+/+ naive CD4+ T cells, although both CD4+ T cell groups expressed equivalent amounts of WSX-1 and gp130. After TCR triggering in the presence of IL-27, Traf5−/− naive CD4+ T cells expressed increased IL-10 and IL-21 and exhibited enhanced proliferation when compared with Traf5+/+ naive CD4+ T cells. TRAF5 was associated with gp130 of the IL-27R signaling complex. Although Traf5-deficiency led to increased activation of the IL-27R signaling pathway in CD4+ T cells, Traf5-deficiency resulted in decreased activation of the GITR pathway in CD4+ T cells, indicating that TRAF5 differentially regulates signals mediated by these receptors in CD4+ T cells (Figure 3) [19].
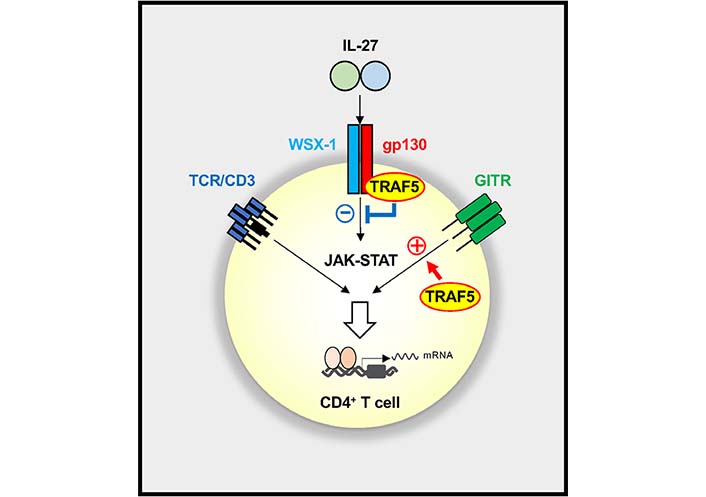
Reciprocal regulation of IL-27R and GITR signaling pathways by TRAF5 in CD4+ T cells. TRAF5 constitutively associated with IL-27R (gp130 and WSX-1) limits the activation of the JAK-STAT pathway mediated by IL-27, whereas TRAF5 inducibly recruited to engaged GITR promotes the activation of GITR signaling in CD4+ T cells. The balance between positive and negative activities of TRAF5 for those signaling pathways controls the outcome of the CD4+ T cell response. A plus symbol denotes an activating signal, and a minus symbol shows an inhibitory signal
Traf5−/− mice showed exacerbated delayed-type hypersensitivity (DTH) mediated by IL-27, whose response is driven by pro-inflammatory Th1 cells [71, 72]. Traf5-deficiency promoted IL-27-mediated differentiation of Th1 cells in vitro. To evaluate the functional significance of TRAF5 for IL-27-driven differentiation of Th1 cells in vivo, Traf5+/+ and Traf5−/− mice were immunized and challenged with methyl bovine serum albumin (mBSA) to induce DTH responses, and IL-27 was additionally injected into these mice at early phases of T cell development. Traf5−/− mice injected with IL-27 displayed significantly enhanced footpad swelling as compared with Traf5+/+ mice. Spleen cells from those Traf5−/− mice extensively proliferated and produced significantly higher amounts of IFN-γ in response to mBSA ex vivo, demonstrating that TRAF5 limits the activity of IL-27R signaling that promotes the differentiation of Th1 cells [19].
Conclusions
Understanding of biological and pathophysiological functions of TRAF5 in lymphocytes and other cell types has progressed over the past few decades. TRAF5 plays important regulatory roles in pro-inflammatory signaling pathways in several members of the TNFR, the TLR/IL-1R, and the IL-6R superfamilies that are major targets for therapeutic intervention for inflammatory diseases. Thus, it is important to elucidate the molecular functions of TRAF5 that are responsible for the aberrant activation of lymphocytes. TRAF5 can interact with multiple intracellular proteins, thereby increasing or decreasing the activity of signaling outcomes in a context-dependent manner (Figure 4). It still needs to fill the current knowledge gaps of how TRAF5 negatively regulates pro-inflammatory signaling mediated by members of the IL-6R and the TLR/IL-1R families in the context of chronic inflammation. Although Traf5-deficiency in mice does not cause severe inflammation at a steady state, those mice display enhanced inflammatory phenotypes in the context of a pro-inflammatory milieu. This means that the higher level of TRAF5 in B- and T-cells may be important for the suppression of hyper-activation of cellular signaling in lymphocytes by restraining the pro-inflammatory activity of IL-6R and/or TLR signaling. Reducing the expression of TRAF5 which can be originated from gene mutations, inflammatory episodes and aging may enhance the potential autoreactivity of lymphocytes. If this is correct, enhancing TRAF5 expression in lymphocytes or administration of a compound that can augment TRAF5 expression would lead to the control of aberrant inflammatory disease responses (Table 1), leading to therapeutic benefits. Further studies will be needed to uncover the anti-inflammatory function of TRAF5 that may be responsible for the development and pathogenesis of autoimmune and inflammatory diseases.
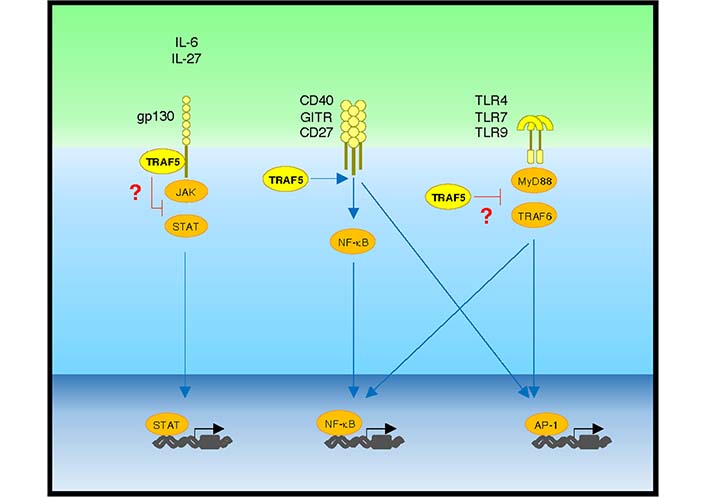
Positive and negative regulatory activity of TRAF5 in pro-inflammatory pathways. IL-6 or IL-27 binding to gp130 activates the JAK-STAT pathway, which is restrained by gp130-associated TRAF5. Upon ligand binding, the TNFR family molecules CD40, GITR, and CD27 activate NF-κB and activator protein-1 (AP-1) pathways via TRAF family adaptor proteins including TRAF5. Upon binding to microbe-derived and host-derived ligands, TLRs including TLR4, TLR7, and TLR9 activate NF-κB and AP-1 pathways via MyD88 and TRAF6. TRAF5 limits TLR signaling activity. Blue lines show activating signals, and red lines show inhibitory signals
TRAF5 and diseases in mice and humans
| Species | Diseases | Phenotypes | References |
|---|---|---|---|
| Human | AS | Enhanced methylation of CpG island of TRAF5 gene promoter and decreased TRAF5 gene expression in PBMCs | [31] |
| RA | A SNP mapping upstream of TRAF5 gene and affecting a putative transcription factor binding site | [32] | |
| Uveitis | SNPs in TRAF5 gene | [33] | |
| SS | Decreased TRAF5 gene expression in the temporal bulbar conjunctiva | [35] | |
| SLE | Enhanced TLR7 signaling caused by decreased expression of TRAF5 in B cells (pre-plasma cells) | [36] | |
| Coronary heart disease | Decreased TRAF5 gene expression in the blood | [21] | |
| Obesity with a high cardiovascular risk | Decreased TRAF5 gene expression in blood leukocytes and adipocytes | [20] | |
| NAFLD | Decreased TRAF5 protein expression in the liver | [23] | |
| Mouse | Allergic asthma | Enhanced Th2 responses caused by Traf5-deficiency | [18] |
| EAE | Enhanced Th17 responses caused by Traf5-deficiency | [13] | |
| Wound healing | Increased pDC-mediated wound repair inflammatory responses caused by Traf5-deficiency | [27] | |
| Atherosclerosis | Enhanced atherosclerosis caused by Traf5-deficiency | [21] | |
| Diet-induced obesity | Enhanced diet-induced obesity and inflammation in the adipose tissue caused by Traf5-deficiency | [20] | |
| NAFLD | Enhanced hepatic steatosis caused by Traf5-deficiency | [23] | |
| Myocardial ischemia-reperfusion injury | Exacerbated inflammation and cell death after a myocardial ischemia-reperfusion injury caused by Traf5-deficiency | [22] |
PBMCs: peripheral blood mononuclear cells
Abbreviations
| AS: | ankylosing spondylitis |
| BD: | Behçet’s disease |
| CFA: | complete Freund’s adjuvant |
| DSS: | dextran sulfate sodium |
| EAE: | experimental autoimmune encephalomyelitis |
| ERK: | extracellular-signal-regulated kinases |
| GITR: | glucocorticoid-induced tumor necrosis factor receptor family-related protein |
| gp130: | glycoprotein 130 kDa |
| HFD: | high-fat diet |
| IFN-β: | interferon β |
| IgM: | immunoglobulin M |
| IL-6: | interleukin 6 |
| IL-6R: | interleukin 6 receptor |
| JAK: | Janus kinase |
| JNK: | c-Jun N-terminal kinase |
| LNs: | lymph nodes |
| LT-βR: | lymphotoxin-β receptor |
| MAPK: | mitogen-activated protein kinase |
| MCP-1: | monocyte chemoattractant protein-1 |
| MOG35–55 : | amino acids 35–55 myelin oligodendrocyte glycoprotein |
| mRNA: | messenger RNA |
| MyD88: | myeloid differentiation primary response gene 88 |
| NAFLD: | nonalcoholic fatty liver disease |
| NF-κB: | nuclear factor kappa B |
| OSM: | oncostatin M |
| OVA: | ovalbumin |
| OX40: | TNFRSF4 or CD134 |
| pDCs: | plasmacytoid dendritic cells |
| PPs: | Peyer’s patches |
| RA: | rheumatoid arthritis |
| SLE: | systemic lupus erythematosus |
| SNPs: | single nucleotide polymorphisms |
| SS: | Sjögren syndrome |
| STAT: | signal transducer and activator of transcription |
| TCR: | T cell receptor |
| TGF-β: | transforming growth factor-beta |
| Th2: | T helper 2 |
| TLR: | Toll-like receptor |
| TNFR: | tumor necrosis factor receptor |
| TNF-α: | tumor necrosis factor α |
| TRAF5: | tumor necrosis factor receptor-associated factor 5 |
| VCAM-1: | vascular cellular adhesion molecule-1 |
| VKH: | Vogt-Koyanagi-Harada |
| WSX-1: | IL-27Rα or T-cell cytokine receptor |
Declarations
Acknowledgments
We thank the Life Science Research Center, University of Toyama and the Institute for Animal Experimentation, Tohoku University Graduate School of Medicine for technical support. We thank members of Laboratory of Molecular Cell Biology, Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, and Department of Microbiology and Immunology, Tohoku University Graduate School of Medicine for their assistance and help. We also thank Michael Croft and Hiroyasu Nakano for Traf5−/− mice and for their encouragement at the start of the project.
Author contributions
TS and NI: Supervision. TS and MHK: Writing—original draft. TS, NI and MHK: Writing—review & editing. All authors read and approved the submitted version.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
The study was approved by Japan Society for the Promotion of Science KAKENHI [Grants JP15H04640 (T.S.), JP18H02572 (T.S.)]. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2023.