
Abstract
Cardiovascular and respiratory diseases are frequently associated with transient and prolonged hypoxia, whereas hypoxia exerts pro-hypertensive effects, through stimulation of the sympathetic system and release of pressor endocrine factors. This review is focused on the role of arginine vasopressin (AVP) in dysregulation of the cardiovascular system during hypoxia associated with cardiovascular disorders. AVP is synthesized mainly in the neuroendocrine neurons of the hypothalamic paraventricular nucleus (PVN) and supraoptic nucleus (SON), which send axons to the posterior pituitary and various regions of the central nervous system (CNS). Vasopressinergic neurons are innervated by multiple neuronal projections releasing several neurotransmitters and other regulatory molecules. AVP interacts with V1a, V1b and V2 receptors that are present in the brain and peripheral organs, including the heart, vessels, lungs, and kidneys. Release of vasopressin is intensified during hypernatremia, hypovolemia, inflammation, stress, pain, and hypoxia which frequently occur in cardiovascular patients, and blood AVP concentration is markedly elevated in cardiovascular diseases associated with hypoxemia. There is evidence that hypoxia stimulates AVP release through stimulation of chemoreceptors. It is suggested that acting in the carotid bodies, AVP may fine-tune respiratory and hemodynamic responses to hypoxia and that this effect is intensified in hypertension. There is also evidence that during hypoxia, augmentation of pro-hypertensive effects of vasopressin may result from inappropriate interaction of this hormone with other compounds regulating the cardiovascular system (catecholamines, angiotensins, natriuretic peptides, steroids, nitric oxide). In conclusion, current literature indicates that abnormal mutual interactions between hypoxia and vasopressin may significantly contribute to pathogenesis of hypertension.
Keywords
Chemoreceptors, eclampsia, hypertension, hypoxia, heart failure, syndrome of inappropriate antidiuretic hormone secretion (SIADH), vasopressinIntroduction
Transient hypoxia is experienced by healthy subjects whenever they are exposed to reduced oxygen content in the inspired air, however prolonged and recurrent hypoxia is one of the most serious complications of sleep apnea disorders and cardiovascular and respiratory pathologies [1, 2]. It has long been known that hypoxia induces remarkable increases of blood pressure due to stimulation of chemoreceptors [2–5], and more recently, it has been shown that the pressure responses tend to be greater in patients with cardiovascular diseases than in healthy subjects [1, 6–8]. The above findings inspired investigators to search for mechanisms responsible for hypertensive effects of hypoxia.
It is known that hypoxia activates neurons of the autonomic nervous system (ANS) engaged in the regulation of cardiovascular functions [4, 5]. The hypoxic signals alarming the ANS are generated in specialized groups of cells of carotid and aortic chemoreceptor bodies as well as in hypoxia sensitive neurons of the brain and peripheral organs [9–11]. Recently, it has been shown that enhanced activity of the superior cervical ganglion causes sensitization of the carotid body in spontaneously hypertensive rats (SHR), and that this effect results in greater pressor and sympathoexcitatory responses to the carotid body stimulation [9]. In addition, deficit of oxygen may promote release of neuroendocrine factors involved in the regulation of the cardiovascular system, such as vasopressin, oxytocin, angiotensins and aldosterone [12–18].
Majority of mammals synthesize arginine vasopressin (AVP), while the pig and hippopotamus produce lysine vasopressin (lysipressin, LVP) and the non-mammal vertebrates secrete arginine vasotocin (AVT). The AVP gene consists of three exones encoding the N-terminal signal peptide, vasopressin, neurophysin II and C-terminal peptide, known as copeptin. Vasopressin and copeptin are released in equimolar quantities, but AVP shows great variability due to its binding to platelets and therefore copeptin is frequently estimated in biological samples as a surrogate of vasopressin release [19–22].
The present review draws special attention to the role of AVP in the development of hypertension during hypoxia. In particular, it discusses the mechanisms through which hypoxia modulates regulation of blood pressure and release of AVP. In addition, the consequences of enhanced release of AVP for tissues oxygenation and cardiovascular regulation are analyzed.
Central and peripheral vasopressin systems
Experimental and clinical studies combined with immunocytochemical and radioimmunological measurements clearly show that vasopressin is synthesized mainly in the hypothalamus, specifically in the magnocellular (MNC) and parvocellular (PNC) neuroendocrine cells of the paraventricular nucleus (PVN), supraoptic nucleus (SON), and suprachiasmatic nucleus. The prevailing number of the MNC neurons send axons to the posterior pituitary, however some of the axons end in the median eminence (ME) of the hypothalamus. Axons of the MCN neurons form close contacts with capillaries either in the posterior pituitary or in the ME, where AVP is released into the blood [23–25]. The PNC innervate the neighboring hypothalamic neurons. They send also multiple projections to several regions of the central nervous system (CNS), namely to the bed nucleus of the stria terminalis (BNST), the medial amygdaloid nucleus, the dorsomedial hypothalamic nucleus, the locus coeruleus (LC), the vertical diagonal band of Broca and the olfactory bulb, as well as to the nucleus ambiguous, nucleus of the solitary tract (NTS), the lateral habenular nucleus, the basal nucleus of Meynert, the substantia nigra, the ventral hippocampus, the central gray, the rostral ventrolateral medulla (RVLM) and the spinal cord [23, 24, 26–33].
Experimental and clinical studies provide evidence that both peripheral and central release of vasopressin are influenced by a variety of regulatory factors [34–36]. AVP release is considerably elevated during hypernatremia and hyperosmolality, hypovolemia, heamorrhage, hypoxia, hyperthermia, stress and pain and it is significantly decreased during hypoosmolality, hypervolemia and elevation of blood pressure. Mechanisms involved in the regulation of vasopressinergic system by these challenges have been discussed extensively in several previous studies [21, 22, 26, 35, 37–43]. Among factors affecting electrophysiological properties of vasopressinergic neurons and release of vasopressin are noradrenaline, gamma-aminobutyric acid (GABA) and glutamate. It has been shown that neurosecretory cells of SON and PVN nuclei receive inputs from A1 noradrenergic, GABAergic and glutamatergic neurons of the brain stem and forebrain [44–51]. Effectiveness of these neurotransmitters is modulated by several locally acting neuropeptides and by gasotransmitters, specifically by angiotensin II, cytokines, and nitric oxide, and by some other local factors [52–57].
Acting in the brain and peripheral organs, vasopressin exerts several regulatory effects, which are of primary importance for regulation of water-electrolyte balance, blood pressure, breathing, metabolism, behavior, learning and memory, sensitivity to stress and pain [21, 22, 26, 27, 32, 38, 42, 58–65]. As discussed below, hypoxia belongs to potent stimulators of the vasopressinergic systems, and accumulating evidence indicates that vasopressin also plays a significant role in the regulation of breathing [37–39, 66].
Regulatory functions of vasopressin are mediated by V1a receptors (V1aRs), V1bRs and V2Rs, which are present in multiple regions of the CNS and in several peripheral organs (Figure 1) [22, 24, 67–75]. Vasopressin can also interact with oxytocin receptors [21, 76]. Besides, specificity of neuroregulatory effects of vasopressin in the CNS depends on interaction with several neurotransmitters and neuromodulators [26, 27, 33, 42, 60, 62, 63, 77–79]. In peripheral organs, V1Rs have been detected in testis, superior cervical ganglion, carotid bodies, liver, kidney, thymus, heart, blood vessels, lung, spleen, uterus, and breast [58, 70, 80–83].
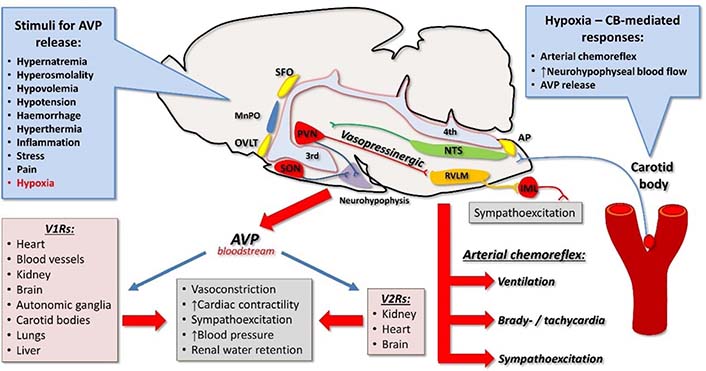
Vasopressin, hypoxia, and cardiovascular system. Vasopressin is released from the neurohypophysis into the bloodstream in response to numerous stimuli, including hypoxia. Carotid body-mediated responses to hypoxia depend on its connections to the CNS via NTS. These responses include activation of arterial chemoreflex, increase in neurohypophysial blood flow, and release of AVP. Vasopressin exerts its cardiovascular actions by binding to V1Rs and V2Rs. Vasopressinergic pathways from the PVN to the brainstem are activated by hypoxia and involve activation of the RVLM and sympathoexcitation. AP: area postrema; CB: carotid body; IML: intermediolateral nucleus; MnPO: median pre-optic nucleus; OVLT: organum vasculosum of the lamina terminalis; SFO: subfornical organ; 3rd: third cerebral ventricle; 4th: fourth cerebral ventricle; ↑: increase
V2Rs were found mainly in the kidney, but there is also evidence for their presence in other organs (Figure 1) [84–86]. Specifically, V2R binding sites were detected in endothelial cells, where they can participate in the regulation of von Willebrand factor secretion [87, 88]. V2R messenger RNA (mRNA) and binding sites have also been identified in the heart and the brain of the rat in early postnatal life [59, 89].
Several studies indicate that secretion of vasopressin, expression of its receptors and its action are significantly affected by multiple homeostatic and pathological challenges such as hypernatremia, hyponatremia, dehydration, hypovolemia, hypervolemia, stress, hypertension, cardiac failure, neurological and psychiatric diseases [35, 69, 90–106].
Role of chemoreceptors in blood pressure regulation in relevance with vasopressin
Arterial chemoreceptors include the carotid bodies strategically located at the bifurcation of the common carotid artery and the aortic bodies situated in the aortic arch. Hypoxia is the primary stimulus for arterial chemoreceptors, especially the carotid bodies (Figure 1). Activation of the arterial chemoreceptors triggers the arterial chemoreflex, which leads to sympathoexcitation, changes in cardiovagal balance, increase in phrenic nerve activity, and pulmonary ventilation and arousal [10, 107, 108]. The involvement of carotid bodies in driving sympathetic activity in hypertension, obstructive sleep apnea, and heart failure is well documented in preclinical and clinical studies. However, the role of aortic bodies is less defined and still requires further investigations.
Increased activity of the arterial chemoreceptors, especially the carotid bodies, has been postulated and found to be causative for experimental arterial hypertension [8, 109–113] and confirmed by clinical studies [114–116]. Increased activity of the arterial chemoreflex is seen before the onset of hypertension in SHR [117], which is in line with findings in normotensive young adults with family history of hypertension, who often manifest increased sensitivity of arterial chemoreflex [8]. There seems to be a selective enhancement of the sympathetic response, with tonically increased activity of the arterial chemoreflex evoked from the carotid bodies as well as increased sensitivity of the chemoreceptors to hypoxic stimuli in hypertension [10, 118, 119]. There is also accumulating evidence that purinergic P2X receptors in the carotid body are involved in this selective drive to the sympathetic system [120–122], thus suggesting that distinct mediators may be involved in specific components of the arterial chemoreflex [10]. The causative role of arterial chemoreceptors in hypertension is further demonstrated by blood pressure lowering effects of removal, denervation, or suppression with hyperoxia of the carotid bodies in animal models of hypertension and in hypertensive patients [110, 112, 114–116].
In this light, it is essential to note that hypoxia increases neurohypophyseal blood flow [123–125] and elevates plasma levels of AVP both in animals and humans [39, 126–128] (Figure 1). The effect of hypoxia on AVP release at least in part depends on the sensory input from the carotid bodies [124, 126]. It has been postulated that the increase in plasma AVP concentration helps in preventing the hypoxia-induced vasodilation, supporting in this way peripheral vascular resistance [129, 130].
Furthermore, AVP released into the circulation may directly target chemosensitive glomus cells of the carotid bodies, which express V1aRs [83] as well as G protein q/11 and phosphokinase C that are key intracellular components of the V1aR signaling [131]. These findings indicate that circulating AVP may affect the glomus cells expressing V1aRs and presumably modulate the activity of the carotid body, thus, providing a potential feedback loop between the hypoxia-mediated release of AVP and activity of the chemoreceptors (Figure 2). In addition, decreased carotid body blood flow activates arterial chemoreflex and has been suggested as a putative mechanism involved in elevated activity of the reflex in hypertension [109, 132]. Thus, it is probable that AVP may also enhance arterial chemoreceptors’ activity by increasing vascular resistance of the carotid body artery thereby decreasing the carotid body blood flow with resultant sensitization of the chemoreceptors.
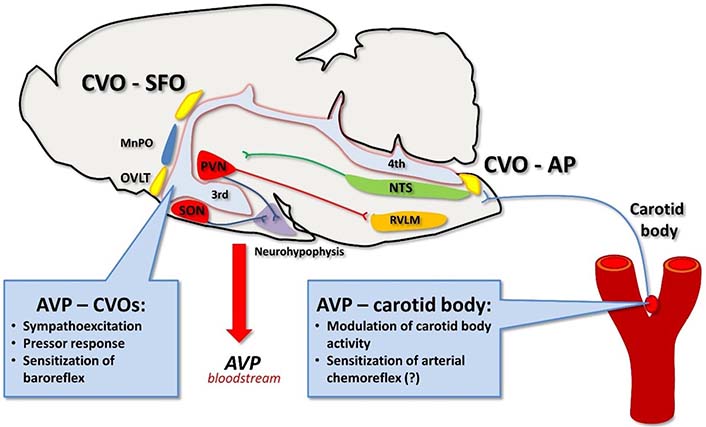
Vasopressin targets circumventricular organs and carotid body. Vasopressin present in the bloodstream contributes to the neural control of cardiovascular system by binding to its receptors in the circumventricular organs in the brain. Vasopressin may also act on V1aRs expressed in the carotid body, however, its sensitizing effect on the carotid body activity and arterial chemoreflex (?) awaits further elucidation, especially under conditions of hypertension. CVO: circumventricular organ
Vasopressin released into the bloodstream also binds to the circumventricular organs that lack the blood-brain barrier. In particular, V1aRs are expressed in the area postrema [133, 134] and binding of AVP to these receptors alters neuronal activity in the surrounding cardiovascular centers [135, 136] and inhibits phrenic nerve activity [133]. In contrast, it has been suggested that AVP acting locally in the carotid bodies may increase its sensitivity [83] and thus counterbalance the inhibitory effects of the neurohormone binding to V1aRs in the area postrema. This action could fine-tune respiratory and hemodynamic responses to hypoxia [38, 83]. In addition, AVP binding to its receptors in the area postrema sensitizes the arterial baroreflex thus, it helps to maintain blood pressure buffering responses [22, 137]. Despite the sensitizing effect on the baroreflex, intracerebroventricular infusions of AVP that allow for binding of AVP with its receptors in the circumventricular organs trigger pressor response under most experimental paradigms [22, 137] (Figure 2). The question arises whether systemically released AVP modulates engagement of chemoreceptors in regulation of blood pressure in normotensive subjects. In this regard, it has been shown that systemic inhibition of V1aRs do not affect pressor response to acutely evoked arterial chemoreflex with cyanide [138]. Thus, it appears that in normotensive rats AVP plays an insignificant role in hemodynamic response to arterial chemoreflex activation. Undoubtedly, the role of AVP and its V1aR in the carotid body under conditions of arterial hypertension awaits further elucidation.
Altered regulation of vasopressin release in cardiovascular and respiratory diseases
Function of vasopressin system in cardiovascular diseases
Hypertension and other cardiovascular diseases are frequently complicated by cardiac failure, which is associated with hypoxia. It has been shown that hypoxia strongly promotes activation of the hypothalamic vasopressinergic neurons and increases release of vasopressin or its surrogate marker copeptin [12, 14, 16, 139, 140].
There is also evidence that heart failure results in significant reorganization of central and peripheral components of vasopressinergic system. It has been found that acute cardiac failure induced by myocardial infarction causes significant elevation of c-Fos protein expression in magnocellular neurosecretory vasopressinergic neurons of the PVN and SON [141, 142] and that the activation results from inappropriate stimulation of these neurons by glutamatergic N-methyl-D-aspartate (NMDA) receptors [143, 144]. It should be noted that hypertension and heart failure are associated with reduced inhibition of the PVN neurons by GABAergic and nitrergic transmission [145, 146]. The study of Kc et al. [16] provided evidence that local hypoxia of PVN neurons stimulates subpopulation of vasopressinergic neurons innervating the RVLM. In addition, the authors found that expression of V1aRs in the RVLM is elevated in rats exposed to chronic intermittent hypoxia and that blockade of V1aRs in the RVLM significantly attenuates blood pressure and heart rate responses to intermittent hypoxia [16].
Experimental studies revealed that hypertension has significant impact on the brain vasopressin receptors in specific brain regions. Hence, AVP mRNA expression and V1aR expression were found to be higher in the PVN of the SHR than in the PVN of Wistar-Kyoto (WKY) rats [147]. In hypertensive renin transgenic TGR(mRen2)27 rats, expression of V1bR mRNA was elevated in the mesencephalon-pontine region [148]. Enhanced engagement of V1aR in regulation of blood pressure was found in SHR, deoxycorticosterone acetate (DOCA)-salt hypertension and transgenic TGR(mRen2)27 hypertension [147–152].
Multiple clinical studies revealed increased plasma AVP concentration in patients with heart failure and reduced ejection fraction [153–155]. Evidence of elevated concentration of AVP was also found in patients with left ventricle hypertrophy and reduced level of atrial natriuretic peptide [156]. It should be noted that in patients with heart failure, plasma AVP level was elevated in spite of normal or even reduced sodium concentration and osmolality—the symptoms which are illustrative for the syndrome of inappropriate vasopressin secretion (SIADH). Presence of water retention, hypoosmolality and hyponatremia in heart failure is usually interpreted as a result of arterial underfilling, which causes unloading of baroreceptors [157] and elevation of non-osmotic release of AVP [158–160].
In majority of vascular beds, AVP elicits vasoconstriction and a decrease of blood flow due to stimulation of V1Rs [42, 161], however, in the pulmonary circulation, it can promote vasodilation [162–164]. Pulmonary vessels are resistant to vasoconstrictory action of vasopressin [164–166] and in vitro experiments on the pulmonary artery provide evidence for its relaxing effect [167]. In the cerebral circulation, vasopressin can cause either increase of the blood flow through an action mediated by V2Rs [85] or restriction of the flow, presumably through stimulation of V1Rs. Interestingly, it appears that the vasoconstrictory action of vasopressin in the cerebral circulation may predominate in hyponatremia, which is an unavoidable attribute of SIADH [168, 169]. The traumatic brain injury which is associated with hypoxia causes significant increase of V1aR in the frontoparietal cortex [104]. Vasopressin interacts synergistically with proinflammatory cytokines and it is likely that its vasoconstrictory action on cerebral vessel may worsen cerebral blood flow in the traumatic shock [104, 170].
Hypoxia and enhanced involvement of vasopressin in regulation of blood pressure in cardiovascular diseases
Several studies provide evidence that hypertension and heart failure are associated with inappropriate regulation of AVP release and its function [42, 90, 94, 171–174]. It has been postulated that elevated concentration of blood AVP in heart failure and hypertension worsens health state due to excessive stimulation of V2Rs which promotes water retention, and because of enhanced stimulation of V1R which causes vasoconstriction and elevation of total peripheral resistance [173–176]. Moreover, chronic intravenous infusion of V1 agonist in normotensive Sprague-Dawley (SD) rats resulted in the development of hypertension [92] and other experimental studies employing V1aR knockout (V1aRKO) mice and mice with myocyte-selective transgenic overexpression of cardiac V1aR suggested that permanent stimulation of these receptors may play an essential role in the development of cardiac hypertrophy [175].
It has been shown that administration of V2R antagonists (tolvaptan, lixivaptan) exerts positive effects in animal and human heart failure, manifested by normalization of electrolytes status, diminution of myocardial infarction-induced fibrosis and macrophage infiltration, as well as by improvement of the left ventricle function [177–184]. Moreover, some long-term trials revealed that administration of tolvaptan improves long-term prognosis in patients with congestive heart failure [185, 186]. However, other authors were not able to prove effectiveness of tolvaptan on the long-term survival of patients with heart failure [160].
It has been reported that administration of conivaptan, which is a dual V1a and V2 vasopressin antagonist, significantly improves hemodynamic parameters and renal functions of patients with heart failure [173, 174, 187–189]. In canine model of tachypacing-induced heart failure, chronic application of pecavaptan (BAY1753011), which is another dual V1aR/V2R antagonist, exerted diuretic effects and significantly improved vital hemodynamic parameters, as shown by elevation of cardiac output and diminution of total peripheral resistance [190]. The above effects were significantly relative to placebo and tolvaptan alone. Affinity of pecavaptan to human and canine V1aR/V2R is similar [190], and therefore it is likely that similar beneficial effects of pecavaptan would occur in human patients with heart failure.
Hypertension is one of the serious complications of eclampsia. Excessive stimulation of vasopressinergic system, associated with hypoxia and inappropriate tissue oxygenation has been found in the pre-eclampsia state [191–193]. Moreover, infusion of vasopressin into pregnant mice resulted in pathological changes, which are typical for pre-eclampsia (hypertension and placental oxidative stress) and which could be prevented by blockade of AVP receptors. Accordingly, it has been suggested that elevated secretion of vasopressin contributes to the development of hypertension in pre-eclampsia [194]. However, exposure of maternal rats to hypoxia increases also expression of V2R in newborn rats [195, 196], whereas stimulation of V2Rs causes dilation of pulmonary vessels [88, 162, 163]. Thus, it is also likely that upregulation of V2R in newborns may belong to mechanisms protecting from negative consequences of maternal hypoxia.
Septic shock is another pathological state which causes inflammatory symptoms, tissue hypoxia and activation of the vasopressinergic system [197, 198]. During experimental model of septic shock, blood AVP concentration initially increases up to 500 pg/mL and subsequently decreases to levels that are disproportionately low in relation to hypotensive effects of shock [199]. Because survival of patients with septic shock is significantly improved by combined infusions of vasopressin and corticosteroids in comparison to vasopressin alone or noradrenaline [198, 200], it is likely that prolonged sepsis may somehow disorganize regulation of vasopressin release and consequently result in defective cardiovascular regulation.
Finally, it is worthy to note that some experimental studies suggest that in emergency states AVP may exert some protective effects in the brain. For instance, in a rat model of asphyxial cardiac arrest, which imitates hemodynamic disturbances occurring during cardiopulmonary resuscitation, intraperitoneal administration of vasopressin was found to reduce neuronal apoptosis in the hippocampus [201].
Conclusions
Hypoxia and elevated blood AVP concentration are frequent attributes of cardiovascular diseases and both exert pro-hypertensive effects. There is strong evidence that hypoxia potently stimulates central and systemic release of AVP and that numerous actions of this hormone determine responsiveness of the cardiovascular system to hypoxia (Figures 1 and 2). Survey of literature carried out in the present review draws attention to mechanisms underlying mutual interactions between hypoxia and activation of systemic and central vasopressin systems in health and cardiovascular diseases. It is concluded that during hypoxia, vasopressin exerts pro-hypertensive effects via intensification of stimulation of chemoreceptors and through interactions with other cardiovascular regulatory factors, mainly catecholamines, angiotensins, natriuretic peptides, steroid hormones and gasotransmitters These effects are potentiated in hypertension and cardiac failure. However, vasopressin may also exert some positive effects through actions mediated by receptors located in specific cardiovascular regions.
Abbreviations
| AVP: | arginine vasopressin |
| CNS: | central nervous system |
| GABA: | gamma-aminobutyric acid |
| mRNA: | messenger RNA |
| NTS: | nucleus of the solitary tract |
| PVN: | paraventricular nucleus |
| RVLM: | rostral ventrolateral medulla |
| SHR: | spontaneously hypertensive rats |
| SON: | supraoptic nucleus |
| V1aRs: | V1a receptors |
Declarations
Acknowledgments
The authors are grateful to Professor Agnieszka-Cudnoch-Jędrzejewska—head of the Department of Experimental and Clinical Physiology and other colleagues of the Department for friendly support during preparation of the manuscript.
Author contributions
ESS conceived conception and design of the paper and wrote the first draft of the manuscript; TŻ contributed to conception of the paper and wrote sections of the manuscript. Both authors contributed to manuscript revision, read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the Medical University of Warsaw Scientific Projects, Poland. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2022.