
Abstract
Whipple’s disease (WD) is a rare systemic disease caused by gram-positive bacillus bacteria that invades multiple organs mainly the intestinal epithelium. Its manifestation is not only limited to the gastrointestinal tract but it also affects the joints, muscle and skin. This is a case of a 54-year-old male patient with a medical history of chronic arthritis presenting with bilateral progressive calves pain, anterior tibial hyperpigmentation, joints pain, anemia and weight loss. He was misdiagnosed as rheumatoid arthritis, for which he was treated by immunosuppressors for several years with no amelioration. After advanced investigations, he was found to have multiple retroperitoneal and mesenteric adenopathies, with an incidental finding of a mesojejunal mass during laparoscopy, from which the biopsies revealed the presence of histiocytosis and numerous intra-cytoplasmic particles with positive periodic acid–Schiff (PAS) suggesting the diagnosis of WD. Endoscopy was done and intestinal histology with polymerase chain reaction (PCR) test confirmed the diagnosis of WD. The patient was then treated with antibiotics (ceftriaxone and trimethoprim-sulfamethoxazole) with a remarkable clinical amelioration. To be aware of WD as a potential etiology behind malabsorption, musculoskeletal and skin abnormalities, is the first step in order to establish the diagnosis and provide adequate treatment, thus, improving the patient’s quality of life. WD is a rare, without antibiotic treatment deadly systemic infectious disease caused by the ubiquitary Gram-positive bacterium Tropheryma whipplei. This article aims to report a case marked with dermatomyositis like presentation that had a missed and delayed diagnosis.
Keywords
Whipple’s disease, myositis, arthralgia, melanoderma, hyperpigmentationIntroduction
Whipple’s disease (WD) was called after George Hoyt Whipple who reported in 1907, the first case of lipid deposits in the intestinal mucosa. WD was named intestinal lipodystrophy. In 1949, Black-Schaffer introduced the periodic acid–Schiff (PAS) stain and demonstrated that the intracellular material was glycoprotein rather than lipid [1]. In 1961, an underlying bacterial etiology was identified [2], and it took around 40 years later to identify 16S ribosomal DNA (rDNA) sequence and establish its relationship with an intracellular organism—the actinomycetes. Tropheryma whippelii was then introduced [3].
This bacterium has been identified in the stool of asymptomatic individuals in Europe and Africa, and gastric aspirates in America [4], suggesting a genetic susceptibility to infection and an association between chronic infection, T-cell function impairment and certain human leukocyte antigen types [5, 6].
WD is a very rare, chronic, systemic infectious disease with an estimated annual incidence of 3 in one million, which may be fatal if not diagnosed and treated appropriately [7, 8]. In a large population-based study conducted in the US, the overall prevalence of WD was 9.8 cases per 1 million people [9].
It affects mainly middle-aged men, around fifty years old [10], and recent studies showed similar rates of affection in men and women [9]. Outdoor workers seem to be more affected by this disease [11]. The reservoir, route of transmission and natural occurrence is not identified and until now under research [12].
According to literature, the most common host of this bacterium is the mucosa of the small intestine in human [13]. The microscopic pathologic findings consist of massive infiltration of foamy macrophages containing dense PAS-positive granules [14].
Polymerase chain reaction (PCR) is increasingly used to identify bacterial 16S rDNA molecules and other targeted repeated sequences of Tropheryma whipplei (T. whipplei, formerly Tropheryma whippelii) [11]. The definitive diagnosis is established in PAS-positive histologic findings along with positive PCR.
WD can affect almost any organ system and is accordingly classified between classic systemic disease and localized or extraintestinal disease [15]. The clinical signs include arthralgia, weight loss, diarrhea and abdominal pain, although the clinical manifestations can vary widely.
In classic intestinal WD, bacterial and inflammatory cells infiltrate small intestinal mucosa and mesenteric lymph nodes, inducing malabsorption with resultant weight loss, diarrhea and abdominal pain and rarely gastrointestinal bleeding [10, 15, 16].
WD is frequently associated with rheumatic manifestations (60% of the cases), which precede the gastrointestinal signs in three fourths of the patients. They are often the first symptoms of the disease [16]. Joints and musculoskeletal manifestations are as following: migratory or additive non-destructive arthralgia involving large joints. Polyarthralgia including symmetric and asymmetric is often intermittent while articular attacks are acute and last for days [17]. Arthralgias and articular attacks precede intestinal symptoms several years.
The use of immunosuppressive drugs for articular treatment (the illness being sometimes misdiagnosed as seronegative rheumatic disease) may accelerate the onset of intestinal or other systemic illness [18].
Myositis is also reported to be associated with WD. Case of bilateral extraocular myositis [19] as well as a case of WD mimicking an autoinflammatory disease with myositis and soft tissue inflammation has been reported [20].
Occasionally central nervous system (CNS) and ophthalmic complications are observed [21]. CNS manifestations are myriad and usually develop in later stages of the illness [22]. Two signs are pathognomonic for CNS WD: oculomasticatory myorhythmia and oculofacial skeletal myorhythmia, which consist of ocular nystagmus and synchronous contractions of the masticatory and proximal and distal skeletal muscles [21–23]. Even in the absence of CNS symptoms, cerebrospinal fluid T. whipplei PCR analysis may be positive [21], in 25% to 47% [24].
Around the malar and orbital hyperpigmentation, purpura and cheilitis are the main skin manifestations of WD. A variety of skin lesions have been described in association with WD and the most common of these is hyperpigmentation or melanoderma that develops in up to 46% of patients in the later stages of the illness. Other characteristic skin abnormalities include subcutaneous nodules, erythema nodosum-like lesions, and inflammatory rashes that may mimic cutaneous lupus, dermatomyositis, psoriasis, or eczema [25]. Finally, fever may occur for years and is mainly low grade and intermittent. Rarely, there may be clinical manifestations of isolated central nervous system disease, endocarditis, or lung, skin, or eye disorders [26].
The most frequently used treatment included a 2-week intravenously cephalosporins followed by oral sulfonamides for 1–2 years. This therapy achieved significantly high success rate with values around 90% [27]. Response is monitored by clinical examination. Microscopic examination is not helpful for treatment follow-up specially that PAS-positive structures may persist for years despite adequate therapy [28]. Without proper antibiotic treatment, the disease invariably culminates in dissemination and is potentially fatal [29].
Herein, a case of WD was reported, with an unusual presentation: the illness was handicapping, and the diagnosis was delayed.
Case report
This is a case of a 54-year-old Lebanese man, a non-smoker, non-alcoholic, admitted to the hospital for handicapping bilateral calves swelling and pain with skin hyperpigmentation over the anterior tibial side of his legs increasing progressively. The patient was also noticed with weight loss of 10 kilograms over 3 months duration with persistent fatigue.
History goes back to 12 years prior to presentation, when the patient started to have chronic migratory peripheral polyarthritis involving the large and small joints of his upper and lower extremities, which was diagnosed as seronegative rheumatic disease treated with methotrexate and folic acid for 12 years (stopped 5 months prior to presentation). Adalimumab was then added to the regimen, taken for one year only and stopped 10 months prior to presentation due to its side effects: pleural effusion and upper respiratory infection. No beneficial effect was noted and symptoms persisted despite antirheumatic drugs. Few months prior to presentation, persistent cramping calves pain (and even pain of proximal extremity—thigh, accelerated by palpation on physical exam with anterior tibial discoloration started to develop, forcing him to ambulate only with assistance. Weight loss as mentioned above was also noted despite good appetite.
The patient is known to have diabetes mellitus type 2 on vildagliptin/metformin mixed combination and repaglinide. No other particular medical history.
He denied any other constitutional, HEENT (head, eyes, ears, nose, and throat), respiratory, cardiovascular, gastrointestinal or genitourinary symptoms, nor bipolar aphthosis. Physical examination revealed a poorly built and cachectic patient with pale skin. He had anicteric sclera with poorly injected conjunctiva. His remaining HEENT examination was unremarkable and his lungs had good bilateral air entry with no added sounds. Regular heart sounds S1 and S2 without a murmur were noted, his abdomen was soft not tender nor distended with positive bowel sounds and his joints showed no signs of arthritis. His lower limbs examination revealed bilateral thigh and calf tenderness with non-pitting swelling (right > left), with homogenous hyperpigmentation of the anterior tibial skin bilaterally. The pain was present at rest (the calves mainly) and exacerbated by active and passive movement. Neurologic exam showed normal mental status, cranial nerves, motor (strength and cerebellar), reflexes and sensations. To note that a complete cerebellar exam or gait examination wasn’t done as the patient couldn’t ambulate without assistance.
The patient denied undergoing any previous surgery and mentioned no family history of any specific disease.
To note that the patient is an outdoor worker. He worked in the nature and was exposed to soil.
Investigations
In addition to the laboratory blood tests in Table 1, lower limbs Doppler ultrasound to rule out a deep venous thrombosis showed calves edema with muscular fascia thickening mainly at right and no evidence of thrombosis.
Laboratory blood tests upon admission
| Lab test | Result | Lab test | Result |
|---|---|---|---|
| Hemoglobin | 100 g/L | Creatinine | 0.76 mg/dL |
| Hematocrit | 0.314 L/L | Calcium | 8.7 mg/dL |
| MCV | 68 fL | Magnesium | 2.1 mg% |
| WBC | 7,700/mm3 | CPK | 19 U/L |
| Neutrophils | 64% (4,928 cells/μL) | CRP | 124 mg/L |
| Lymphocytes | 22% (1,694 cells/mm3) | ESR | 110 mm/h |
| Eosinophils | 6% (426 cells/mL) | TSH | 3.11 mIU/L |
| Platelets | 335 × 109/L |
MCV: mean corpuscular volume; WBC: white blood cells; CPK: creatine phosphokinase; CRP: C-reactive protein; ESR: erythrocyte sedimentation rate; TSH: thyroid-stimulating hormone
Lower limbs magnetic resonance imaging (MRI) to rule out myositis revealed fasciitis and myositis of the two calves of posterior predominance. A musculoskeletal MRI of the whole body was suggested for better assessment of topographic muscular involvement (Figure 1A and 1B). The latter imaging showed, in addition to the fasciitis and myositis at the level of the legs, a fissure surrounding the right semi-membranous and the left biceps at the level of the thigh, with also a suspicion of myositis at the attachment of the right gluteus minimus. The paravertebral muscles at the level of L5 and S1 were also involved. It was also noticed the involvement of the supraspinatus and infraspinatus muscles bilaterally and symmetrically is with a very probable presence of mild myositis of the right triceps muscle.
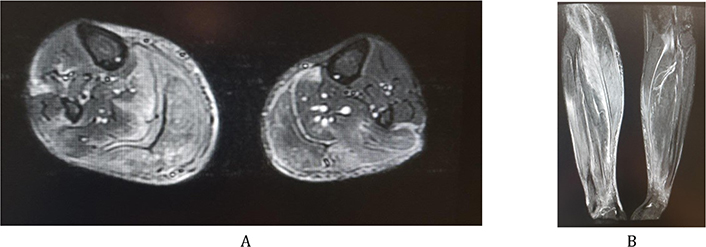
At this point, skin and muscle biopsies (biopsies of right gastrocnemius muscle and of the above skin) were done and immunofluorescence technique was performed, and it confirmed the presence of myositis and fasciitis at that same level. PAS stain was done on both muscle and skin biopsies but did not show any additional finding. Trichinellosis was ruled out.
Moreover, on MRI there was a diffuse and homogenous marrow replacement suspicious for lymphoproliferative disorder.
Therefore, a computed tomography (CT) scan for the chest, abdomen and pelvis with injection was ordered to assess the possibility of any lymphoproliferative disorder, or any other neoplasm ruling out paraneoplastic presentation (Figure 2A and 2B).
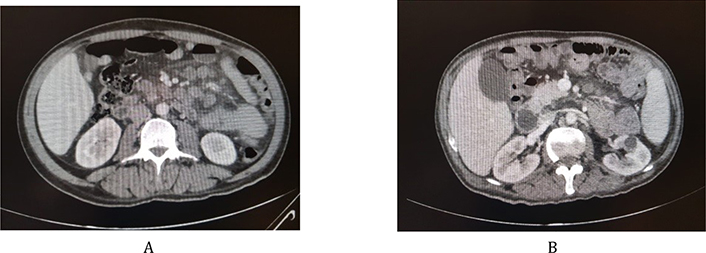
The thoraco-abdomino-pelvis CT scan revealed a minimally prominent right paratracheal node measuring 10 mm, an enlarged spleen measuring 14.5 cm, mildly enlarged retroperitoneal nodes as well as moderately enlarged mesenteric nodes (the largest measures 15 mm), with diffuse increased density and haziness of the mesenteric fat, mainly in the mid abdomen and on the left side. Findings are compatible with lymphoma or metastasis, nevertheless, a benign condition such as inflammatory process cannot be ruled out.
Consequently, tuberculosis, human immunodeficiency virus (HIV) infection, autoimmune diseases, and systemic diseases were ruled out (Table 2).
Blood tests done later in the course
| Lab test | Result | Lab test | Result |
|---|---|---|---|
| ANA | Negative | c-ANCA | Negative |
| p-ANCA | Negative | Anti-CCP | Negative |
| RA test | Negative | Myositis panel | Negative |
| Autoimmune panel | Negative | HIV | Negative |
| ACE | Negative | PPD skin test | Negative |
ANA: antinuclear antibodies; p-ANCA: perinuclear antineutrophil cytoplasmic antibodies; RA: rheumatoid arthritis; ACE: angiotensin converting enzyme; PPD: purified protein derivative; c-ANCA: cytoplasmic antineutrophil cytoplasmic autoantibody; CCP: cyclic citrullinated peptides
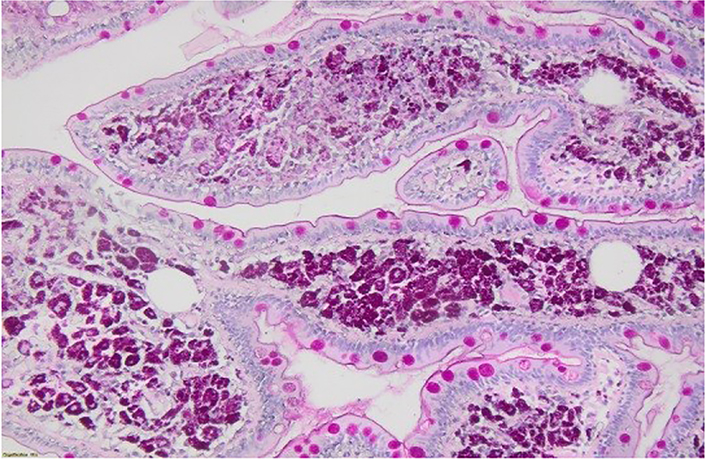
Retroperitoneal lymph nodes biopsy by laparoscopy was done to rule out lymphoma. However, during the laparoscopy, a meso-jejunal mass was identified, from which biopsies were taken and showed a lymph node structure containing multiple histiocytes with foamy cytoplasm (Figure 3A and 3B). Numerous intra-cytoplasmic particles positive for PAS and PAS diastase (Figure 4A and 4B) were identified. This non-Langerhans histiocytosis suggested the diagnosis of WD, but necessitated more investigations to confirm the diagnosis. In front of this finding of histiocytosis, the differential diagnosis was wide including leukemia, lymphoma, myeloma, malignant melanoma, xanthoma, Rosai-Dorman disease, Erdheim-Chester disease, storage diseases and solid tumors.
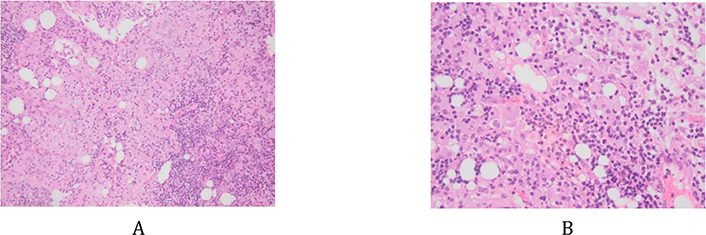
Histiocytes with clear foamy cytoplasm. A: hematoxylin and eosin (HE) stain, ×10; B: HE, ×200

Numerous intra-cytoplasmic particles positive for PAS stain and PAS diastase. A: PAS stain, ×100; B: diastase stain, ×100
The biopsy of retroperitoneal lymph nodes showed normal histological finding, ruling out malignancies.
Meanwhile, the bone marrow aspirate and biopsy were performed to rule out multiple myeloma (MM), myelodysplastic syndromes/myeloproliferative neoplasms (MDS/MPN), or myelofibrosis and showed no evidence of hematologic disease with normal cytogenetics and normal karyotype. A requested Jack2 mutation test revealed negative result.
At this point, gastroscopy and colonoscopy were done, with biopsies taken from stomach, duodenum, jejunum, ileum, and colon. The colon biopsy was normal. The duodenal, ileal and jejunal biopsies revealed WD with hyperplasia of Paneth cells (Figure 5A and 5B) with positive staining for PAS (Figure 6). Consequently, a real-time PCR performed on the biopsy to search for T. whipplei DNA turned positive confirming the diagnosis of WD.

Hematoxylin and eosin (HE) stains of the duodenal biopsy showing multiple foamy histiocytes in the axis of the villi. A: HE, ×100; B: HE, ×200
In view of the diagnosis, the patient was treated with a traditional ceftriaxone followed by a trimethoprim-sulfamethoxazole (TMP-SMX) regimen. He received IV Rocephin for 14 days and was discharged home on oral (per os: PO) TMP-SMX (one double-strength tablet [160 mg TMP/800 mg SMX] twice a day) for one year. He had rapid improvement of symptoms and all clinical signs within 2–3 weeks.
Discussion
The diagnosis of WD is challenging in that it allows specific treatment and difficult due to its rarity, to the diversity of clinical manifestations and to its slow progressive course that can mimic autoinflammatory, autoimmune or malignant diseases.
WD usually presents with weight loss and gastrointestinal symptoms and diagnosis is confirmed by histology duodenal biopsy [7]. More rarely WD can affect the musculoskeletal system, most often as oligo- or polyarthritis, mainly of the large joints [8] and more rarely as monoarthritis or with axial involvement [13, 21]. Exceptionally it can cause inflammation of muscles and soft tissues [16, 26].
Herein, the presentation of a WD case presenting with as a dermato-polymyositis like syndrome.
In WD, once the disease is suspected, it is important to rule out other differential diagnoses such as inflammatory bowel disease, other infectious causes, connective tissue diseases, immunosuppression and hyperthyroidism [30]. All the tests were done to our patient and were negative.
The diagnosis is usually made with endoscopic procedures and biopsies of the affected tissue. However, in 30% of cases the duodenal biopsies can be normal [31].
In this presented case, the middle-aged patient was diagnosed with seronegative polyarthritis more than twelve years ago, for which he sought multiple internists and rheumatologist. He started on multiple courses immunosuppressors (methotrexate, corticosteroids and adalimumab) without medical benefit, even worsening his medical condition. Immunosuppressive therapy may contribute to the progression of the infection and it may represent a ‘second hit’ in the pathogenesis of the disease [31]. It is reported in that immunosuppressants therapy may induce and accelerate the gastrointestinal symptoms in WD [27], yet it is debatable whether exposure to these agents represents an independent risk factor or is a clinical misstep before diagnosis.
Diagnosis in our case was difficult when gastrointestinal symptoms were absent. Clues for WD were lack of response to anti-inflammatory and antirheumatic disease treatment (methotrexate, adalimumab and folinic acid), weight loss, signs of chronic inflammation (high CRP), signs of malabsorption (low hemoglobin) and occupational exposure.
During his hospital stay, multiple features and findings helped us to establish the diagnosis. In fact, despite extensive list of tests, the patient did not receive a clear diagnosis. What reinforced the hypothesis is that this was not only a joint disease and it could be related to a rare systemic disease mimicking polymyositis. Physical findings of skin hyperpigmentation over both lower limbs guide us toward a systemic disease. The patient had persistent elevation of inflammatory markers (ESR110, CRP124) and normocytic anemia. Muscle biopsy even was not diagnostic but it showed unspecific myositis without inclusions bodies which could be associated with WD.
Imaging was helpful to identify moderately enlarged mesenteric lymph nodes (laparoscopy has been helpful to guide biopsy) with further endoscopy showing typical duodenal changes: duodenitis and villous atrophy with subsequent biopsy revealing PAS-positive granules compatible with WD.
A similar reported case describes bilateral WD-associated extraocular muscle myositis in a 38-year-old Caucasian female who presented with acute-onset diplopia and previously had intermittent fevers, arthralgia, rash and fatigue [17]. Two cases of WD were reported, in which the manifestation was diffuse abdominal and retroperitoneal lymphadenopathies [27]. Another case in Rio de Janeiro in 2014 showed WD manifested as difficult-to-diagnose polyarthralgia in a 45-year-old male patient [17]. And another similar case has been reported in a 58-year-old female patient with a one-year history of markedly elevated inflammatory markers and progressive anemia and seronegative polyarthritis turned to have WD [17].
Screening for WD by duodenal biopsies with PAS stain plus PCR test (or immunohistochemistry) and the final diagnosis is made if it meets one of the following criteria: a positive PAS staining in duodenal tissue, two different positive tests of the same tissue or two positive tests of different tissues [32].
In this case, biopsy had a key role to achieve the diagnosis, since both soft tissue mass and digestive endoscopy duodenal biopsies were conclusive and diagnostic (two positive tests of different tissues). The patient had a firm diagnosis of WD following PAS-positive macrophages demonstrated on mass biopsy and confirmed with positive PCR. The sensitivity of the PAS staining of small bowel biopsies ranges from 71% to 78%. In some cases, PCR is needed to confirm the diagnosis given its higher sensitivity and specificity, however PCR is not available in all centers [33]. PCR is an efficient tool for diagnosis of WD, even if PAS is negative on examination of biopsies. This was the case of a 41-year-old woman, diagnosed as WD only with PCR analysis on duodenal and mesenteric adenopathy biopsies, while PAS staining was normal [34].
The patient started on Rocephin for 14 days followed by the classical 1-year treatment with TMP-SMX. He experienced rapid improvement. The recommended gold standard treatment is with TMP-SMX 160/800 mg orally twice daily for a least 1 year. Preceded by a 2-week parenteral therapy of ceftriaxone (2 g per day).
There are different alternative treatments, depending on the toxicity and tolerance of the patients to certain antibiotics. Clinical improvement begins to be evident from days 7–21.
Relapses are largely responsible for a higher morbid-mortality, meaning the prognosis worsens with relapse and it commonly occurs after treatment. However, patients can present a relapse up to 30 years after the treatment stopped [35].
In the reported case, the patient was treated with the classical 1-year treatment and responded adequately to the pharmacological management.
WD is a rare systemic infection with T. whipplei, which is still a challenge to diagnose as its clinical manifestations are not specific, resembling many other systemic diseases and may manifest late during the course of the disease. Several years usually elapse between symptom onset, characteristic clinical manifestations and the diagnosis. WD should be taken into account because of its wide variety of clinical manifestations and because of its potential fatal outcome in the absence of treatment. The risk of sequelae and mortality rate are high if it’s not diagnosed and treated in a timely manner.
To avoid misdiagnosis, clinicians should be aware of this unusual presentation of WD, mimicking an autoimmune disease with dermatomyositis-like presentation that should be a part of the list of the differential diagnosis, as delay in diagnosis may be life-threatening. Knowing that clinicians are able to treat this rare disease with adequate antibiotherapy once the diagnosis is established avoiding therapeutic measures that can be detrimental to the patient.
Abbreviations
| CNS: | central nervous system |
| CRP: | C-reactive protein |
| CT: | computed tomography |
| MRI: | magnetic resonance imaging |
| PAS: | periodic acid–Schiff |
| PCR: | polymerase chain reaction |
| T. whipplei: | Tropheryma whipplei |
| TMP-SMX: | trimethoprim-sulfamethoxazole |
| WD: | Whipple’s disease |
Declarations
Author contributions
JF, RC and NS: Writing-original draft. JN and JM: Data collection. JA: Biopsies endoscopy. CG: Interpretation of biopsy results. All authors contributed to revision of manuscript, read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This manuscript has been approved by the Institutional Review Board of Lebanese Hospital Geitaoui.
Consent to participate
Written informed consent was obtained from the patient.
Consent to publication
Not applicable.
Availability of data and materials
All relevant data are contained within the manuscript.
Funding
Not applicable.
Copyright
© The Author(s) 2022.