
Abstract
Aging and age-associated diseases (AADs) are growing risk factors in societies worldwide. During aging, there is an accumulation of excessive oxygen free radicals [reactive oxygen species (ROS)] and nitrogen free radicals [reactive nitrogen species (RNS)] due to dysfunctional mitochondria, dysregulated catalytic activities of cytochrome P450 (CYP), nicotinamide adenine dinucleotide (NAD) phosphate [NADP(H)] oxidase (NOX), cyclooxygenases, and nitric oxide synthases (NOS) over the threshold of physiological levels, creating oxidative stress (OS). Excessive ROS and RNS oxidize, break, denature, and sometimes cause aggregations of key cellular components including DNA, proteins, and lipids. Normally, these denatured molecules and their aggregates are eliminated by autophagy (AP) and ubiquitin-proteosome system (UPS). However, these two proteostatic mechanisms are impaired as age progresses. As a result, these abnormal molecules turn into damage-associated molecular patterns (DAMPs), recognized as non-self by immune cells, leading to systemic chronic inflammation (SCI), which together with OS are the major causes of aging and AADs. Therefore, instead of trying to prevent and cure AADs individually, the logical approach should be the restoration of redox homeostasis by the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway which can prevent the damaging effect of OS and the upregulation of AP and UPS to eliminate DAMPs, and together they attenuate SCI to lessen the effect of inflammation. The central regulators, adenosine monophosphate-activated protein kinase (AMPK), sirtuin 1 (SIRT1), and Sestrins, synergistically control the restoration of the redox homeostasis by activating Nrf2, the upregulation of AP-UPS, and the inhibition of SCI. The activation of these central regulators can be achieved by exercise, caloric restriction (CR), and intakes of certain CR mimetic natural compounds. Consequently, the activations of these regulators may lead to the prevention and/or attenuation of the AADs.
Keywords
Oxidative stress, redox homeostasis, autophagy, inflammation, age-associated diseasesIntroduction
As countries throughout the world are progressing to full-fledged aging societies, the aging of the population is becoming a serious risk factor. Inevitably, aging is accompanied by age-associated diseases (AADs) that place a heavy burden on the elderlies’ incomes and overall quality-of-life, while increasing public health expenditure. The prevalence of AADs is also rising world-wide, including metabolic-related syndromes like obesity, dyslipidemia, nonalcoholic fatty liver, hypertension, diabetes, cardiovascular disorders, autoimmune diseases, neurodegenerative diseases [notably Alzheimer’s disease (AD) and Parkinson’s disease (PD)], various forms of dementia, and a wide range of cancers. The longevity or life span (due to attenuation of the aging process) and health span (due to attenuation of AADs) are intricately linked, and aging is inevitably accompanied by AADs. Therefore, instead of trying to prevent aging and cure AADs individually, the logical preventive-therapeutic approaches are to find and attenuate common denominators that cause both aging and AADs.
Both genetics and the environment contribute to human aging, longevity, and AADs. Environmental conditions and lifestyle factors appear to contribute more at younger ages, while genetic factors are more crucial in enabling an individual to reach extraordinary old age, with the occurrence of AADs late in life. Specific genes that have been clearly identified to be associated with human longevity are forkhead box protein O3 (FOXO3), insulin-like growth factor (IGF)-1 receptor (IGF-1R), sirtuin 1,3 (SIRT1,3), apolipoprotein E (APOE), and interleukin-6 (IL-6) [1]. Remarkably, FOXO3, IGF-1R, and SIRT1,3 genes are key regulators in nutrient-energy-stress sensing pathways (as shown in Figure 1). In particular, FOXO3 is active when insulin/IGF signaling is reduced and the upregulation of this gene provides stress resistance by activating nuclear factor erythroid 2-related factor 2 (Nrf2) and autophagy (AP). Incidentally, the deficiency or levels of expression of these genes (as described later) also affect the occurrence and severity of AADs. Other genes are involved in growth-factor pathways [e.g., epidermal growth factor receptor (EGFR)] as well as the immune system and inflammation (e.g., IL-6). In addition, genome-wide association studies (GWASs) [2], which compare genes in long-lived (e.g., centenarian) individuals with those of normal (controls) individuals, confirm that genes in nutrient-energy-stress sensing pathways are critical for human longevity. GWASs have also yielded a few more significant gene candidates apart from genes with already known functions, which include cholinergic receptor nicotinic α 3/5 (CHRNA3/5), lipoprotein A (LPA), and new genes involved in immune regulation in humans immunity such as major histocompatibility complex, class II, DQ α 1 (HLA-DQA1) and HLA, DR β 1 (HLA-DRB1). However, the roles of other genes in relation to longevity as discovered by GWASs have not been clearly delineated, implying that human longevity is the result of a complex interaction of many genes, some with unknown functions. Taken together, human genetic studies confirm the nutrient-energy-stress sensing genes and possibly genes controlling the immune system, as well as genes involved in maintaining genome stability, are major players in extending longevity and preventing the early onset of AADs. However, the expressions of the above-mentioned genes are influenced by environmental and epigenetic factors which together impact human aging and AADs.
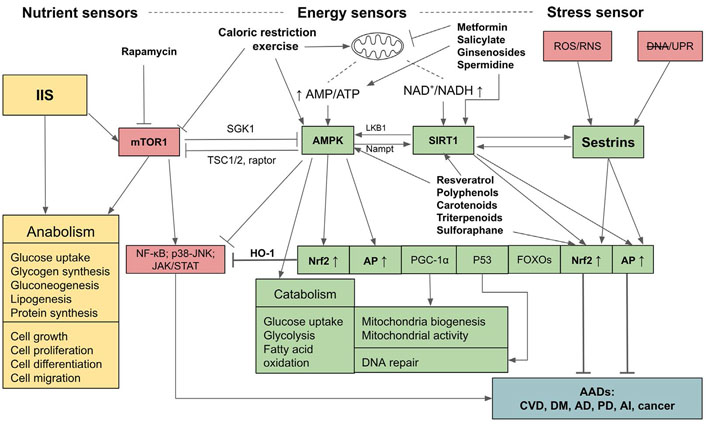
Relationships and interactions between adenosine monophosphate-activated protein kinase, SIRT1, Sestrins, insulin/insulin-like growth factor signaling, and mammalian targets for rapamycin pathways in sensing nutrient and energy statuses and controls over anabolic and catabolic processes and the outcomes of their activations. mTOR1: mammalian targets for rapamycin 1; AMPK: adenosine monophosphate (AMP)-activated protein kinase; JAK: Janus kinases; STAT: signal transducer and activators of transcription; NF-kb: nuclear factor-kappa B; IIS: insulin/insulin-like growth factor signaling; AI: autoimmune and inflammatory diseases; CVD: cardiovascular diseases; DNA: impaired DNA; LKB1: liver kinase B1; Nampt: nicotinamide phosphoribosyl transferase; HO-1: heme oxygenase-1; SGK1: serum- and glucocorticoid-regulated kinase 1; DM: diabetes mellitus; RNS: reactive nitrogen species; UPR: unfolded protein response; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator-1α; TSC1/2: tuberous sclerosis protein 1/2; P53: tumor protein 53 or transformation-related protein 53;  : activation;
: activation;  : inhibition; ↑: upregulation; ↓: downregulation
: inhibition; ↑: upregulation; ↓: downregulation
One major nongenetic factor that affects aging and AADs is oxidative stress (OS). The human body undergoes aging from a continuous build-up of oxidative free radicals that are generated internally from metabolic processes and externally from environmental pollution and contaminants in food. Pollutants from the external environment can directly cause free radical accumulation in the body or indirectly through catabolic processes by detoxifying organs such as the liver. Free radicals are also generated internally by the ongoing metabolic processes in the body. The increase of free radicals over the physiological threshold level causes OS. It is now accepted that mitochondria are the main generator of reactive oxygen species (ROS) from the uncoupling reactions in the electron transport chain (ETC) and the oxidative phosphorylation (OP) processes [3], and as aging progresses mitochondrial DNA (mtDNA) is damaged by OS and mutated, causing mitochondria to become more dysfunctional and, in turn, generating more ROS to accelerate the aging process and the occurrence of AADs. Furthermore, excessive free radicals that accumulate and increase over time from mitochondria, as well as from catalytic activities of cytochrome P450 (CYP), nicotinamide adenine dinucleotide (NAD) phosphate [NADP(H)] oxidase (NOX), and other sources [4], can elicit inflammation which exacerbates the aging and AADs by causing additional cellular damage, leading to tissue and organ dysfunction. Additionally, OS worsens with aging since the anti-oxidation capacity mediated by Nrf2 signaling decreases over time. Thus, OS and inflammation are the major factors that cause genomic instability, telomere attrition, epigenetic alterations, mitochondrial dysfunction, loss of proteostasis, deregulated nutrient-sensing, cellular senescence, stem cell exhaustion, and altered intercellular communication, which are the hallmarks of aging [5]. Additionally, impaired and dysfunctional antioxidation systems (mediated by Nrf2) and AP contribute to the occurrence and severity of AADs.
OS, dysregulated AP, and inflammation
The initiating cause of aging and AADs is believed to be OS caused by an excessive level of free radicals, including ROS and RNS. ROS and RNS are generated by routine cellular metabolic processes, especially from mitochondria where leakage of electrons occurs during the ETC which is incompletely coupled with OP [3], catalytic inactivation of exogenous xenobiotic as well as endogenous compounds and drugs by CYP in liver cells [4], catalytic activities of NOX, cyclooxygenases, and nitric oxide synthases (NOS), particularly by innate immune cells through inducible NOS (iNOS) system during the process of fighting infection [6]. Normally, at low or physiological levels free radicals are essential as redox signaling that controls many physiological processes. Among ROS, hydrogen peroxide (H2O2) plays the most prevalent role in redox signaling by reversibly oxidizing cysteine residues which form parts of catalytic or active sites of many enzymes and transcription factors that mediate most essential pathways that control cellular metabolism, cell growth, cell proliferation, cell differentiation, and cell death [7, 8]. As well, RNS, especially nitric oxide (NO), at a physiologically low level, acts as a trigger for many key redox-mediated pathways that control essential homeostatic processes, including the activation of AMPK [9], the induction of vasodilatation and angiogenesis by NO generated from the catalytic action of endothelial NOS (eNOS) in endothelial cells (ECs) [10]; while NO generated from inflammatory signaling through the catalytic action of iNOS in phagocytic and antigen-presenting cells is involved in killing infecting microbes and recruitment of immune cells [11].
As age progresses mitochondria become dysfunctional, leading to increased uncoupling of ETC and OP, loss of adenosine triphosphate (ATP) production, releasing more ROS that overwhelms the redox homeostasis which is normally regulated by Nrf2 signaling. This exacerbates the OS, while the processes of mitophagy and mitochondrial biogenesis become imbalance and cells cannot attain an optimal number of this energy-generating organelle [3]. External pollutants and contaminants in the environment, food, as well as the body’s inflammatory responses to these toxic compounds also generate free radicals. Excessive ROS and RNS oxidize, break, and denature key cellular components including DNA, proteins, and lipids [12–14]. As a result, these abnormal molecules, especially denatured and aggregated proteins, and lipids turn into antigens designated as damage-associated molecular patterns (DAMPs) [15], recognized as non-self by innate immune cells, leading to inflammation which is mediated by NF-kB-inflammasome [16–19], P38-c-jun N-terminal kinase (JNK) mitogen-activated protein kinase (MAPK) [20], and JAK-STAT [21, 22] pathways. Because of the decline of the autophagic process and ubiquitin-proteosome system (UPS) during aging [23], due partly to the oxidative damage to their lipid and protein components, DAMPs cannot be completely eliminated, and this result in the sustained formation and release of DAMPs that continuously stimulate immune cells and switches inflammation from acute to systemic chronic inflammation (SCI) [15]. OS can also alter nucleotides, the building blocks of DNA, resulting in mutations at both single nucleotides and chromosomal levels [12, 14] which alter the translation process resulting in the abnormal proteome, affecting cell growth, cell division, cell differentiation, cellular signaling, and general cellular functions, resulting in mutated and cancerous cells. Chronic inflammation (CI) also generates damage—repair cycles whereby cells are continuously dividing, mutating, and dying. This excessive rate of cell division has a high probability of DNA miscopying, causing mutations that are the initiating causes of cancer and other AADs. Taken together, OS, SCI, dysfunctional redox homeostasis, and AP-UPS, are the root causes of aging and AADs.
Redox homeostasis, antioxidation, and anti-inflammation
Normally, cells counteract OS and restore redox balance by activating the Nrf2 pathway [24] that induces the synthesis and release of antioxidant molecules, such as glutathione and thioredoxin, and antioxidant enzymes including superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), peroxiredoxin (Prx), and HO-1 [25–27]. SOD converts superoxide anion, which arises from the addition of free electrons leaked from ETC-OP uncoupling to molecular oxygen, to yield H2O2, which in turn is converted to dihydrogen monoxide (H2O) and oxygen (O2) that are nontoxic by CAT, GPx, and Prx [25–27]. Glutathione and thioredoxin supply hydrogen from NADP(H) to help restore GPx and Prx to their reduced state, rendering them ready for another cycle of H2O2 break down [28]. HO-1 oxidizes heme to generate biliverdin, carbon monoxide (CO), and ferrous ion (Fe2+). CO has an anti-inflammatory action by inhibiting NF-kB, iNOS, eNOS, as well as p38-JNK, which down-regulates the production of proinflammatory cytokines, including tumor necrosis factor-α (TNF-α), IL-1β, IL-6, 17, 21 as well as cyclooxygenase-2 (Cox2) and NO, while upregulating the anti-inflammatory cytokines [29–32]. Biliverdin is converted to bilirubin by the catalytic action of biliverdin reductase. Bilirubin can act as a strong antioxidant, and in cooperation with CO, it can also inhibit iNOS and eNOS and attenuate the production of RNS [31, 32]. Fe2+ stimulates the production of ferritin which has an antioxidant and cytoprotective effect [32]. CO can also inhibit the JAK-STAT pathway by binding directly to STAT and preventing its phosphorylation and dimerization. Thus, STAT fails to enter the nuclei of immune cells to activate inflammatory genes [32]. Apart from inhibiting inflammation at the level of the innate immune cells HO-1 also down-regulates adaptive immune response by delaying dendritic cell maturation and differentiation, disrupting its activation of down-stream T cells, including T helper type 17 (TH17) and the cluster of differentiation 4+ (CD4+) T cells. Alternatively, HO-1 promotes regulatory T (Treg) cells as well as upregulates proinflammatory cytokines, including transforming growth factor-β (TGF-β) and IL-10, as well as induces the shifting of macrophage from macrophage 1 (M1, pro-inflammatory phenotype) to M2 (anti-inflammatory phenotype) [31]. Therefore, the net results of Nrf2 activation are to provide an anti-oxidation system to restore redox balance and protection against OS, as well as suppress inflammation and promote tolerogenic conditions via the expression of HO-1. Additionally, Nrf2 can upregulate AP [33] which can get rid of abnormal molecules, preventing the generation of DAMPs and attenuating inflammation. Likewise, AP can also upregulate Nrf2 signaling through chaperone-mediated AP of its inhibitory molecule, Kelch-like epichlorohydrin-associated protein 1 (Keap1) [34]. As aging progresses both Nrf2 and AP-UPS systems decline, partly due to OS that damage their components [23, 35], rendering them to be less effective in counteracting OS and inflammation.
The upstream regulators that synergistically control the upregulations of Nrf2, AP, and the down-regulation of inflammation comprise sensors of energy and stress levels. These sensors are composed of AMPK which detects and regulates the energy status through the ratio of AMP to ATP to maintain energy homeostasis [36, 37], and SIRT1 which is activated by increased NAD+ to NADH ratio, also indicating the low energy status [38–40]. The stress sensor is Sestrins which sense cellular stresses including OS, endoplasmic reticulum (ER) stress (UPR), hypoxia, DNA damage, genotoxic stress, and long endurance exercise [41–44]. AMPK, SIRT1, and Sestrins provide positive feedback and thus enhance the activities of each other, and synergistically activate the downstream pathways including Nrf2, AP, FOXOs, P53, PGC1-α, while inhibiting NF-kB signaling and other inflammatory pathways [42, 45]. On the other hand, these pathways are reversely controlled by mTOR1 which detects nutrient sufficiency status. The IIS also detects nutrient sufficiency status and activates mTOR1, and consequently its downstream targets, eukaryotic initiation factor 4 (eIF4) and ribosomal protein S6 kinase 1 (S6K1), to facilitate protein synthesis and anabolic processes. Upregulated IIS signaling enhances mTOR activity via AKT/protein kinase B (PKB) [46] and accelerates the aging process due to the inhibition of AMPK, Nrf2, and AP [46, 47], and the activation of NF-kB signaling [48]. Conversely, AMPK, SIRT1, and Sestrins inhibit the mTOR1 complex via two distinct pathways, either directly by phosphorylating the regulator-associated protein of the mTOR (raptor), an upstream regulatory component of the mechanistic target of rapamycin complex 1 (mTORC1), or through the phosphorylation of TSC2, which subsequently suppresses the activity of mTOR1 [36, 37, 39]. As well, AMPK can suppress IIS pathways and indirectly inhibit mTOR1 by phosphorylating insulin receptor (IR) substrate-1 (IRS-1) at serine 794 (Ser794) [49]. Therefore, AMPK-SIRT1-Sestrins and IIS-mTOR pathways are mutually inhibitory. If the balance of these two arms of controls is deregulated, i.e., if IIS-mTOR signaling prevails, the aging process will accelerate and the risk of AADs will increase. In contrast, if the AMPK-SIRT1-Sestrins and their downstream signaling pathways, especially Nrf2 and AP, are activated, aging and AADs will be attenuated (see Figure 1 for a summary).
OS and inflammation cause AADs
Recently, many studies reported that OS and SCI are the major contributors to AADs including neurodegenerative diseases such as AD, PD, amyotrophic lateral sclerosis (ALS), Huntington’s diseases (HDs), metabolic syndromes, non-alcoholic fatty liver disease (NAFLD), DM, and CVD, as well as cancer [50]. In this review, we devote our discussion to the etiologic causes due to OS, SCI, dysfunctional Nrf2, and AP on five AADs, namely, AD and PD, DM, CVD, and cancer. Finally, we also propose approaches that could be adopted to prevent and/or attenuate these AADs and some related diseases.
Neurodegenerative diseases
Despite differing triggering events, a common feature of neurodegeneration is the presence of high levels of reactive free radicals intrinsically derived from mitochondrial dysfunction, infection, and local inflammatory reactions in the glial cells and neurons, or extrinsically from systemic inflammatory cytokines, environmental toxins, and pollutants that pass the blood-brain barrier (BBB) into the neural tissue. Because of its continuous activity and relatively high metabolic rate compared with other tissues, the brain consumes large amounts of oxygen and glucose, thus generating more ROS than other tissues. Furthermore, the nerve cells’ membranes and myelin sheath contain large amounts of unsaturated fatty acids, which are readily oxidized by ROS into lipid peroxides which are themselves highly active free radicals capable of conferring oxidative damage to cell membranes and organelles. Incidentally, the neural tissue also possesses a low antioxidant system and less effective Nrf2 signaling which also decline as age progresses. This limits the brain’s counteractive actions against ROS and lipid peroxides [51, 52]. Furthermore, after birth, the brain has low cellular regenerative ability due to a limited number of stem cells, hence after neurons and glial cells succumb to injury by OS and inflammation they cannot be readily replaced.
OS induces neurons to produce unfolded or misfolded proteins followed by the formation of β-structures with a tendency to self-aggregate. Notable examples include β-amyloid and TAU, the microtubule-associated protein in AD, α-synuclein in PD, SOD in ALS, and huntingtin in HDs. These protein aggregates cause slow progressive dysfunction and loss of neurons and their axons in the central nervous system (CNS). OS and denatured proteins also activate astrocytes and microglia, with the latter as the resident macrophages of nervous tissue, to undergo the process of gliosis and secrete proinflammatory cytokines that stimulate chronic neuroinflammation in the CNS. Consequently, innate immune cells including monocytes, neutrophils, and lymphocytes are chemo-attracted to the inflammatory site. Invariably, aggregates of misfolded and abnormal proteins, such as β-amyloid plaque or α-synuclein, also cause damage and eventually neuron death (see Figure 2 for a summary).
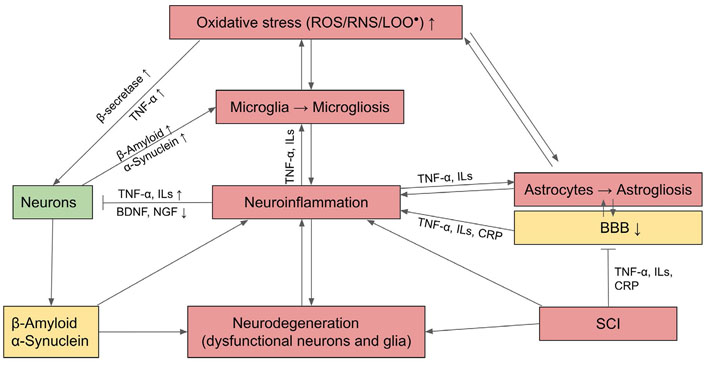
Mechanism of neurodegeneration through OS and activation of inflammatory astrocytes and microglia which undergo gliosis and cause neuroinflammation. BDNF: brain-derived neurotrophic factor; CRP: C-reactive protein; NGF: nerve growth factor; LOO•: lipid peroxide; : activation; : inhibition; ↑: upregulation; ↓: downregulation
AD
AD is the most prevalent form of dementia. While the true etiology of this disease remains unknown, it was found that the characteristically associated histo-pathological sign is the accumulation of β-amyloid plaque in the hippocampus, a brain area involved in memory, as well as in other areas of the cerebral cortex causing neuronal death and deterioration of memory and other mental capacities. It is believed that the initiating cause of AD is the over-production and the accumulation of free radicals generated by defective mitochondria in neurons of the involved brain areas, which can be due to genetic or induced damage by OS and β-amyloid [53, 54]. This results in the over-production of ROS which leads to OS [55]. OS activates β-secretase which cleaves amyloid precursor protein (APP), a membrane protein of still unclear function, into β-amyloid which is a sticky product that clumps together to form amyloid plaque [56]. In turn, this abnormal protein aggregate, together with ROS, causes lipid peroxidation that disrupts the structure and function of the cell and organelle membranes leading to the failure of homeostatic controls and eventually apoptosis of neurons. Furthermore, these factors also cause a hyper-phosphorylation of TAU, leading to their abnormal aggregation into the so-called neurofibrillary tangle, which affects the structure of neuronal processes and synapses, resulting in failures of the axonal transport, neurite outgrowth, as well as synaptic transmission [56]. In the affected neurons, autophagic mechanisms are less effective or even suppressed, such that proteostatic recycling of abnormal proteins could not be carried out effectively, contributing to neuronal death [57, 58].
In addition, ROS and β-amyloid oligomers can activate microglia and astrocytes to release pro-inflammatory cytokines and also more free radicals so that these result in neuroinflammation that contributes to the degeneration of neurons [59]. The β-amyloid or the misfolded proteins themselves could also have a direct effect on the death of the neurons. Concurrently, the misfolded proteins and activated microglia can downregulate BDNF and NGF which are the trophic factors that promote the survival of neurons [60–62].
Apart from direct action on neurons through gliosis and neuroinflammation, the ROS and SCI also occur in the so-called neurovascular unit, namely, the astrocytes and the ECs lining the neural vasculature, which are the compositions of BBB. As the result of oxidation and inflammation, gliosis is generated in the surrounding astrocytes; this together with EC dysfunction leads to the disruption of BBB that affects the transport of water, electrolytes, nutrients, and wastes products into and from the brain. Thus, the microenvironment and homeostasis of the brain tissue could no longer be regulated, and this exacerbated conditions that lead to neuron death [63].
In addition to locally generated reactions from OS and neuroinflammation, neurodegeneration can be exacerbated by SCI from the persistently high levels of serum inflammatory mediators, including CRP, IL-6, and TNF-α. These circulating proinflammatory cytokines from SCI can permeate the impaired BBB and activate microglia to undergo microgliosis, which contributes to neuroinflammation and ensuing AD (Figure 2) [64, 65]. AD could be ameliorated by the upregulation of Nrf2 signaling which can counteract OS and activate AP of β-amyloid [66].
PD
PD is second only to AD in terms of prevalence and deleterious effects on the socio-economic and health status of the population. The main pathological sign of this disease is the degeneration and death of dopaminergic neurons in the region of the brain stem, called substantia nigra, due to the aggregation of the misfolded protein named α-synuclein [classically called Lewey bodies (LBs)] [67, 68]. The disease results in a motor deficit, rigidity, tremor, and inability to maintain a normal posture. In addition, the patient will progressively become demented and depressed.
PD could be both genetic and environment dependent. At least mutations of ten genes could be involved in the pathogenesis of PD, namely, G protein subunit gamma 3 (GNG3), MAPK1, formyl peptide receptor 1 (FPR1), ATP synthase F1 subunit 5β (ATP5B), guanine nucleotide-binding protein G subunit gamma 2 (GNG2), protein kinase cyclic AMP-dependent α catalytic subunit (PRKACA), Harvey murine sarcoma virus oncogene (HRAS), heat shock protein family A member 8 (HSPA8), presenilin-associated protein (PSAP), and γ-aminobutyric acid type B receptor subunit 2 (GABBR2) [69]. Specifically, the mutations of a series of Parkinson-associated genes (PARK), such as leucine-rich repeat kinase 2 (LRRK2), caused mitochondria dysfunction as well as accumulation of free radicals leading to the formation of misfolded protein, α-synuclein, whose aggregation forms LBs [70]. Free radical accumulation also inhibits the functioning of the proteasome, chaperone-mediated AP (CMA), blocking clearance of the abnormal protein. As well, mutations of Parkin/phosphatase and tensin homolog induced putative kinase 1 (PINK1) genes disrupt mitophagy processes. Thus, mitochondrial biogenesis and recycling mechanisms of abnormal proteins are disrupted. Aggregated α-synuclein species, if not cleared in time by AP, can also lead to the formation of large aggregates of LBs, which can be physically obstructive to neuronal function and eventually lead to neuronal death [71]. Environmentally, the causative agents of PD could be pollutants, toxins, and certain agrochemicals used for insecticides and weed killers, such as 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP), rotenone, paraquat, glyphosate [72, 73]. These chemicals can pass BBB into neurons where they can disrupt mitochondrial function in producing ATP, and instead generate more ROS and suppress AP, such that neurons lose their homeostatic control over protein recycling and undergo cell death [74, 75]. Furthermore, α-synuclein itself is a toxic oxidative molecule that causes lipid peroxidation, disrupting cell membrane structure and function, thus contributing further to neuron death [67, 76]. Like in the case of AD, neuroinflammation, and neurodegeneration in PD could be alleviated by the activation of Nrf2 and AP signalings [77].
DM
OS and SCI have been demonstrated to be the major causes of both type 1 diabetes mellitus (T1DM) as well as T2DM, and both are entailed by microvascular, cardiovascular, kidney, retinal and neural damages [78]. T1DM is an autoimmune disease where insulin is recognized as a non-self-antigen by dysregulated B cells which produce anti-insulin antibodies to attack pancreatic β-cells [79, 80]. Subsequently, macrophages and T-cells are drawn to the islet by these auto-antibodies. ROS and RNS from macrophages and excessive amounts of proinflammatory cytokines released by the immune cells damage β-cells as well as adjacent cells, resulting in insulitis, which eventually leads to dysfunction and death of β-cells and pancreatic islet atrophy [81] which causes T1DM.
In comparison to T1DM, T2DM is more common and accounts for over 90% of worldwide diabetic cases. T2DM is a multifactorial disease, where a mixture of polygenic effects and environmental factors are interacting to cause the disease. The genetic factors are believed to be key predisposing conditions in individuals susceptible to T2DM [82]. From the GWAS, it was found that there could be up to 70 genes whose variations at single nucleotide polymorphism (SNP) level or mutation predispose individuals to develop T2DM [83]. Some of these genes may be involved in maintaining the physiology of β-cells while others in controlling the secretion of insulin. Other genes may encode proteins in the insulin signaling pathway, such as the IR and the IRS, or other transducing proteins downstream in the insulin signaling pathway [84]. Defect or functional impairment of one or more of these genes can initiate insulin resistance, such as a mutation in IR and its IRS, and impairments of insulin signaling at the level of IRS1-associated phosphoinositide 3-kinase (PI3K) activity and Akt/glycogen synthase kinase-3 (GSK-3), FOXO1 phosphorylation [85]. Once insulin resistance occurs, β-cells try to compensate by producing and secreting a larger amount of insulin, which leads to β-cell overwork. Excessive ROS from increased mitochondrial activity accompanying heightened β-cell activity builds up OS that causes more damage to β-cell [86]. Additionally, β-cell functions in insulin production and secretion gradually deteriorate due to a persistently high amount of glucose (hyperglycemia) and free fatty acids (hyperlipidemia) in the circulation, which over-stimulate insulin secretion. Once hyperglycemia and hyperlipidemia become overt, the β-cell’s function progressively deteriorates. In the diabetic state, hyperglycemia and hyperlipidemia elicit OS that increases the suppressions of insulin synthesis and secretion, and finally leads to β-cell apoptosis and death [84]. Since the expression levels of antioxidant enzymes in β-cells are very low compared to other cells and tissues, β-cells are more easily damaged by OS. Consequently, it is likely that chronic exposure of β-cells to high glucose and fatty acid concentrations in genetically susceptible individuals finally leads to β-cell failure and T2DM.
Another cause of IR is the activation of NOX by high glucose levels that leads to excessive ROS production and ensuing inflammation and rising level of proinflammatory cytokines, especially TNF-α, interferon-γ (IFN-γ), and IL-1β that cause β-cell dysfunction and death [87]. Habitually high glucose level also activates the protein kinase C (PKC) pathway via diacylglycerol (DAG) which increases the hexosamine pathway that leads to the production of advanced glycation-end product (AGEs) and increased activity of the polyol pathway [88, 89]. ROS is generated in the polyol pathway when aldose reductase reduces glucose to sorbitol. Excessive glucose enhances this pathway leading to over conversion of NADP(H) to NADP, resulting in the exhaustion of NADP(H), which is needed for the regeneration of antioxidants to counteract OS [89]. Accumulated ROS as well as AGEs can activate inflammatory pathways.
IR is driven intracellularly at a molecular level through the inhibition of IR and its associated transducing proteins by inflammatory factors, leading to the interruption of the insulin signaling cascade (see Figure 3 for a summary). Under normal circumstances, glucose transport into cells is mediated by insulin and its receptor signaling. Rising blood glucose after a meal induces the secretion of insulin from β-cells. In turn, insulin binds to the transmembrane IR, which is a tyrosine kinase receptor in a dimeric form, with each monomer comprising α and β subunits linked by disulfide bonds. The binding of insulin to the extracellular domain of the α subunit causes the two monomers to dimerize and auto-phosphorylate each other at the tyrosine residues. Then the signals are transmitted through two pathways. In the first pathway an IR-associated protein, insulin IRS-1 is phosphorylated. Activated IRS-1 binds to and phosphorylates PI3K which, in turn, phosphorylates and converts the membrane-bound phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 is a secondary messenger that activates downstream kinases, including phosphoinositide-dependent kinase 1 (PDK1) which, in turn, phosphorylates PKB also called AKT. PKB induces the translocation of vesicles bearing glucose transporter 4 (GLUT-4) from the cytoplasm to join the cell membrane where GLUT-4 facilitates the transport of glucose into the cells. PKB also inhibits GSK-3, which is an inhibitor of glycogen synthase, an enzyme that catalyzes glycogen synthesis from glucose. In addition to facilitating the entry of glucose into cells and mediating metabolic control over glucose utilization, insulin also stimulates glycogen synthesis and lipogenesis. PKB also stimulates eNOS and mTOR which stimulates protein synthesis, while lipolysis and FOXOs are suppressed. Thus, insulin signaling favors anabolism (also see Figure 3) [90]. The second pathway of IIS is mediated through IR-adaptor protein Src homology 2 (SH2) domain containing transforming protein (SHC) with SH2 domain-Son of Sevenless (SOS)- rat sarcoma virus protein (Ras)-MAPK krasin (MEK)-MAPK/extracellular signal-regulated kinase 1/2 (ERK 1/2) pathway which promotes cell growth, proliferation, differentiation, motility, and survival [91].
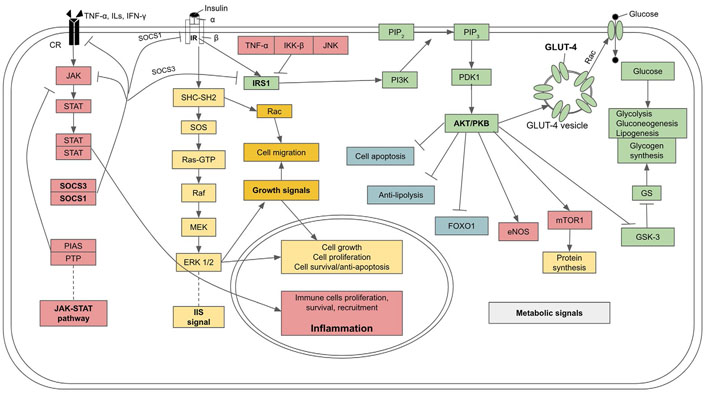
IIS pathway, its metabolic effects, and molecular mechanism of insulin resistance and diabetes due to the inhibitory effect of cytokines from SCI on IIS. GS: glycogen synthase; Rac: ras-related C3 botulinum toxin substrate; PTP: protein tyrosine phosphatase; PIAS: protein inhibitor of activated STATs; SOCS3: suppressors of cytokine signaling 3; IKK-β: inhibitor of kappa B kinase; Raf: rapidly accelerated fibrosarcoma kinase; : activation; : inhibition
Inflammation, through upregulation of proinflammatory cytokines including TNF-α, monocyte chemoattractant protein-1 (MCP-1), CRP, and ILs, can interfere with and disrupt certain steps of the insulin signaling cascade and cause IR. In particular, TNF-α mediates phosphorylation of IRS-1 at Ser307 residue and obstructs its signaling action, disrupting GLUT-4 translocation to the plasma membrane and decreasing glucose transport into the cells [92]. As well, IRS-1 can be phosphorylated at Ser307 by kinases of inflammatory pathways, including IKK-β and JNK, leading to a decrease in insulin signaling [93–95]. Furthermore, certain ILs, especially IL-6, and an over expression of JAK-STAT signal can promote IRS degradation by upregulating expressions of SOCS1/3 [96, 97], which dampens JAK-STAT and induce ubiquitylation of IRS proteins, leading to the degradation of the complex by the proteasome (also see Figure 3) [97].
Invariably, T2DM is closely associated with obesity [91, 98]. Obesity is due mainly to the increasing number of white adipose cells overloaded with excessive triglycerides and free fatty acids, resulting in ROS production, oxidation of fatty acids, and the release of DAMPs that attract innate immune cells, particularly macrophages, which initiate inflammatory reaction [99]. Thus, in obese persons recurrently high levels of proinflammatory cytokines released from overloaded white adipose tissue can interfere with the insulin signaling pathway and lead to T2DM.
Atherosclerosis and CVD
Atherosclerosis (AS) and CVD are the results of several risk factors including overweight and obesity, physical inactivity, unhealthy diet, smoking, consumption of alcohol, pollution, as well as aging. All these factors finally lead to the accumulation of OS and inflammation which are the presumptive causes of AS and CVD [100]. The major precipitating cause of vascular oxidation and inflammation is dyslipidemia characterized by increased circulating levels of low-density lipoproteins (LDLs), cholesterol, triglycerides, and decreased level of high-density lipoproteins (HDLs). High levels of LDLs and cholesterol have been regarded as the most prominent etiologic cause of AS and CVD. In the situation where there are OS and LDLs, at a high level both locally and systemically, will be oxidized by ROS, resulting in the formation of lipid oxidized LDLs (oxLDLs), including malondialdehyde (MDA), thiobarbituric reactive substances (TBARS), lipid hydroperoxides (LH), and 4-hydroxynonenal (4-HNE), all of which are highly reactive [100–102]. In blood vessels, oxLDLs damage ECs which release macrophage colony-stimulating factor (M-CSF), which attracts the initial set of monocytes. These cells, in turn, release MCP-1, which attracts more monocytes and other immunocompetent cells into the region. Receptors for MCP-1 [C-C chemokine receptor type 2 (CCR2)] become highly expressed on monocytes, ECs, as well as vascular smooth muscle cells (VSMCs), during the early phase of plaque formation [103]. In addition, oxLDLs and truncated lipids that activate inflammation pathways in the ECs, and induce them to express adhesion molecules, including vascular cell adhesion molecule-1 (VCAM-1), E-selectin, and P-selectin that slow down the circulating monocytes and facilitate their transmigration into the subendothelial layer, where they differentiate into macrophages. These macrophages phagocytose oxLDLs through the scavenger receptors (SRs) and CD36 and transform into enlarged foam cells, and once overloaded they undergo apoptosis and form a plaque, consisting initially of debris from the disrupted cells and residual oxLDL [103]. The ECs and VSMCs in the tunica media layer, immersed in intense inflammatory mileu, produce pro-inflammatory molecules including TNF-α, IL-1β, and MCP-1 which attract neutrophils and more monocytes to the inflammatory site [103, 104]. Neutrophils, in turn, engulf cellular debris and oxLDLs while also enhancing pro-inflammatory effects. Monocytes recruited to the site of plaque formation are activated by toll-like receptors (TLRs), and INF-γ present in the micro-environment of a growing plaque to become M1 subtype [104, 105] which secrete more pro- inflammatory cytokines such as IL-1β, IL-6, TNF, IL-12, and IL-23 along with ROS and RNS that sustain the ongoing inflammation [105]. Injured ECs also release inflammatory cytokines as well as von Willebrand factor (vWF) from their Weibel-Palade bodies, further promoting inflammation, while vWF induces platelet adhesion and aggregation by binding with platelet glycoprotein-1b (GP-1b), contributing to the growing plaque. Under normal circumstances, the ECs produce inhibitors of thrombin activity (including NO, prostaglandins, and thrombomodulin), which prevent platelets’ aggregation. However, at the site of vascular injury and inflammation, the dysfunctional ECs transform into a pro-thrombotic phenotype and secrete coagulation factors, including vWF, P-selectin, and angiopoietin-2. The endothelial-derived vWF induces platelet activation and coagulation around the plaque and contributes to its growth [106, 107]. The VSMCs in tunica media at the lesion site synthesize collagen, elastin, and other extracellular proteoglycans that form fibrous cap around the plaque and contribute to intimal hyperplasia [108]. Some VSMCs may undergo dedifferentiation and transform into mesenchyme-liked cells that also engulf oxLDLs. M1-type macrophages also recruit Th1 and natural killer cells to the growing plaque, further exacerbating the injury due to uncontrolled inflammation [109]. M1 macrophages secrete several matrix metalloproteinases (MMPs), comprising MMP-1, MMP-2, MMP-8, MMP-9, MMP-13, MMP-14 which degrade collagen types I, III, IV, proteoglycans, and elastin in the vessel wall, leading to the destruction of the basement membrane and the extracellular matrix [109, 110]. Local increases of cytokines, especially IL-1β, TNF-α, and IFN-γ by activated macrophages and T cells at the lesion site induce apoptosis of ECs and VSMC and block collagen production [111]. Myeloperoxidase produced by neutrophils generates hypochlorous acid, causing more ECs and VSMC apoptosis. Loss of collagen, apoptotic ECs, and VSMCs disrupt the vessel wall. The direct damaging effect of a stable plaque, that turns into an artheroma, is the blockage of the vessel’s lumen that disrupts the blood supply to the surrounding tissues and distal organs. Inadvertently, an atheroma may become unstable and loosened from its original site, due to erosion of ECs and VSMCs, and circulate as an embolus to the heart, where if it is large enough may cause blockage of the coronary artery or its subsidiary branches, resulting in local ischemia, cardiac infarction; and if the affected area is large cardiac failure ensues. Likewise, an atheroma may circulate to the brain where it may block an arterial vessel supplying the brain tissue, resulting in neural ischemia, stroke, neurodegeneration, and even death if the blockage occurs in a critical area of the brain [112]. Therefore, oxLDLs, the main product of ROS and lipid peroxidation, especially in the case of obesity, contribute to the oxidation-inflammation cycle which is a major cause of AS, CVD, and related complications [113].
Cancer
Many types of cancers can develop from OS. Excessive ROS/RNS, produced by overactive normal cells or dysfunctional mitochondria, can oxidize and alter nucleic acids, proteins, and lipids. In particular, the hydroxyl radical (OH•) oxidizes DNA at purine and pyrimidine bases and the deoxyribose-phosphate chains to generate single-strand and double-strand breaks and cause mutation. Furthermore, ROS selectively interacts with guanine to produce 8-oxo-7,8-dihydro-deoxyguanine (8-oxo-dG) molecules which cause G:C to T:A transversions and mutation, as well as activates oncogenes and suppress tumor suppressor genes (including P53), giving rise to carcinogenesis [12, 14, 114]. Oxidation of lipids gives rise to lipid peroxides that generate highly mutagenic MDA and HNE [13]. Both MDA and HNE form stable bonds with nucleic acids, proteins, and phospholipids, disrupting signaling pathways, dysregulation of oncogenes and tumor suppressor genes that turn normal cells into cancer cells with uncontrollable cell division, resistance to apoptosis, and invasive migration [115, 116]. Because of their high rate of proliferation and growth, cancer cells require vast amounts of energy and thus consume very high amounts of glucose and oxygen compared to normal cells. Consequently, their mitochondria are overactive and generate a very high level of ROS during ATP synthesis through ETC and OP processes. This contributes even more to OS which causes DNA modification and disruption, leading to genetic instability and mutation. When DNA damage occurs in normal cells the expression of P53 is induced and Nrf2 signaling is upregulated so that ROS and RNS can be controlled, and damage DNA can be repaired. In pre-cancerous cells both pathways are down-regulated, thus ROS-damaged DNA accumulates. As a result, there is an amplification of genomic instability and loss of tumor suppressor genes. This vicious cycle of OS build-up and more genomic instability eventually cause the cells to become full-fledged cancer [114–116].
ROS can also accumulate as a consequence of the metabolism of exogenous toxic substances the body receives from aerial pollution (e.g., PM2.5), food contaminants, and drugs, as well as endogenous compounds such as hormones, which are catabolized by the CYP system in liver cells, leading to the production of ROS and byproducts or metabolites that are more reactive, toxic and mutagenic than the original compounds. A good example is benzo(a)pyrene (BaP) which is a polycyclic aromatic hydrocarbon derived from incomplete combustion of diesel engines, smoke from wood burning, coal tar, tobacco smoke, grilled meat, and chicken. When the body uptakes the substance through the respiratory and digestive systems, this compound is catabolized by the CYP system of the liver cells, giving rise to a highly reactive byproduct, a BaP diol epoxide (BPDE), which binds and reacts to DNA and yield guanine adduct, leading to mutation and eventually cancer [117]. Cancer frequently arises in the two systems whose organs are directly exposed to BaP and its metabolite, including lung cancer, colon cancer, and liver cancer [117]. Another example is cancer induced by over consumption of ethanol. Upon entry into the body, ethanol is metabolized by the CYP system that generates a copious amount of ROS which results in the oxidation of DNA, proteins, and lipids, leading to genomic instability, mutation, and cancer as mentioned earlier. Moreover, the generated lipid peroxides interact with DNA and create etheno-DNA adducts, resulting in mutation that causes hepatocarcinoma (HCC) [118]. In addition, ROS generated by CYP can trigger inflammation and NAFLD which, if severe, can lead to the death of liver cells, liver tissue damage, fibrosis, and eventually HCC [119]. Therefore, OS and mutagens arising from exogenous toxicants and endogenous compounds being catabolized by the CYP system are one precipitating cause of cancer development.
CI from persistent exposure to environmental pollutants, infection [e.g., from Helicobacter pylori (H. pylori)], and tissue damage that continues to release DAMPs (e.g., from an unhealed wound and obesity), and autoimmune condition, is another root cause of cancer [117, 120]. Reciprocally, during inflammation, innate immune cells can generate excessive amounts of ROS that cause cancer via the aforementioned mechanisms. Moreover, ROS can activate the MAPK pathways with the upregulations of ERK, JNK, and p38, the signaling proteins that induce cell proliferation-cell apoptosis cycle during CI, such as in unhealed wounds [120]. Such a vicious cycle of cell death and cell renewal at the site of CI results in continuous compensatory cell division with an increased chance of having daughter cells with mis-copied and mutated DNA that eventually become cancer cells [120–122]. An obvious example is a patient with a gastric ulcer, due to persistent infection with H. pylori, which can develop into stomach cancer [123, 124]. Patients with habitual and uncured gastric flux which creates non-healing wounds at the gastro-esophageal region can develop esophageal cancer [125]. Patients who have chronic inflammatory bowel diseases (IBDs). For example, ulcerative colitis and Crohn’s disease can succumb to colon cancer [126, 127].
Prevention and attenuation of AADs
As evident from the above discussion that OS and inflammation are the root causes of AADs, it could, therefore, be surmised that the logical approach that may prevent or attenuate the AADs is the restoration of redox homeostasis by activation of the antioxidation pathway while suppressing the inflammatory pathways. This can be achieved by activating Nrf2 which induces the expressions of antioxidant molecules and enzymes including SOD, CAT, GPx, Prx, and HO-1. The latter is crucial in providing strong antioxidation as well as inhibition of inflammation as mentioned earlier. There have been extensive studies on the effects of the upregulation of Nrf2-HO-1 which could alleviate and prevent AADs, particularly inflammatory and autoimmune diseases, including multiple sclerosis (MS), IBDs, systemic lupus erythematosus (SLE), and rhumatoid athritis [32]. Additionally, several AADs and related diseases have also been reported to be alleviated by the upregulation of Nrf2-HO-1, for example, obesity, NAFLD, related metabolic syndromes including hypertension, arthrosclerosis, and CVD [128–132]. Nrf2 activation counteracts OS, inhibits inflammation, and promote AP; thus, the Nrf2-HO-1 system is critical for preventing the onset of DM and its entailed vascular, cardiac, and kidney complications [133–136]. Furthermore, neurodegenerative diseases including AD and PD have also been reported to be alleviated by Nrf2-HO-1 upregulation, especially with the ability of Nrf2 to activate AP and UPS systems, which help to eliminate misfolded proteins (β-amyloid and α-synuclein) and attenuate the neuroinflammation caused by OS [137–140]. For cancer, the role of Nrf2 is paradoxical: in normal cells, Nrf2 expression is moderate to protect DNA and other molecules from damage by OS and carcinogens and prevent the cells from becoming cancerous. However, once cancer cells become established Nrf2 is overexpressed, which allows cancer cells to withstand high OS, develop drug resistance, promote angiogenesis and cancer stem cells, as well as metastasis [141–144]. Apart from AADs, Nrf2-HO-1 activation also alleviates and prevents diseases of other organ systems, including kidney diseases, such as acute kidney injury (AKI) [145, 146], lung diseases [147], and even bone from osteoporosis [148].
As mentioned above, the activations of Nrf2 and AP are regulated by a set of synergistically acting energy and stress sensors, i.e., AMPK-SIRT1-Sestrins. These three sensors auto-stimulate each other to promote catabolism to restore energy deficit and counteract OS through the activation of the Nrf2 and AP pathways, while also directly inhibiting inflammation (see Figure 1). There have been a number of studies indicating that by activating these central regulators, together with their downstream targets, especially Nrf2-HO-1 and AP, many AADs can be prevented or ameliorated. The most effective and intensively studied is the activation of Sestrins which could effectively attenuate and prevent AADs. By synergistically acting with AMPK and the inhibition of mTORC1 Sestrins can control glucose homeostasis and prevent insulin resistance [149]. Sestrins activation could prevent or alleviate CVD and related complications, including AS, acute myocardial infarction (AMI), heart failure, hypertension, myocardial hypertrophy, atrial fibrillation, and myocardial fibrosis [150–152]. Sestrins can attenuate insulin resistance and are considered a target for diabetes therapy [153, 154]. Sestrins prevent AD and PD as well as ALS through their ability to activate Nrf2 to counteract OS and upregulate AP to eliminate misfolded proteins, which are the initiating causes of these neurodegenerative diseases [155–158]. Sestrins can suppress cell growth and proliferation and thus attenuate colorectal cancer, lung carcinoma, and endometrial cancer [159–163]. Apart from AADs, recent studies have demonstrated the preventive function of Sestrins in musculoskeletal system diseases, such as intervertebral disc degeneration (IDD), osteoarthritis (OA), and sarcopenia [164]. Sestrins could prevent various liver diseases, including NAFLD and ensuing liver cancers, such as HCC [165, 166]. The hepato-protective effect of Sestrins is dependent on its upregulation of the Nrf2-HO-1 pathway which mitigates oxidative damage to hepatocytes [166]. Apart from AADs, recent investigations have demonstrated that Sestrins protect against many OS-generated respiratory diseases, including chronic obstructive pulmonary disease (COPD), asthma, acute respiratory distress syndrome (ARDS), obstructive sleep apnoea (OSA), and cigarette-induced emphysema [167–169]. Sestrins activation could prevent AKI, glomerular parietal epithelial cells (PECs) injury, glomerular mesangial cell (MC) damage, and diabetic kidney disease (DKD) [170].
Apart from Sestrins, activation of AMPK with Sestrins and/or SIRT1 can help protect and attenuate many AADs. Co-activation of AMPK and Sestrins could prevent metabolic diseases, including obesity, NAFLD, and diabetes [149]. Activation of AMPK, SIRT1, and Nrf2 can help preserve bone mass and prevent osteoporosis [171]. Activation of Sestrins and AMPK together with inhibition of mTOR1 can prevent metabolic disorders, neurodegenerative diseases, CVD, cancer, as well as musculoskeletal diseases [172]. Therefore, one key approach in preventing or attenuating AADs is to find means or agents that can activate AMPK-SIRT1-Sestrin signalings, with strong upregulations of Nrf2-HO-1 and AP.
Recent studies indicated that this goal can be achieved by simple strategies through exercise and caloric restriction (CR) which act through mitochondria by enhancing their metabolism, leading to increased AMP/ATP and NAD+/NADH ratios and direct activation of AMPK and SIRT1 signaling. Exercise increases glucose uptake facilitated by the translocation of the GLUT-4 to the plasma membrane via activation of the IIS/PI3K/AKT signaling pathway (see Figure 2). Increased AMPK-SIRT1 activation and rising intracellular calcium levels also induce GLUT-4 translocation independent of the IIS pathway [173]. Furthermore, AMPK-SIRT1 activation increases PGC-1α gene expression and directly phosphorylates the protein. Phosphorylated PGC-1α activates Nrf1/2 that resulting in the expression of mitochondrial transcription factor A (mtTFA), which stimulates replication and transcription of mtDNA, leading to mitochondrial biogenesis and mitochondrial OP process to satisfy the energy demand [174, 175]. Additionally, exercising muscle also releases myokines, including muscle-derived IL-6 and irisin [176, 177], as well as β-hydroxybutyrate, a ketone metabolite [178, 179], all of which synergistically suppress inflammation in muscle as well as in other organs, thus helping to alleviate AADs, such as in liver (NAFLD, liver fibrosis, HCC), adipose tissue and pancreas (insulin resistance, diabetes), blood vessels (AS, hypertension, and CVD) and brain (AD and PD) [180, 181]. Mechanistically, β-hydroxybutyrate can activate AMPK and Nrf2 as well as inhibit inflammation by suppressing inflammasome formation and endoplasmic reticulum stress [179, 180]. Furthermore, it has been demonstrated that BDNF is produced by strenuously contracting muscle which is transported to CNS to promote neurogenesis and prevent neurodegenerative diseases, including AD, PD, and various forms of dementia [182, 183]. The synthesis and transport of BDNF in neural tissue to improve brain function is also initiated and facilitated by the rise of β-hydroxybutyrate from heightened muscle metabolic activity [183]. Interestingly, it was recently proposed that β-hydroxybutyrate may be considered a key metabolite that can attenuate aging as well as AADs [184]. Exercise also activates Sestrins perhaps through the build-up of free radicals from the increased metabolism of muscle cells [43, 185]. Thus, beneficial outcomes of exercise could also be mediated through Sestrins acting together with AMPK, which can attenuate AADs through their combined ability to activate Nrf2-HO-1 and AP [186]. Exercise, particularly resistance exercise, also maintains and increases muscle mass by promoting protein synthesis via Akt/mTOR pathway. The mTOR activates translation and protein synthesis via the translation regulators p70 ribosomal protein S6 kinase (p70s6k) and the eIF4E complex [176].
CR with adequate nutrition can extend longevity and health span in many species including humans, and protects against the development of obesity and AADs [187]. A key cellular response elicited by CR is AP which is induced to compensate for nutrient deprivation and, consequently, plays a beneficial role in alleviating AADs. CR upregulates AP by activating regulators of energy sensors while inhibiting nutrient sensors. CR, like exercise, is a mild stress that acts on mitochondria by enhancing their metabolism to meet energy demand, which leads to increased AMP/ATP and NAD+/NADH ratios that directly activate AMPK and SIRT1 signaling [188]. In turn, AMPK inhibits mTOR, an AP repressor. SIRT1 and AMPK auto-stimulate and enhance the activity of each other and, consequently, amplify the effect of CR. The net result of this signaling loop activation is the upregulation of AP [189]. Apart from direct activation of AP by AMPK, SIRT1 deacetylates nuclear microtubule-associated proteins 1A/1B light chain 3B (LC3B), stimulating its interaction with the nuclear protein, the diabetes- and obesity-regulated protein (DOR), resulting in its export to the cytoplasm, where it acts as a key initiator of AP [190]. As well, SIRT1 deacetylates mitofusin-2, a protein tethered to the mitochondrial membrane, facilitating SIRT1-induced mitophagy [191]. Apart from the activation of AP, AMPK-SIRT1 signaling also stimulates the up-regulation of Nrf2-HO-1. The combined effects of these two processes are the main factor through which CR alleviates and prevents AADs.
The other approach that can alleviate and prevent AADs is the intake of natural products as nutraceuticals, notably, polyphenols [e.g., berberine, quercetin, myricetin, caffeic acid (CA), and CA ethanolamide (CAEA), CA phenyl ester (CAPE), epigallocatechin-3-gallate (EGCG), gallic acid (GA), curcumin], flavonoids (e.g., resveratol), terpenoids (e.g., ginsenoside and triterpene glycosides), carotenoids, xanthophyll (e.g., astaxanthin), spermidine, and sulforaphane, all of which could act as CR mimetics. Most of these natural compounds have high antioxidation properties, and they can also directly stimulate AMPK (e.g., curcumin, EGCG, spermidine, GA, and CAPE) or SIRT1 (e.g., berberine, EGCG, quercetin, and myricertin) or both (e.g., resveratol, CA, and CAEA), and consequently causing the upregulations of Nrf2 and AP (also see Figure 1) [29, 32, 171, 172, 192–195]. Some natural compounds can also suppress inflammation (e.g., curcumin, EGCG, and spermidine) [192]. Furthermore, it was found that certain drugs [e.g., metformin, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), and salicylate/aspirin] can activate AMPK by acting as a mild toxic agent against mitochondria to increase AMP/ATP ratio, in a process known as mitohormesis [171, 192]. Alternatively, a certain drug like rapamycin can inhibit mTOR1 [29, 192], hence allowing AMPK and its ancillary pathways (SIRT1 and Sestrins) to become highly active, which results in similar outcomes.
In summary, exercises, CR, intakes of natural products, and certain mitohormetic drugs like metformin can activate AMPK-SIRT1-Sestrins signaling, and consequently their downstream target genes, particularly those that are involved in Nrf2 and AP upregulations, which are the final common pathways that attenuate aging as well as AADs. However, further investigations are needed to reveal details of molecular mechanisms of these approaches and actions of the CR mimetic natural compounds as well as their bioavailability, biotransformation, and dosage-toxicity, so that they can be developed into efficient, non-toxic strategies or drugs that can be used alone or as adjunctive or combinatorial treatments to prevent and/or cure AADs. Stringent clinical trials in humans are also necessary before these compounds could be declared safe for adoption as nutraceuticals.
Conclusions
As age progresses there is an accumulation of excessive ROS and RNS over the thresholds of physiological levels, leading to OS which oxidizes and denatures key cellular components, including DNA, proteins, and lipids. Due to age-impaired AP and UPS these denatured molecules and some of their aggregates cannot be eliminated, turning them into DAMPs, recognized as foreign by immune cells, causing SCI, which together with OS are the major causes of AADs. To prevent and cure AADs redox balance should be restored by activation of Nrf2 and HO-1 which can prevent the damaging effects of OS and SCI, as well as upregulate AP and UPS to eliminate DAMPs and misfolded proteins. The central regulators, AMPK, SIRT1, and Sestrins, synergistically control the Nrf2 pathway, upregulation of AP-UPS, and inhibit inflammatory pathways and SCI. Consequently, the activations of these central regulators may lead to the prevention or attenuation of AADs. Recent studies indicate that these goals are achievable through exercise, CR, intakes of nutraceuticals and natural products which are CR mimetic, including polyphenols, flavonoids, resveratrol, terpenoids, carotenoids, spermidine, sulforaphane, and certain mitohormetic drugs (e.g., metformin and salicylate) which activate AMPK-SIRT1-Sestrins and, consequently, their downstream target genes, especially those that upregulate Nrf2 and AP which are the major factors that attenuate aging and AADs. Alternatively, there are certain drugs, e.g., rapamycin, that can inhibit mTOR1 and allows AMPK-SIRT1-Sestrins to prevail and achieve similar outcomes. The molecular mechanisms that exercise, CR, and natural products act on the central regulators should be explored in detail. As for natural products, their bioavailabilities, biotransformations, dosage-toxicity, and clinical trials should be pursued so that they can be adopted safely for human use as preventive and therapeutic measures against AADs.
Abbreviations
| AADs: | age-associated diseases |
| AD: | Alzheimer’s disease |
| ALS: | amyotrophic lateral sclerosis |
| AMP: | adenosine monophosphate |
| AMPK: | adenosine monophosphate-activated protein kinase |
| AP: | autophagy |
| AS: | atherosclerosis |
| ATP: | adenosine triphosphate |
| BBB: | blood-brain barrier |
| BDNF: | brain-derived neurotrophic factor |
| CAT: | catalase |
| CI: | chronic inflammation |
| CNS: | central nervous system |
| CO: | carbon monoxide |
| CR: | caloric restriction |
| CRP: | C-reactive protein |
| CVD: | cardiovascular diseases |
| CYP: | cytochrome P450 |
| DAMPs: | damage-associated molecular patterns |
| DM: | diabetes mellitus |
| ECs: | endothelial cells |
| EGCG: | epigallocatechin-3-gallate |
| eNOS: | endothelial nitric oxide synthases |
| ERK 1/2: | extracellular signal-regulated kinase 1/2 |
| ETC: | electron transport chain |
| FOXO3: | forkhead box protein O3 |
| GLUT-4: | glucose transporter 4 |
| GPx: | glutathione peroxidase |
| GSK-3: | glycogen synthase kinase-3 |
| GWASs: | genome-wide association studies |
| H2O2: | hydrogen peroxide |
| HCC: | hepatocarcinoma |
| HNE: | hydroxynonenal |
| HO-1: | heme oxygenase-1 |
| IFN-γ: | interferon-γ |
| IIS: | insulin/insulin-like growth factor signaling |
| IL-6: | interleukin-6 |
| iNOS: | inducible nitric oxide synthases |
| IR: | insulin receptor |
| IRS-1: | insulin receptor substrate-1 |
| JAK: | Janus kinases |
| JNK: | c-jun N-terminal kinase |
| LDLs: | low-density lipoproteins |
| M1: | macrophage 1 |
| MAPK: | mitogen-activated protein kinase |
| MCP-1: | monocyte chemoattractant protein-1 |
| MDA: | malondialdehyde |
| MMPs: | matrix metalloproteinases |
| mTOR1: | mammalian targets for rapamycin 1 |
| NAD: | nicotinamide adenine dinucleotide |
| NADP(H): | nicotinamide adenine dinucleotide phosphate |
| NAFLD: | non-alcoholic fatty liver disease |
| NF-kB: | nuclear factor-kappa B |
| NO: | nitric oxide |
| NOS: | nitric oxide synthases |
| NOX: | nicotinamide adenine dinucleotide phosphate oxidase |
| Nrf2: | nuclear factor erythroid 2-related factor 2 |
| OP: | oxidative phosphorylation |
| OS: | oxidative stress |
| oxLDLs: | oxidized low-density lipoproteins |
| P53: | tumor protein 53 or transformation-related protein 53 |
| PD: | Parkinson’s disease |
| PGC-1α: | peroxisome proliferator-activated receptor gamma coactivator-1α |
| PI3K: | phosphoinositide 3-kinase |
| PIP3: | phosphatidylinositol 3,4,5-trisphosphate |
| PKB: | protein kinase B |
| Prx: | peroxiredoxin |
| RNS: | reactive nitrogen species |
| ROS: | reactive oxygen species |
| SCI: | systemic chronic inflammation |
| SIRT1: | sirtuin 1 |
| SOCS3: | suppressors of cytokine signaling 3 |
| SOD: | superoxide dismutase |
| STAT: | signal transducer and activators of transcription |
| T1DM: | type 1 diabetes mellitus |
| TNF-α: | tumor necrosis factor-α |
| UPS: | ubiquitin-proteosome system |
| VSMCs: | vascular smooth muscle cells |
| vWF: | von Willebrand factor |
Declarations
Author contributions
PS: Conceptualization, Writing—original draft. GS: Formal analysis, Investigation, Writing—review & editing. SW: Validation, Writing—review & editing, Supervision. All authors have read and agreed to the submitted version of the manuscript.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2023.