 Review
Review

Affiliation:
1Department of Anatomy, Faculty of Science, Mahidol University, Bangkok 10400, Thailand
Email: prasert.sob@mahidol.ac.th; prasertsobhon@gmail.com
ORCID: https://orcid.org/0000-0003-4018-5487
Affiliation:
2NIST International School, Bangkok 10110, Thailand
Affiliation:
3Department of Obstetrics and Gynecology, Bangkok Hospital Udon, Udon Thani 41000, Thailand
ORCID: https://orcid.org/0000-0002-6763-2040
Explor Med. 2023;4:127–156 DOI: https://doi.org/10.37349/emed.2023.00129
Received: October 23, 2022 Accepted: December 16, 2022 Published: April 17, 2023
Academic Editor: Calogero Caruso, University of Palermo, Italy
The article belongs to the special issue Determinants of Exceptional Longevity
Oxygen free radicals [reactive oxygen species (ROS)] and nitrogen free radicals [reactive nitrogen species (RNS)] are generated by mitochondria during adenosine triphosphate synthesis, and catalytic activities of cytochrome P450, nicotinamide adenine dinucleotide phosphate oxidases (NOXs), cyclooxygenases, and nitric oxide synthases during drug catabolism, phagocytosis, and acute inflammation. Under normal circumstances, low levels of ROS and RNS provide redox signalings that control many essential physiological processes. As age progresses ROS and RNS increase excessively due to dysfunctional mitochondria, dysregulated NOX, and other free-radical generating sources, leading to oxidative stress, which causes oxidation and denaturation of key cellular components including DNA, proteins, and lipids, which become abnormal, constituting damage-associated molecular pattern (DAMP), recognized as ‘non-self’ by immune cells, leading to inflammation which is mediated by nuclear factor kappa B-inflammasome, p38-c-Jun N-terminal kinase and Janus kinase-signal transducer and activator of transcription pathways. DAMPs are continuously released from damaged and senescent cells, causing an otherwise normally transient inflammation turning into systemic chronic inflammation, the root cause of aging and age-associated diseases (AADs). Cells restore redox balance by activating the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway that induces the synthesis and release of antioxidation molecules and enzymes including haem oxygenase-1, which also inhibits the three inflammatory pathways. Furthermore, upregulation of autophagy (AP) can get rid of abnormal molecules, prevent the generation of DAMPs, and attenuate inflammation. Both AP and Nrf2 signalings decrease with age. The upregulations of Nrf2, AP, and downregulation of inflammation are controlled by sensors of energy and stress levels, i.e., adenosine monophosphate-activated protein kinase, silent information regulator 1, and Sestrins, as well as the extracellular matrix, while mammalian targets for rapamycin complex 1, a nutrient sensor, act in the opposite direction. If the balance of these sensor systems becomes dysregulated, aging process accelerates, and the risk of AADs increases.
Demographically, the world is facing a problem of aging population: in 2022, approximately 10% of people globally are aged 65 or over. By 2030 and 2050 the proportion of this age group is projected to be 12% and 16%, respectively. In 2022, Europe and Northern America have almost 19% of the population aged 65 or over, followed by Australia and New Zealand with 16.6%. By 2050, one in every four persons in these developed regions would be aged 65 years or over. For Latin America and the Caribbean, the proportion of the population aged 65 years or over could increase from 9% in 2022 to 19% by 2050. Additionally, the proportion of people aged 65 or over in Eastern and South-Eastern Asia, including Thailand, is projected to double from around 13% in 2022 to 26% in 2050. Sub-Saharan Africa, despite having a relatively young population, is still predicted to face population aging with the percentage of people aged 65 or over being estimated to rise from 3% in 2022 to 5% in 2050 [1]. As the world progresses to a full-fledged aging society, the prevalence of age-related diseases is also rising, including metabolic-related syndromes like obesity, dyslipidemia, nonalcoholic fatty liver, hypertension, diabetes, cardiovascular disorders, autoimmune diseases, neurodegenerative diseases (notably Alzheimer’s and Parkinson’s), various forms of dementia, and a wide range of cancers.
The root causes of aging and age-related diseases are oxidation by free radicals and inflammation, which are reciprocally generated. Inflammation (especially chronic inflammation) caused by oxidation, obesity, and infection induces immune cells to be continually active, resulting in chronic damage—repair cycles whereby cells are continuously dividing, mutating, and dying. This accelerated rate of cell division has a high probability of DNA miscopying, causing mutations which are the immediate causes of cancer and other age-related diseases.
Free radicals are generated by routine cellular metabolic processes: a leakage of electrons from the electron transport chain (ETC) which is coupled with oxidative phosphorylation in mitochondria, catabolism of exogenous compounds and drugs by liver cells, and the process of fighting infection by innate immune cells. External pollutants and contaminants in the environment, food, and the body’s immune reactions to these challenges also generate free radicals. These results in the oxidation of lipids and proteins, the key components that form and control the structure and functions of cell membranes, directly affecting cellular homeostasis. Oxidation can also alter nucleotides, the building blocks of DNA, resulting in mutations at both single nucleotides and chromosomal levels which affect cell division, cell differentiation, cellular signaling, and general cellular functions, resulting in mutated, cancerous cells. Furthermore, free radicals can denature or cross-link proteins causing misfolding, endoplasmic reticulum (ER) stress, mitochondrial damage, and loss of function in adenosine triphosphate (ATP) production while exacerbating the production of reactive oxygen species (ROS). Oxidation and excess free radicals also generate inflammation. Taken together, oxidation and inflammation are the primary initiating factors that cause aging and age-related diseases. Developing a holistic approach to mitigate the effects of aging and age-related diseases is the principal aim of a newly emerging discipline called Geroscience.
Oxygen free radicals, or ROS, and the reactive nitrogen species (RNS), which provide redox signalings at a low level and create oxidative stress (OS) at an excessively high level, are derived from exogenous as well as endogenous sources. The exogenous sources are environmental pollution, especially PM 2.5 from air pollution, smoking, ionizing radiation, UV radiation, household chemicals, and agricultural chemicals, especially herbicides (e.g., paraquat and glyphosate) and insecticides which are widely used in rural Thailand. Endogenously, ROS are derived from the mitochondria, ER-associated-cytochrome P450 (CYP), membrane-bound nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX 1–5), and peroxisomes. In mitochondria, the oxidative phosphorylation process consumes O2 for the synthesis of ATP which is coupled with the ETC. However, as electrons pass through the ETC, some electrons can escape and are captured by O2 to form the ROS. ETC sites where electron leakage occurs and ROSs are being formed include complexes I, II, III, and IV while the terminal complex V is not involved. ROS can damage mitochondrial components, affect the membrane potential, exacerbate mitochondrial dysfunction, and cause more ROS production [2–4]. As aging progresses, mitochondria become increasingly dysfunctional and the rate of electron loss from ETC increases, resulting in the generation of more ROS and exacerbating deleterious effects of OS on various tissues and organs [4].
The second major source of ROS is the mammalian CYP-dependent microsomal electron transport system. CYP enzymes (especially those associated with smooth ER of liver cells) catalyze the oxygenation of exogenous and endogenous organic substrates, including drugs, pollutants, and hormones, and turn them into more soluble and non-toxic forms, which are removed by bile and blood circulating through the kidney. If oxygen transfer to a substrate is not tightly controlled, uncoupling of the reaction occurs, leading to the formation of ROS [5–7].
The third major source of ROS is the membrane-bound NOXs. Innate immune cells, comprising neutrophils and macrophages, employ potent oxygen-dependent mechanisms that generate ROS to kill invading microorganisms through the catalytic activity of the membrane-bound NOX 2 [8]. In addition to the membrane of phagocytic cells, NOX is present in cells of many tissues, as it is needed to generate a low amount of ROS for cell signaling that governs a number of physiological processes. Aside from the above-mentioned sources, the presence of redox-active transition metals such as Fe and Cu also contributes to ROS generation. In the presence of Fe2+ and Fe3+, hydroxyl radical (OH•) can be generated from hydrogen peroxide (H2O2) through the Fenton reaction [9]. Overall, the generated ROSs consist of superoxide anion (–O2•), H2O2, OH•, hydroperoxyl radical (HOO•), and peroxyl radical (ROO•). Though transient in nature, these ROS are extremely active due to the presence of free electrons. In addition to ROS, RNSs are generated from endogenous as well as exogenous sources. RNSs comprise nitric oxide (NO), nitrogen dioxide (NO2), peroxynitrite (ONOO•), dinitrogen trioxide (N2O3), and nitrous acid (HNO2) [10]. NO is generated by catalytic actions of at least three isoforms of nitric oxide synthases (NOSs) that convert L-arginine to L-citrulline with the release of NO. If NO is generated in excessively high amount, it interacts with ROS to form many species of RNS that contribute to the damaging OS.
ROS and RNS at low or physiological concentrations act as second messengers regulating many redox-sensitive signaling pathways essential for controlling cell metabolism, growth, differentiation, and death. Among ROS H2O2 at a physiological concentration, ranging from 1–100 nmol/L, is the most prevalent and active free radical that exercises its redox signaling by reversible oxidation of thiol groups in cysteines within target proteins which include enzymes and transcription factors that control many essential signaling pathways [11]. For examples, insulin signaling which controls glucose transport and utilization and anabolic metabolism in cells; adenosine monophosphate (AMP)-activated protein kinase (AMPK) and Sirtuin signalings which are involved in sensing energy and stress levels and maintaining energy homeostasis; nuclear factor erythroid 2-related factor 2 (Nrf2) which senses changes in cellular redox status through its cysteine-rich adaptor Kelch-like ECH-associated protein 1 (KEAP1) which, if oxidized by H2O2, frees up Nrf2 to enter the nucleus and activate the production of antioxidant molecules and enzymes; the nuclear factor kappa B (NF-kB) which mediates inflammatory responses; hypoxia-inducible factor 1α (HIF-1α) which regulates cellular responses to decreased oxygen levels; forkhead box O (FoxO) which are critical regulators of the physiological stress response [12]. Other enzymes and transcription factors that are regulated by H2O2 are c-Jun N-terminal kinase (JNK), signal transducer and activator of transcriptions (STATs) [13], and target-of-rapamycin (TOR) [14]. On the other hand, if H2O2 reaches a high concentration above 100 nmol/L, it will denature biomacromolecules that are involved in cellular structure as well as cellular regulation [11]. Physiologically, NO, at a physiologically low level, acts as a signaling molecule that triggers many key cellular pathways that control important homeostatic processes, including the activation of AMPK [15], the induction of vasodilatation and angiogenesis by the catalytic action of endothelial nitric oxide synthase (eNOS) in endothelial cells [16], and inflammatory signaling through the catalytic action of inducible nitric oxide synthase (iNOS) which is present in phagocytic and antigen-presenting cells (APCs) [17].
The redox balance between the generated ROS/RNS and the antioxidant defense system, especially antioxidant molecules and enzymes induced by Nrf2 signaling (to be discussed in details later) help to maintain cellular redox homeostasis. On the contrary, a redox imbalance will occur if there are unphysiologically high concentrations of ROS/RNS that overwhelm the capacity of the antioxidant system; and as a result, the excessive amounts of free radicals will cause OS, leading to the disruptions of redox signalings and controls. Furthermore, OS is a key factor that elicits inflammation as will be described later. Reciprocally, inflammation is also a major generator of ROS. During inflammatory phase, lipoxygenases, cyclooxygenases (COXs), xanthine oxidase, and NOXs present in phagocytic cells, such as macrophages and neutrophils, catalyze the reactions that generate ROSs that are released locally as well as systemically [18]. These enzymes are activated by pro-inflammatory cytokines, particularly tumor necrosis factor α (TNFα) and interleukin 1β (IL1β), and inflammatory transcription factors including AP-1, HIF-1α, and NF-kB that are simultaneously expressed in phagocytic cells [19]. Thus, inflammation can induce ROS generation and contribute substantially to OS, especially if the inflammation becomes chronic [20]. Reciprocally, ROS can induce certain inflammatory signaling transductions, including p38-JNK, and NF-kB pathways which can activate the assembly and activation of inflammasome to enhance the effect of inflammatory process [20, 21].
The direct damaging effects of excessive ROS and RNS on biomolecules are extensive and result in deleterious outcomes. Excessive ROS/RNS can oxidize and alter nucleic acids, proteins, and lipids. The OH• radical oxidizes DNA at purine and pyrimidine bases and the deoxyribose-phosphate chains to generate single-strand and double-strand breaks which cause mutation. Furthermore, ROS selectively interacts with guanine to produce 8-oxo-7,8-dihydro-deoxyguanine (8-oxo-dG) which is highly mutagenic as it causes G:C to T:A transversion [22, 23]. On the other hand, oxidation of lipids gives rise to lipid peroxides that generate highly toxic and mutagenic malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE). MDA, through its aldehyde group (-CHO), forms stable bonds with -NH2 amine groups in nucleic acids, proteins, and phospholipids, to produce inter- and intramolecular bridges of amino-3-iminopropene. MDA binding to nucleotide bases, especially guanine and cytosine, creates bridges between the DNA strands that block the replication process and induce mutagenic effects. HNE reacts with the -SH groups of cysteines, and the -NH2 groups of lysine and histidine, causing crosslinking of proteins and disrupting their structure and functions [24, 25]. The denaturation of proteins and lipids damage cell and organelle membranes, leading to their disruption and dysfunction and eventually cell death. As the result of DNA oxidation and mutation, there is a mis-replication and mis-translation, leading to the production of abnormal proteins and proteomes. Furthermore, through the process of carbonylation and crosslinking, proteins undergo abnormal folding, aggregation, or even breakdown. These result in the releases of abnormal proteins and lipids which become parts of damage-associated molecular patterns (DAMPs) recognized by our immune cells as non-self. Under a normal circumstance, DAMPs could be eliminated by autophagy (AP) which comprises two processes: microautophagy by proteasome, and macroautophagy through phagophore, a membrane-bound AP [26]. Depending on their sizes, small abnormal proteins are labeled by ubiquitin and then transposed and digested by proteasome, whereas larger abnormal proteins are dealt with by chaperon-mediated macroautophagy, and finally the end products are recycled [27]. During aging, there is a decrease in activities of both proteasome and macroautophagy, resulting in continuous release of abnormal proteins, thus increasing the quantity of DAMPs, the major contributor to systemic chronic infection (Figure 1) [28, 29].
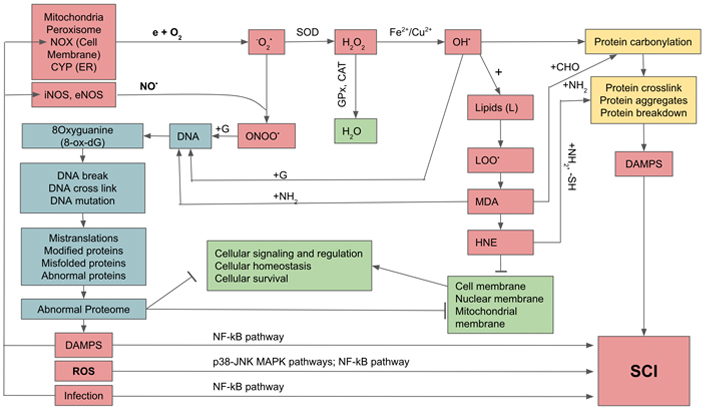
Generation of oxygen and nitrogen free radicals and their effects on cellular constituents and activation of systemic chronic inflammation (SCI). e: free electron; G: guanine; GPx: glutathione peroxidase; LOO•: lipid hydroperoxides; MAPK: mitogen-activated protein kinase; →: activation;  : inhibition
: inhibition
Acute inflammation (AI) is the process whereby the body takes care of infection and injury which is evolutionarily conserved throughout the animal kingdom. AI consists of two phases which could be called inflammatory and resolution phases, or inflammatory and anti-inflammatory phases, which are mediated by protein and lipid mediators. When infections occur, the microbes release molecules called pathogen-associated molecular patterns (PAMPs), which are recognized by innate immune cells including phagocytic cells and other APCs [30, 31]. In the case of tissue injury and failure of autophagic processes to eliminate denatured molecules arising from oxidative process, the damaged and abnormal molecules are released from the injured cells which are collectively called DAMPs [32]. These two types of molecules act as ligands which bind to the pattern recognition receptors (PRRs) including toll-like receptors (TLRs), particularly TLR4, which are located in the membrane of phagocytic cells. As a result, the PRRs are activated to undergo the process of inflammation which releases pro-inflammatory cytokines, comprising major components such as the IL1β, IL6, IL12, IL18, TNFα and others [30]. The recruited innate immune cells, including macrophages, neutrophils, and APC, phagocytose, digest, and clean the infection or debris from damaged tissues. Once the cleaning process has been completed, there are molecular cues designated as specific proresolving mediators (SPMs) which are composed of lipids that signal the resolution of the inflammation to begin the clearance-healing phase together with the release of protein anti-inflammatory cytokines. These include IL10, IL37, tumor growth factor β (TGFβ), and also the free receptors of TNFα and IL1R and others from macrophages which are switched from the pro-inflammatory phenotype (M1) to the anti-inflammatory phenotype (M2) [33].
In addition to protein cytokines, the proinflammatory phase of inflammation is also mediated by lipid mediators generated from arachidonic acid (ARA) through catalytic actions of COXs and lipoxygenases to generate prostaglandins, thromboxane, and leukotrienes, with prostaglandin E2 being a key lipid inflammatory cytokine. Once the proinflammatory phase is completed, there is a lipid signaling that initiates the anti-inflammatory phase comprising lipoxins, resolvins, maresins, and protectins which are derived from eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) [31, 33]. Hence, ARA with its derivatives mediate proinflammation while EPA, DHA, and their derivatives mediate anti-inflammation.
The intracellular transduction pathways that lead to the synthesis and release of the above-mentioned pro-inflammatory cytokines are chiefly controlled by the NF-kB and inflammasome. In the basal state, NF-kB is bound to and inhibited by the inhibitor of nuclear factor kappa B (IkB) inhibitor. When a PAMP or DAMP ligand is bound to the TLR, which is a part of PRR, there is a cascade of kinase enzymes which phosphorylate inhibitor of nuclear factor kappa B kinase (IKK). In addition, IKK is also activated by cytosolic H2O2 generated by mitochondria. IKK itself is also a kinase that phosphorylates IkB, resulting in NF-kB being freed and passing into the nucleus where it turns on the genes controlling the syntheses of the precursors of protein and lipid pro-inflammatory cytokines and also the inflammasome receptor, NOD-like receptor family pyrin domain containing 3 (NLRP3). NLRP3 signals the assembly and becomes a part of the inflammasome, the platform that activates Caspase1 that processes the precursors of the IL1, IL18, and other IL to become mature peptides which are then released from innate immune cells to act as pro-inflammatory cytokines. On the other hand, there are also external signals as well as damage-signaling molecules from mitochondria that act as NLRP3 ligands or agonists. Mitochondria, especially aging mitochondria, release a large amount of ROS through the uncoupling of oxidative phosphorylation and the loss of electrons during ETC at complex I and II as mentioned earlier. These free radicals are very active in stimulating the assembly of NLRP3 inflammasome. At the same time the cell attempts to remove the damaged mitochondria by initiating the process of mitophagy whereby the damaged mitochondria are phagocytosed and destroyed by the macroautophagy [30, 34].
In addition to the NF-kB pathway, there are two alternative pathways that can elicit as well as reinforce inflammation, i.e., p38-JNK MAPK and Janus kinase (JAK)-STAT pathways. In p38-JNK MAPK pathway proinflammatory cytokines, including TNFα, ILs, interferon (IFN)-γ, ROS, and cellular stresses, can bind to corresponding receptors and activate receptor-associated or adaptor proteins, such as tumor necrosis factor receptor-associated factor (TRAF) and RAC. These proteins sequentially phosphorylate upstream kinase [mitogen-activated-kinase-kinase kinase (MAP3K/MEKK)], then mid-stream kinases [mitogen-activated-kinase kinase (MAP2K/MKK)], and finally downstream kinase, p38-MAPK and JNK-MAPK, which enter the nucleus and activate inflammatory genes which control the expressions of the proinflammatory cytokines. The activities and intensities of both p38 and JNK could be negatively controlled by mitogen-activated protein kinase phosphatases (MPKs) (Figure 2) [21, 35, 36]. For JAK-STAT pathway, proinflammatory cytokines (TNFα, ILs, IFN-γ) can bind to corresponding cytokine receptors (CTRs) which phosphorylate receptor-associated or adaptor protein, JAK, that in turn phosphorylates STAT protein and causes dimerization of the latter. The dimerized STAT enters the nucleus and induces genes that control the proliferation, survival, and recruitment of immune cells, as well as the genes controlling the expressions of proinflammatory cytokines. In turn, the activity and intensity of JAK-STAT are negatively controlled by suppressor of cytokine signaling 1,3 (SOCS1,3) and other inhibitor proteins including protein inhibitor of STAT (PIAS) and protein tyrosine phosphatases (PTPs) (Figure 2) [37, 38].
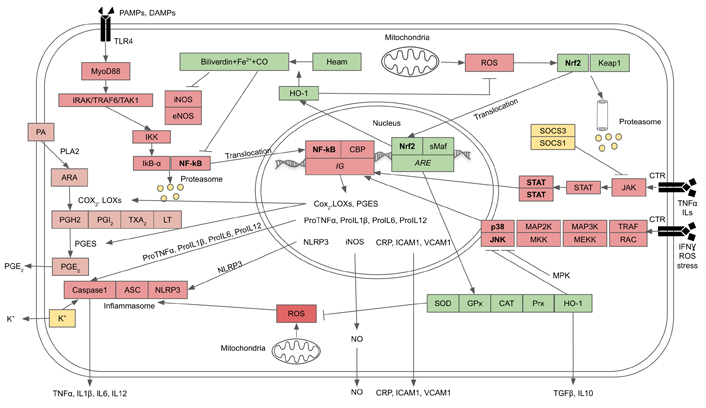
Crosstalks between inflammation and anti-oxidation pathways. The three inflammation pathways comprise NF-kB, p38-JNK MAPK, and JAK-STAT, and the antioxidation pathway is Nrf2. Inflammation and antioxidation pathways are functionally antagonistic towards each other (see text for description). ARE: antioxidant response element; ASC: apoptosis-associated-speck-like protein containing a caspase recruitment domain or inflammasome adaptor protein; CBP: cyclic AMP response element binding protein or NF-kB cofactor protein; CO: carbon monoxide; CRP: C-reactive protein; HO-1: haem oxygenase-1; ICAM1: intercellular adhesion molecule 1; LOX: lipooxygenase; IRAK: IL1 receptor-associated kinase; LT: leucotriene; MyoD88: myeloid differentiating primary response gene 88; PA: membrane phospholipids; PGE2: prostaglandin E2; PGES: prostaglandin E synthase; PGH2: protaglandin H2; PGI2: prostaglandin I2; PLA2: cytosolic phospholipase; Prx: peroxiredoxin; sMaf: Nrf2 cofactor protein; TXA2: thromboxane; VCAM1: vascular cell adhesion molecule 1; →: activation; : inhibition
In aging persons, there is an increasing number of senescent cells with dysfunctional mitochondria, abnormal molecules including misfolded proteins and oxidized lipids which cannot be cleared due to decreased AP. These senescence cells thus become senescence-associated secretory phenotypes (SASPs) that continuously release DAMPs and proinflammatory cytokines which generate continuous low-grade inflammation known as age-related SCI or inflammaging [32]. The increase of ROS from dysfunctional mitochondria during aging as mentioned above can also directly activate immune cells to induce the upregulation of inflammatory-mediating transcription factors, including AP-1, NF-kB, p38, JNK, and activate NOX system to generate more ROS that exacerbate the effect of OS [39, 40].
Chronic inflammation, which is generated by DAMP, could also be generated by obesity. When there are excessive intakes of carbohydrate and fat, white adipose tissue (WAT) and cells are overloaded with triglycerides. Some overloaded adipose cells break down releasing triglyceride and free fatty acids and other DAMPs that could attract macrophages and other immune cells into WAT and release proinflammatory cytokines including monocyte chemoattractant protein-1 (MCP1), attracting more macrophages and other innate immune cells. Thus, obesity sustains inflammatory signaling contributing to SCI and OS [41, 42].
The redox homeostasis can be restored by anti-oxidation mechanisms which include both enzymatic and nonenzymatic entities. Antioxidant enzymes consist of superoxide dismutases (SODs), GPxs, catalase (CAT), and Prxs-thioredoxin (Trx) system which cooperate in the destruction of ROS [43]. SODs in mammals comprise two isoforms: the cytosolic CuZn-SOD localized in both extracellular and intracellular (cytoplasmic) compartments, and mitochondrial-bound Mn-SOD. Both isoforms of SOD make up the first line of defense against ROS and are initially activated by ROS itself; in turn, they catalyze the transformation of extracellular as well as intracellular superoxide into H2O2 [44]. Subsequently, GPxs and CAT catalyze the breakdown of H2O2 into water and oxygen. As mentioned earlier, mitochondria are the main source of intracellular ROS generated from ETC and oxidative phosphorylation, especially if they become dysfunctional during cellular senescence. Excessive amount of ROS oxidizes lipids and proteins, damaging mitochondria membranes and eliciting the release of apoptotic signaling molecules including cytochrome c and other pro-apoptotic factors such as Bax, which activate cytoplasmic caspases, causing cell apoptosis. Mitochondria counteract this excessive ROS by catalyzing and transforming them into H2O2 using Mn-SOD [45].
The breakdown of H2O2, the end product of SOD catalytic activities, is further catalyzed by GPxs, CAT, and Prxs, all of which are highly conserved in mammals [46]. GPxs use glutathione (GSH), a cytoplasmic non-enzymatic antioxidant molecule, as an electron acceptor, to turn H2O2 and simultaneously occurring lipid peroxides into neutral water and corresponding alcohols and molecular oxygen [47]. GPxs comprise at least four isoforms localized in different cellular compartments: GPx1 in the cytoplasm and mitochondria, GPx2 in the cytoplasm and nucleus, GPx3 in extracellular compartment, and GPx4 is membrane-bound which protects membranes from ROS attack [46]. Another key anti-oxidation enzyme is CAT which is present in all cells particularly in peroxisomes. CAT catalyzes the breakdown of H2O2 derived from the conversion of ROS by SOD as well as those generated in peroxisomes to less-reactive molecular oxygen and water, thus preventing cellular damage [48]. Prxs, a family of peroxidases, are enzymes that hydrolyze H2O2 through their cysteine-containing active sites. Mammalian Prxs comprise six isoforms that are distributed over various cellular compartments. The most abundant isoforms are cytosolic Prx1 and Prx2 which remove H2O2 using the two conserved Cys residues present in their catalytic sites which become oxidized [49]. The Trx/thioredoxin reductase (TrxR) system then reduces the disulfide bond of post-catalytic Prxs to free thiol and restores their reduced state. The Trx antioxidant system consists of NADPH, TrxR, and Trx acting in concert. TrxR is the dimeric FAD-containing enzyme that catalyzes the NADPH-dependent reduction of the oxidized Trx (Trx-S2) to yield the reduced form, Trx-(SH)2 [50]. Thus, the GSH and Trx systems provide additional supports to GPx and Prxs for the removal of H2O2 generated from the catalytic activity of SOD [51].
The key signaling factor which directly regulates the expressions of genes encoding antioxidant enzymes is the Nrf2. When unactivated, Nrf2 binds with KEAP1. In their basal state, KEAP and Nrf2 undergo degradation and turnover by proteasome. In response to rising OS, ROS, especially H2O2, can oxidize redox-sensitive cysteine residues on KEAP1 in the Nrf2-KEAP1 complex, resulting in KEAP1 being released from Nrf2. The freed Nrf2 then is translocated into the nucleus and forms a heterodimer with a cofactor, a small MAF protein, to bind to a DNA sequence named AREs within the regulatory regions of multiple antioxidant genes. Nrf2 regulates the expressions of genes encoding antioxidant proteins and enzymes, including SODs, CAT, GPxs, Prxs, glutathione-s-transferase (GST), and HO-1 [52, 53], all of which are synergistically counteracting OS as aforementioned.
The HO-1 system, in particular, can oxidize heme and generate biliverdin, CO, and Fe2+. CO exercises additional beneficial action as it has an anti-inflammatory function: the inhibitions of NF-kB, iNOS, eNOS, as well as p38-JNK, which down-regulates the production of proinflammatory cytokines, while upregulating the anti-inflammatory cytokines [54]. It is still unclear how HO-1 inhibits p38 and JNK. One possibility is that it may act through MPKs, a well-known p38-JNK inhibitor. However, this remains to be proven. Biliverdin is converted to bilirubin by catalytic action of biliverdin reductase. Bilirubin can act as a strong antioxidant, and in cooperation with CO, it can also inhibit iNOS and eNOS and attenuate the production of NO and ONOO• (Figure 2) [55–59]. Fe2+ stimulates the production of ferritin which has antioxidant and cytoprotective effects [55]. HO-1 can also inhibit JAK-STAT pathway by binding directly to STAT and preventing its phosphorylation and dimerization. Thus, STAT fails to enter the nuclei of immune cells to activate inflammatory genes [58]. Therefore, the net results of Nrf2 signaling are to provide an anti-oxidation system for protection against OS and restoration of redox homeostasis, as well as suppressing or attenuating inflammation via the expression of HO-1.
It has been widely accepted that the hallmarks of aging comprise genomic instability, telomere attrition, epigenetic alterations, mitochondrial dysfunction, loss of proteostasis, deregulated nutrient-sensing, cellular senescence, stem cell exhaustion, and altered intercellular communication [60]. Mechanistically, the most widely accepted theory of aging is the free radicals-OS theory first proposed by Harman [61, 62], now considered in combination with the mitochondrial dysfunction theory proposed by Miquel et al. [63] and the systemic chronic inflammatory theory, collectively termed oxy-inflamm-aging theory proposed by De la Fuente [64]. This combined theory hypothesizes that mitochondria are the main generator of ROS from the uncoupling reactions in the ETC and the oxidative phosphorylation processes, and as aging progresses mitochondrial DNA (mtDNA) is damaged and mutated, causing mitochondria to become more dysfunctional and, in turn, generating more ROS. The progressive degeneration of ROS-damaged mtDNA leads to more dysfunctional mitochondria and more ROS production which accelerates the aging process. Furthermore, ROS/RNS that accumulate and increase over time from mitochondria, as well as from catalytic activities of CYP, NOX, and others, can elicit inflammation which reinforces the aging process by causing additional cellular damage and senescence, leading to tissue and organ dysfunction, as well as age-associated diseases (AADs) [65]. Additionally, OS worsens with aging since the anti-oxidation system controlled by Nrf2 signaling is downregulated over time [66, 67].
ROS oxidizes both nuclear and mtDNA, particularly at guanine residue, and converts it into 8-oxo-dG, directly causing genomic instability and telomeric attrition. These processes are exacerbated by MDA and HNE, the metabolites from oxidation of lipids by ROS, which also crosslink DNA and proteins [25]. Oxidized nuclear DNA results in nucleotide alterations, incorrect code on, chromosomal breakages, deletions, mutations and dysregulated translation, and abnormal proteome which disrupts various signaling and metabolic pathways, affecting cellular signaling, metabolism, and homeostasis, culminating in cell apoptosis (Figure 1). Oxidation of lipids and proteins also damages nuclear membranes, indirectly causing DNA and chromosome instability, because nuclear membranes and their associated nuclear lamina serve as anchor points for chromatin and DNA, as well as controlling the nuclear shape which directly affects the transcription and translation processes.
Similarly, oxidation of mtDNA results in nucleotide alterations, deletion, mutations, and syntheses of abnormal mitochondrial proteins that cause dysfunctional mitochondria. ROS, through peroxidation of membrane phospholipids, oxidation, and carbonylation of membrane proteins, also directly affects the structural integrity of mitochondrial membranes, causing increased permeability and loss of membrane potential, leading to mitochondrial dysregulation, increased uncoupling of oxidative phosphorylation, generation of additional ROS, and increased dysfunctional mitochondria [68]. Mitochondrial dysfunction and the accumulation of ROS also lead to telomere attrition and genomic instability as mentioned above [69].
OS also dysregulates proteostasis which accelerates aging. ROS causes carbonylation and crosslinking of intramembranous and cytosolic proteins, leading to their denaturation and loss of function, as well as damaged cell organelles [70]. In normal cells, the optimal numbers of organelles and proteostasis are maintained by AP and ubiquitin-proteasome system (UPS). These processes help cells to remove damaged organelles as well as maintain their optimal numbers, break down denatured proteins, and recycle their constituents for further usage by the cells. There are three major types of AP, namely, macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Macroautophagy (generally referred to as AP) involves the enclosing of relatively large targeted structures, like damaged or aging cell organelles, or large aggregates of denatured molecules, by specialized membranes called phagophores which turn into autophagosomes. Finally, autophagosomes fuse with lysosomes and the sequestered materials are digested by lysosomal acid hydrolases and their constituents recycled into the cytoplasm for reuse [71]. In microautophagy, the lysosomal membrane is invaginated into tiny pits that pinocytose large aggregates of abnormal molecules into the lysosomal interior where they are broken down by lysosomal enzymes [72]. In the CMA, moderately large abnormal molecules are directly transported across lysosomal membrane by chaperone proteins into the lysosomal matrix for further digestion by lysosomal enzymes [73]. The UPS degrades small misfolded proteins, as well as regulatory and signaling proteins, to control the duration of their actions, by marking them with polymeric ubiquitin tags. These ubiquitin-labeled targets are recognized and passed through the hollow core of cylindrical-shaped proteasomes where they are degraded by proteases [74]. Both AP and UPS are present in every kind of cell and their activities decline as aging progresses, partly due to decreased effectiveness from oxidative actions of ROS and RNS on their lipid and protein components [75, 76]. Hence, during aging, cells lose their regulative capacities to control proteostasis and maintain optimal numbers of organelles. This phenomenon is most evident in the case of mitochondria where ROS causes disruption of mitochondrial membranes and loss of membrane potential. These dysfunctional mitochondria express PINK1 and PARKIN markers on their outer membranes that stimulate mitophagy [77, 78], which removes damaged mitochondria and stimulates mitochondrial biogenesis to maintain optimal numbers of fully functional mitochondria [79]. As aging progresses, the cell’s ability to control the number of mitochondria through mitophagy declines. The dysregulation as well as the decrease of AP and UPS activities by ROS lead to diminished proteostasis, a key cause of aging and many AADs, notably neurodegenerative diseases, such as Alzheimer disease (AD) and Parkinson disease (PD), where aging neurons with decreased AP and UPS fail to clear misfolded proteins, such as beta amyloid in AD and alpha synuclein in PD [80]. AP plays a dual role in carcinogenesis: initially preventing normal cells from becoming cancerous by eliminating abnormal proteins. However, if conditions destabilize and the cells become cancerous, AP is employed by definitive cancer cells to acquire extra energy for their survival and proliferation [81].
Aging cells expressing SASP are the result of OS. ROS induces senescent cells to undergo mitotic arrest by activating retinoblastoma protein (pRB) which upregulates cyclin-dependent kinase inhibitors (CDKIs). In turn, CDKI inactivates cyclin-dependent kinases, leading to irreversible cell-cycle arrest. All normal fully differentiated and aging somatic cells stop dividing after a finite number of cell divisions and enter a state of permanent cell cycle arrest, either in the G1 or G2/M phase, termed replicative senescence or Hayflick limit [82]. Such replicative senescence is partly due to telomere attrition or shortening by ROS as discussed earlier [69]. As a consequence of ROS-induced senescence, the aging cells assume SASP or inflammatory phenotype by secreting pro-inflammatory cytokines, including IL1α, IL1β, IL6, and IL8 which, in turn, upregulate inflammatory pathways, leading to SCI and its ensuing deleterious effects as mentioned earlier. Likewise, as aging progresses, the body’s innate and adaptive immunities decline, leading to a state known as immuno-senescence, wherein the immune system cannot readily clear senescence cells which leads to their accumulation in various tissues and organs. This leads to further increased levels of pro-inflammatory cytokines both locally and systemically, exacerbating SCI that is the root cause of progressive aging as well as generation of AADs.
Another hallmark of aging is stem cell exhaustion, which affects tissue regeneration and organ function as aging progresses. Most tissues possess unipotent undifferentiated adult stem cells which undergo self-renewal to maintain their pools, while also differentiating into definitive fully mature cells to replace the aging and dying cells in those tissues. Additionally, it is believed that in many tissues, the tissue-specific adult stem cells are backed up by pools of mesenchymal stem cells (MSCs) that reside in the tissues themselves, or by MSC derived from bone marrow which is the main reservoir of MSC. During aging, the populations of these two groups of stem cells decrease due to the reduction of their self-renewal capacities [83]. Several studies demonstrated that AP and mitophagy, together with compensating mitochondrial biogenesis, and the ability to induce antioxidant enzymes, through Nrf2 signaling, are necessary to maintain viability and regenerative capacities of both types of stem cells [84–86]. Furthermore, FoxO3 is a key factor that helps maintain the pools of stem cells in mammals through its positive controls over AP, induction of antioxidant enzymes, as well as notch-1 and notch-2 that maintain stem cells in a quiescent state, for example, muscle satellite cells [87, 88], adult neural stem cells [89], and hematopoietic stem cells [90, 91].
Finally, OS can damage and deregulate nutrient- and energy-sensing pathways (Figure 3). During nutritional abundance (especially amino acid and glucose), nutrient-sensing pathways stimulate anabolism, i.e., biosyntheses of proteins, carbohydrates, lipids and their storages, whereas during nutrient scarcity and energy depletion energy-sensing pathways restore homeostasis by catabolizing internally stored nutrients to meet energy requirements. If these nutrient- and energy-sensing pathways are deregulated, aging accelerates and AADs occur simultaneously. The energy- and nutrient-sensing pathways are composed of AMPK and mammalian targets for rapamycin complex 1 (mTOR1) which detect and regulate the statuses of energy and nutrient levels available to cells [92, 93]. Living cells use ATP as their primary energy source: hydrolysis of ATP to ADP, and AMP provides energy for most biological processes. The ratio of AMP to ATP is, therefore, detected by cells as an indicator of cellular energy status, if rising will activate AMPK which acts as a master regulator of catabolism to restore and maintain energy homeostasis [94]. Downstream, AMPK activates AP, FoxOs, p53, Nrf2, peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α), and inhibits NF-kB signaling and the deleterious effects of inflammation [95]. AMPK acts in synergy with silent information regulator 1 (SIRT1) which is activated by increased nicotinamide adenine dinucleotide (NAD+) to reduced nicotinamide adenine dinucleotide (NADH) ratio which also indicates energy status in the same direction as AMP to ATP ratio [93, 96, 97]. Mammalian SIRT1 is a class III protein deacetylase targeting histones and multiple transcription factors [98]. SIRT1 restores energy depletion by activating the same targets as AMPK, i.e., AP, FoxOs, p53, Nrf2, PGC-1α, and inhibiting NF-kB signaling [99]. It has been demonstrated that activated AMPK stimulates SIRT1 by increasing the intracellular concentration of NAD+ through the enhanced expression of nicotinamide phosphoribosyl transferase (Nampt) which subsequently increases the level of NAD+ and thus potentiates SIRT1 activity [100]. Reciprocally, SIRT1 can deacetylate and activate liver kinase B1 (LKB1) kinase, an upstream activator of AMPK. Thus, SIRT1 provides positive feedback activation of AMPK to potentiate the action of each other (Figure 3) [101]. Another positive feedback of AMPK activation is Sestrins, a stress-inducible metabolic regulator which senses cellular stresses including OS, ER stress [unfolded protein response (UPR)], hypoxia, DNA damage, genotoxic stress, and long-term endurance exercise. The expression and activation of Sestrins are induced by a complex interaction of FoxO and p53 [102–105]. Sestrins further enhances AMPK activity by inhibiting mTOR1 and stimulates AP to counteract the incurring cellular stresses [106]. In addition, when stimulated by OS, Sestrins also induce Nrf2 signaling to activate the anti-oxidation system and prevent cell apoptosis [104]. mTOR1, a conserved Ser/Thr protein kinase, counteracts against AMPK-SIRT1-Sestrins by being a potent inhibitor of AMPK and AP. mTOR senses abundant nutrients (e.g., amino acids, glucose) and sufficient energy status, and is synergistically connected with insulin/insulin-like growth factor (IGF) signaling (IIS) pathways whose protein kinase B (PKB) activates mTOR1 while simultaneously represses AP and FoxOs. AMPK inhibiting mTOR1 complex (mTORC1) via two distinct pathways, either directly by phosphorylating the regulator-associated protein of mTOR (Raptor), an upstream regulatory component of mTORC1, or through the phosphorylation of tuberous sclerosis protein 2 (TSC2), which subsequently suppresses the activity of mTOR1 [107]. The IIS pathway also activates mTOR1 and consequently its downstream targets, eukaryotic initiation factor 4 (eIF4) and ribosomal protein S6 kinase 1 (S6K1), to facilitate protein synthesis. Upregulated IIS signaling enhances mTOR1 activity via AKT/PKB kinase [108] and accelerates the aging process due to inhibition of AMPK and AP [109], and activation of NF-B signaling [110]. In turn, AMPK can suppress IIS pathways and indirectly inhibit mTOR1 by phosphorylating insulin receptor substrate 1 (IRS1) at Ser794 [111]. Therefore, IIS-mTOR and AMPK-SIRT1-Sestrins signaling pathways are mutually inhibitory. As aging progresses, AMPK signaling declines [112, 113], impairing catabolic regulation, reducing Nrf2 and AP activities, leading to increased OS and reduced autophagic clearance of abnormal molecules and damaged organelles in cells, and allowing the release of DAMPs that activate innate immunity and cause chronic low-grade inflammation. The Figure 3 illustrates the balanced, yet antagonistic relationships between AMPK, SIRT1, and Sestrin on one hand and IIS and mTOR on the other, their intertwining pathways in sensing nutrient and energy statuses, their controls over the energy and nutrient homeostasis, and the outcomes of their activations. Upregulation of AMPK, SIRT1, and Sestrins activate AP, Nrf2, FoxOs while inhibit NF-kB, leading to attenuation of aging and preventing AADs. By contrast, upregulation of IIS pathway and mTOR1 inactivate AP, Nrf2, and FoxOs, and activate NF-kB, leading to accelerated aging and occurrence of AADs.
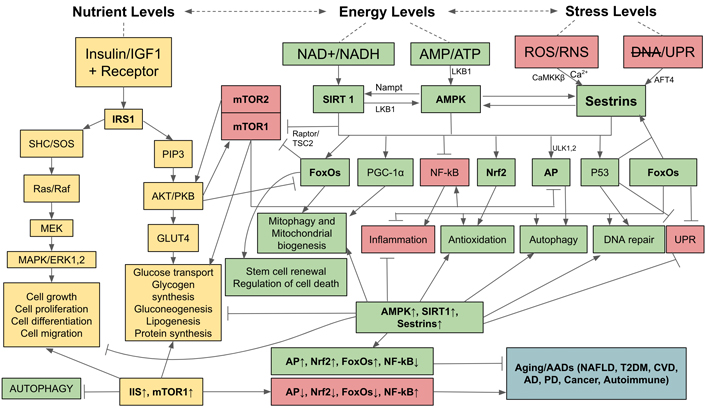
Relationships and interactions between AMPK, Sirtuin 1, Sestrins, IIS, and mTOR pathways in sensing nutrient and energy statuses, their controls over anabolic and catabolic processes, and the outcomes of their activations. AFT4: activating transcription factor 4; CaMKKβ: Ca2+/calmodulin-dependent protein kinase kinase β; CVD: cardiovascular disease; ERK: extracellular signal-regulated kinase; GLUT4: glucose transporter 4; MEK: mitogen-activated protein kinase kinase; NAFLD: nonalcoholic fatty liver disease; PIP3: phosphatidyl-inositol-3,4,5 triphosphate; SHC/SOS: Src homology 2 domain-containing transforming protein/son of sevenless; T2DM: type 2 diabetes; ULK1,2: Unc-51-like autophagy activating kinase 1,2; →: activation; : inhibition; ↑: upregulation; ↓: downregulation
Except highly mobile blood cells, a majority of cells constituting human body are surrounded and fixed in their corresponding tissues by extracellular matrix (ECM) which serves to join the cells together. Furthermore, in case of epithelial and endothelial cells, in addition to binding to their neighbors through ECM on lateral aspects while resting on the basement membrane and supporting ECM on the basal aspects, their apical aspects, which are adorned with cilia and microvilli, are in close contact with flowing liquid or semi-solid materials. The ECM, through its modulative action on cytoskeleton, plays important roles in controlling cell shape, motility, proliferation, and differentiation, positioning of cell organelles, and many intracellular processes including AP. Recently, the influence of ECM on regulation of AP is recognized as a very important process physiologically and pathologically, and this key research area is under intense investigation. Though details on the involved mechanism are not completely known, the evidence gathered so far indicate that this is a major regulator of AP as well as mitochondrial structure and function, which has been intensively reviewed [114, 115]. It is well-known that AP is activated by a variety of chemical cues and stresses, such as hypoxia, DNA damage, inflammation, and metabolic stress such as starvation and exercise. A lesser known and yet very important process is that AP is also activated by mechanical forces and stresses exerted by surrounding ECM whose physical properties (i.e., composition, density, and rigidity) are undergoing transient changes physiologically, pathologically, and chronologically. Furthermore, the cells’ three-dimensional parameters (e.g., size, shape, curvature) give rise to tension in the plasma membrane and its underlying cortical zone of cytoplasm enmeshed with actin filaments. The tensive force is also exerted by the neighboring cells’ shape, physical activities, causing cell-cell shear forces. As well, tension is also generated by the forces from the interstitial fluids that can exert osmotic pressure to induce cell swelling or shrinking. In addition, epithelial and endothelial cells are subjected to shear force from the flow of liquids or semi-solid materials on their free surface in the lumen of tubular organs that they are lining (e.g., intestinal tract, heart and blood vessels, reproductive and urinary tracts), while cells in extensible organs such as in the muscle, heart, blood vessels, lungs, and digestive tract, are subjected to stretching and compressing forces during expansion and contraction cycle.
Cells perceive these mechanical forces from the ECM and their immediate microenvironment by mechanoreceptors residing in their plasma membranes that are usually coupled with ECM on the external surface and the cytoskeleton which are anchored on the internal surface. Through this arrangement, the ECM can interact with mechanosensors and exercise control over the dynamic organization of the cytoskeleton that regulate cells’ external and internal structures, organelle distribution, intracellular transport, and the formation and distribution of autophagic organelles. These mechanosensors comprise intramembranous and interstitial protein complexes, primarily aggregates of integrin in the form of focal adhesions that bind adjacent cells, hemidesmosomes that bind epithelial and endothelial cells to basement membranes and underlying ECM, cadherins and associated proteins and glycoproteins that bind neighboring cells through junctional complexes, mechanosensitive ion channels in the plasma membrane that are opened to ion influx by changing tension in plasma membrane and associated cortical cytoplasm, and the primary cilia.
Focal adhesions are composed of clusters of transmembrane integrin proteins interacting at the internal surface of plasma membrane with actin-binding proteins (talin, vinculin), signaling molecules [e.g., focal adhesion kinase (FAK), Src, phosphoinositide 3-kinase (PI3K)], adaptor proteins (e.g., paxillin) and actin linker proteins (e.g., filamin, alpha-actinin), collectively called adhesomes [116]. Externally, adhesomes interacts with the components ECM to sense its rigidity. Thus, through adhesomes, ECM exerts its influence on cytoskeletal organization, modulating cellular behaviors, including motility, migration, and activation of AP which maintains cellular homeostasis as well as assists cells to face metabolic and OSs. Several studies demonstrated how the ECM and adhesome trigger AP via FAK and integrin-linked kinase (ILK) [117, 118]. Additionally, ECM constituents can also act as activators or inhibitors of AP, for example, decorin, collagen VI, kringle 5, perlecan, and endostatin are activators [119–121], whereas laminin a2 is an inhibitor [122].
Physical properties of the ECM such as stiffness can affect the autophagic process via its influence on cytoskeleton dynamics and actin-associated signaling proteins, such as the Rho small GTPase and its downstream effector Rho-associated protein kinase (ROCK), which regulate the formation of double-membrane phagophore and autophagosome [123], and via Yes-associated protein (YAP)/transcriptional co-activator with PDZ-binding motif (TAZ) tension-sensing system [124, 125]. YAP and the TAZ signaling are regulated by ECM stiffness, cell density, and cell shape. When cells are at low density or grown on a stiff matrix, YAP and TAZ are activated and transferred from cytoplasm into the nucleus, where they interact with the DNA-binding transcription factor TEAD to promote the expression of several proliferation and growth-related genes that eventually induce cell division [126, 127]. Conversely, when cells are at high-cell density or grown on a soft matrix, YAP/TAZ is inactivated in the cytoplasm [126, 128] causing inhibition of cell proliferation. Such an event is a part of contact inhibition phenomenon. YAP/TAZ directly modulates AP by controlling the transcription of actomyosin genes (MLC2, MYH10, MYH14) essential for phagophore and autophagosome formation [124], and the expression of the RAB7 GTPase-activating protein Armus, a factor that is essential for the autophagic pathway, especially autophagosome-lysosome fusion [125]. Reversely, AP regulates integrin-mediated adhesion by controlling focal adhesion turnover through a mechanism involving LC3, paxillin, and Src [129], thus directly affecting cells’ attachment to ECM and cell migration. Despite these important observations, the exact details of molecular mechanisms and sequelae of how these mechanosensors control AP in response to physical properties of the ECM need more research.
In addition to ECM, cells in a tissue are physically adhered to other cells through junctional complexes that are either coupling the cells electrically (e.g., gap junction, tight junction) or physically (adherens junction and desmosomes). Among these junctional complexes, adherens junctions are shown to be the force-sensitive complex which perceives and transmits force between the cadherin in the ECM within the intercellular space and the actin cytoskeleton. Similar to focal adhesion such a complex also comprises structural as well as signaling proteins which is collectively termed the cadhesome [130]. Tension on adherens junctions and its associated cadherin induces a conformational change in cadherin-associated proteins, catenins, located on the internal face of the plasma membrane, enabling them to bind with vinculin, an actin associated protein, resulting in modulation of actin filament dynamics. Tension-induced vinculin can activate acto-myosin interaction, resulting in localized actin polymerization [131, 132]. Thus, vinculin functions as an integrator between external mechanical force and internal transduced signaling that controls the cytoskeletal dynamics. It has also been demonstrated that the application of force to E-cadherin upregulated AP via LKB1 activation, which mobilizes AMPK, an AP-activating regulator, to the E-cadherin complex [133], which initiates the autophagic process. Reversely, there are turnovers of cadherin and catenin from the cell membrane to the cytosol where they are subsequently degraded and recycled by the autophagosome [134].
The molecular details on how the above-mentioned mechanosensors activate AP remain incomplete and thus need more research. However, it has been shown that mechanical forces transmitted into cells via focal adhesion and cadhesomes activate mTORC2 [135] which inhibits mTORC1 [136], which negates the inhibition of the latter on ULK1, thus allows it to be free and initiate AP [137]. Evidently, the decrease of mechanical forces at the focal adhesions, which may occur upon detachment of the cells from the substrate or due to changes in structural properties of the ECM, induces FAK dissociation from the focal adhesion complex [138]. Soluble FAK phosphorylates and activates mTORC2 which, consequently, initiates AP due to its inhibition of mTORC1 as mentioned. Additionally, mTORC2 can also activate AKT, which has inhibitory action on mTORC1 [139]. Hence, the ECM can act on adhesome and cadhesome to initiate AP via FAK, mTORC2 and AKT which inhibit mTORC1 and allows ULK1 to become active and binds to ATG13 and FIP200 to form ULK1 complex that initiates AP. Subsequently, there is a recruitment of specific ATGs that mobilize actin filaments to repolymerize under the membrane at the site of phagophore formation at the existing ER or nuclear membrane to cause curving of the membrane into omegasome, the forerunner of phagophore. Additional recruitment of membrane constituents by ATGs, particularly phospholipids, enables the elongation of phagophore that progresses into autophagosome [140].
For epithelial and endothelial cells, shear force from the flowing fluid can induce AP by activating the Rho GTPases (Rac1, RhoA, and Cdc42) and Bin/amphiphysin/Rvs (BAR) proteins that acts as tension sensors in the plasma membrane [141], to cause upregulation of Beclin-1, ATG5, ATG7 and LC3 which initiate autophagic process [142]. Another mechanism of tension sensing may be perceived by the actin-binding protein filamin which crosslinks actin filaments together [143]. Such filamin A crosslinks of actin can provide stability at the focal adhesions and the plasma membrane as well as the associated cortical zone to counteract force generated by fluid flow; this action can also prevent cells from apoptosis [144]. Stretching forces on cells can induce AP which happens during muscular activity in both skeletal and cardiac muscles [145, 146]. The muscle-stretch sensor LIM protein [MLP; also known as cysteine and glycine-rich protein 3 (CSRP3)] is present at the sarcomeric Z-disc, and it responds to stretching forces by initiating AP [147].
Stretch- or tension-generated force imposed on the plasma membrane causes a conformational change of ion channels, particularly calcium channels, including mechano- and voltage-sensitive transient receptor potential (TRP) channel, polycystin 1 (PC1), and pore-forming Piezo channel [148]. These channels are opened when plasma membrane is stretched or compressed during cell migration or when the cells are faced with shear force of fluid flow. Likewise, these channels also respond to the osmotic force which increases membrane tension during cellular swelling. Calcium ions influx through these opened calcium channels can initiate autophagic process via AMPK and mTOR pathway [149].
Primary cilium is present in most cell types. This cilium lacks a central microtubule doublet and is non-motile, but it can sense extracellular chemical and mechanical stimuli. Shear forces of fluid flow cause bending of this cilium and induce calcium entry into the cell via PC2 and transient receptor potential vanilloid 4 (TRPV4) channels [150]. The calcium influx triggers AP [151, 152] through the LKB1-AMPK-mTOR signaling pathway [153], with LKB1 being localized in the axoneme [151]. And more recently, folliculin (FLCN) has been demonstrated to act upstream to LKB1 in controlling AMPK-mTOR activity, and AP [152]. Subsequently, the autophagic process is initiated and modulated by activation of ULK1, Beclin-1 and PI3K/VPS34 signalings [154]. The generated AP is cytoprotective by inhibiting cell apoptosis and cellular senescence, as well as inhibiting inflammation [155], which in case of endothelial cells can contribute to the attenuation of artheroma formation and CVD complications. Reversely, the process of ciliogenesis which controls assembly and extension of primary cilium length is modulated by AP as evident by the presence of several components of the autophagic machinery in the cilium’s basal body [156].
During aging, the ECM becomes stiffer and senescence cells also become more rigid due to less pliable cytoskeleton and change in nucleus structure due to stretching. These changes unavoidably affect mechano-transduction. As aging progresses there is an increased synthesis of collagen fibrils [157, 158] as well as their cross-linkings by lysyl oxidase-mediated reaction between the lysine or hydroxylysine residues [159], resulting in ECM becoming stiffer. Similarly, fibronectin increases significantly during aging due to overexpression of ILK, a cytoplasmic protein regulating fibronectin–matrix assembly [160], contributing further to ECM stiffness. Senescence cells also become more rigid due to modification of plasma membrane’s accumulations of polysaturated fatty acids and cholesterol in place of more flexible polyunsaturated fatty acids. Such replacement of fatty acids and increased lipid peroxidation enhance plasma membrane rigidity, resulting in a decrease in cell flexibility [161]. Cytoskeleton also becomes less pliable, as there are increases of F-actin polymerization [162] and density of microtubules’ network. The nuclear lamina component, especially lamin B1 (LB1), decreases dramatically as cells become senescent, and its depletion results in changes of nuclear structure and nuclear membrane architecture, causing chromatin disorganization [163]. As there are direct connections between the nuclear lamina and the cytoskeleton network, stiffening in the ECM and cytoskeleton causes stretching of the nucleus, resulting in chromatin conformation turning into a decondensed (euchromatic) form that promotes gene expressions, including the abnormal genes [164]. Thus, the stiffening of ECM coupled with the more rigid cytoskeleton and change of nuclear shape as a result of aging disrupts the mechano-transduction pathways and causes dysregulation of gene expressions in the cells, the details of which are intensively reviewed by Ferrari et al. [165].
In addition to the effect of ECM stiffness on disrupting mechanotransduction of AP and gene expressions, it has been demonstrated that that ECM rigidity can affect mitochondrial morphological dynamics, their function and redox homeostasis in both normal and cancer cells. For example, when neonatal rat cardiac muscle cells were cultured on fabricated stiffness-tunable fibronectin plate, the growing cells showed increased oxygen consumption in correlation with the ECM stiffness, implying that mitochondrial function was regulated by ECM elasticity, whereas the ability of tissues to adapt to metabolic stress is regulated by both ECM elasticity and tissue alignment on the fibronectin substrate [166]. Through integrin receptor, ECM stiffness also regulates mitochondrial dynamics and function, especially mitochondrial fusion and fission in response to stress, which is mediated via two distinct signaling mechanisms, namely kindlin 2-dependent up-regulation of mitochondrial fusion and PINCH-1-dependent suppression of dynamin-related protein 1 (DRP1) expression which mediates mitochondrial fission. As such, ECM can regulate cellular energy production, metabolism, survival, and cell behavior [167]. Furthermore, it was demonstrated that breast cancer cells cultured on a soft ECM display increased peri-mitochondrial F-actin, promoted by Spire1C and Arp2/3 nucleation factors, and increased DRP1- and MIEF1/2-dependent mitochondrial fission, leading to increased production of mitochondrial ROS and activation of the Nrf2 antioxidant pathway, and increased cystine uptake as well as GSH metabolism. These responses enable cancer cells to become resistant to chemotherapeutic drugs that kill cancer cells through generation of OS [168].
From above discussion, it is now clear that apart from controlling cell growth, proliferation, differentiation, and behaviors, the ECM also plays very important roles in controlling intracellular processes, including mitochondrial dynamics and function which affect cell metabolism, and the key cellular regulative processes, i.e., AP and possibly antioxidation pathway (Nrf2). In the near future, research on the roles of ECM in controlling the activation or inhibition of central regulators including AMPK-SIRT1-Sestrins and mTOR should be pursued to give more insights into the working and coordination of these important regulators in controlling cellular activities, especially with regard to aging and diseases.
Evidently, from above discussion, OS and inflammation are the root causes of aging and AADs; therefore, a logical approach that may attenuate aging, extend longevity, and prevent AAD is the restoration of redox homeostasis by activation of the antioxidation pathway while suppressing the inflammatory pathways. This can be achieved by activating Nrf2 which induces the expressions of antioxidants molecules and enzymes including SODs, CAT, GPxs, Prxs, and HO-1. The latter is crucial in providing strong antioxidation as well as inhibition of inflammation as mentioned earlier. Nrf2 signaling also upregulates AP which is the key process that attenuates aging and extends longevity, as well as preventing AADs. The research on the positive role of AP in anti-aging has been extensive. The findings were reported first in yeast [169], later in mammals [170–172], and then in humans [173] while its effects on anti-AAD as well as against a wide variety of other diseases have been extensively studied and recognized as described in an intensive review [174]. By contrast, only recently have there been studies on the effects of the upregulation of Nrf2-HO-1 in extending longevity [175], while its roles in alleviating and preventing AAD have been long investigated and well recognized, including its ameliorating effects against inflammatory and autoimmune diseases, such as multiple sclerosis (MS), inflammatory bowel diseases (IBDs), systemic lupus erythematosus (SLE), rheumatoid athritis [176], obesity, and metabolic diseases, including NAFLD, hypertension, arthrosclerosis, and CVD [177–180]. Furthermore, neurodegenerative diseases including AD and PD have also been reported to be alleviated by Nrf2-HO-1 upregulation, especially with the ability of Nrf2 to activate AP and UPS systems, which help to eliminate misfolded proteins (beta amyloid and alpha synuclein),and attenuate the neuroinflammation caused by OS [181–184].
As mentioned above (Figure 3), the activations of Nrf2 and AP are regulated upstream by a set of synergistically acting energy and stress sensors, i.e., AMPK-SIRT1-Sestrins. These three sensors auto-stimulate each other to promote catabolism to restore energy deficit and counteract OS through the activation of the Nrf2 and AP pathways, while also directly inhibit inflammation. There have been a number of studies indicating that activation of these central regulators leads to upregulation of their downstream targets, especially Nrf2-HO-1 and AP, so that aging and AAD can be attenuated. The most intensively studied is the activation of AMPK and SIRT1 which could effectively attenuate aging and extend longevity [185–189], as well as prevent AADs [190, 191]. AMPK and SIRT1 exert their effects through activations of AP and Nrf2 [185, 187, 189].
Recently, research on Sestrins’ roles in anti-aging began to gather momentum [192, 193], while their roles in attenuating and preventing AADs have long been recognized [194–197]. It has been demonstrated that Sestrins’ signaling also upregulates Nrf2 and AP, as in the cases of AD and PD where it activates Nrf2 to counteract OS and upregulate AP to eliminate misfolded proteins, which are the initiating causes of these neurodegenerative diseases [195–197]. Co-activation of AMPK and Sestrins could extend longevity and prevent metabolic diseases, including obesity, NAFLD, and diabetes [194]. Activation of AMPK and Sestrins together with the inhibition of mTOR1 can prevent neurodegenerative diseases, CVDs, cancer, as well as musculoskeletal diseases [191]. Therefore, one key approach in extending longevity and preventing or attenuating AADs is to find means or agents that can activate AMPK-SIRT1-Sestrin signaling, which leads to strong upregulations of Nrf2-HO-1 and AP.
Recent studies indicated that this goal can be achieved through exercise which acts through mitochondria by enhancing their metabolism, leading to increased AMP/ATP and NAD+/NADH ratios and direct activation of AMPK and SIRT1 signaling [198]. Furthermore, AMPK-SIRT1 activation increases PGC-1α gene expression which stimulates replication and transcription of mtDNA, leading to mitochondrial biogenesis and mitochondrial oxidative phosphorylation process to satisfy the energy demand [199, 200]. Additionally, exercising muscle also releases myokines, including muscle-derived IL6 and irisin [201, 202], as well as β-hydroxybutyrate, a ketone metabolite [203, 204], all of which synergistically suppress inflammation in muscle as well as in other organs, thus helping to extend longevity and alleviate AADs. Mechanistically, β-hydroxybutyrate can activate AMPK and Nrf2 as well as inhibit inflammation by suppressing inflammasome formation and ER stress [204]. Interestingly, it was recently proposed that β-hydroxybutyrate may be considered as a key metabolite that can extend longevity as well as attenuate AADs [205, 206]. Exercise also activates Sestrins perhaps through the build-up of free radicals from increased metabolism of muscle cells [207]. Additionally, exercise induces muscle contraction and stretching which can activate mechanoreceptor in the muscle that directly activates AP [145–147]. Thus, benefits of exercise could also be mediated through Sestrins acting together with AMPK and SIRT1, which can extend longevity and attenuate AADs through their combined ability to activate Nrf2-HO-1 and AP [208].
Caloric restriction (CR) with adequate nutrition can extend longevity and health span in many species including humans, and protect against the development of AADs [209]. A key cellular response elicited by CR is AP which is induced to compensate for nutrient deprivation and, consequently, plays beneficial role in extending longevity and alleviating ADDs. CR, like exercise, is a mild stress that acts on mitochondria by enhancing their metabolism to meet energy demand, which leads to increased AMP/ATP and NAD+/NADH ratios that directly activate AMPK and Sirtuin 1 signaling [210]. In turn, AMPK inhibits mTOR1, an AP repressor. SIRT1 and AMPK auto-stimulate and enhance the activity of each other and, consequently, amplifies the effect of CR. The net result of this signaling loop activation is the upregulation of AP [211]. Apart from activation of AP, AMPK-SIRT1 signaling also stimulates the up-regulation of Nrf2-HO-1. The combined effects of these two processes are the main factor through which CR extends longevity and alleviates AADs.
The other approach that can extend longevity and alleviate AADs is the intakes of natural products as nutraceuticals, notably, polyphenols [e.g., berberine, quercetin, myricetin, caffeic acid (CA), caffeic acid ethanolamide (CAEA), caffeic acid phenyl ester (CAPE), epigallocatechin 3-gallate (EGCG), gallic acid (GA), curcumin], flavonoids (e.g., resveratrol), terpenoids (e.g., ginsenosides, triterpene glycosides), carotenoids (e.g., lycopene), xanthophyll (e.g., astaxanthin), spermidine, and sulforaphane, all of which could act as CR mimetics. Most of these natural compounds have high antioxidation property, and they can also directly stimulate AMPK (curcumin, EGCG, spermidine, GA, CAPE) or SIRT1 (berberine, EGCG, quercetin, myricetin) or both (resveratrol, CA, CAEA), and consequently cause the upregulations of Nrf2 and AP [176, 212–216]. Some natural compounds can also suppress inflammation (curcumin, EGCG, spermidine) [214]. However, all these natural antioxidants and free radicals, including ROS and NO, induce hormesis or biphasic dose-responses with regard to Nrf2-HO-1 activation, where only optimally low doses elicit positive outcomes while high doses induce negative or even toxic outcomes [217, 218]. It has been demonstrated that hormetic activation of Nrf2 effectively extends longevity and reduce the occurrence and severity of a wide range of human-related pathologies, including AD, PD, stroke, age-related ocular damage, chemically induced brain damage, renal nephropathy, as well as obesity and diabetes, indicating that activation of the Nrf2-HO-1 pathway and AP are probably the final, integrative, and underlying mechanism that function to limit age-related damages, the progression of numerous disease processes, and chemical- and radiation-induced toxicities [219, 220]. Furthermore, it was found that certain drugs (e.g., metformin, salicylate/aspirin) at low doses can activate AMPK by acting as mildly toxic agents against mitochondria to result in the increase of AMP/ATP ratio, in a special hormesis process called mitohormesis [213, 214]. Alternatively, certain drug like rapamycin can also hormetically inhibit mTOR1 [212, 214], hence allowing AMPK and its ancillary pathways (SIRT1 and Sestrin) to become highly active, which results in similar outcomes.
In summary, exercises, CR, intakes of natural products, and certain mitohormetic drugs can activate AMPK-SIRT1-Sestrins signaling, and consequently, their downstream target genes particularly those that are involved in Nrf2, AP upregulations, which are the final common pathways that attenuate aging as well as AADs. However, further investigations are needed to reveal details of molecular mechanisms of these approaches and actions of the CR mimetic natural compounds as well as their bioavailabilities, biotransformations, hormetic properties with regard to dosage-toxicity, so that they can be developed into efficient, non-toxic strategies or drugs that can be used alone or as adjunctive or combinatorial treatments to extend longevity, prevent, and/or cure AADs. Stringent clinical trials in humans are also necessary before these compounds could be declared safe for adoption as nutraceuticals.
Free radicals are generated in normal metabolism from mitochondria during ATP synthesis, and catalytic activities of CYP, NOX, COXs, and NOSs during drug catabolism, phagocytosis, and AI. At low levels free radicals, especially H2O2, elicit redox signalings that control many essential physiological processes. As age progresses free radicals increase excessively due to dysfunctional mitochondria, dysregulated NOX and other free-radical generating sources, which overwhelm antioxidation capacity mediated by Nrf2 signaling that also declines with aging, leading to OS, which causes oxidation and denaturation of cellular molecules that turn into DAMPs which stimulate SCI. Both OS and SCI contribute to the aging processes and AADs. There may be two direct approaches that could be adopted to mitigate both aging and AAD: upregulating the Nrf2 pathway to neutralize OS and restore redox balance, and AP to restore protein and organelles homeostases, both of which can downregulate the inflammatory pathway to attenuate or inhibit SCI. Alternatively, the central regulators, i.e., AMPK together with SIRT1 and Sestrin may be activated which can lead to activations of Nrf2 signaling and AP and inhibition of inflammation, which can extend longevity as well as prevent the occurrence of AADs. Recent studies indicate that these goals are achievable through exercise, CR, nutraceuticals, and natural products (e.g., polyphenols, flavonoids, terpenoids, carotenoids, xanthophyll, sulforaphane, spermidine) and certain mitohormetic drugs (e.g., metformin) which activate AMPK-SIRT1-Sestrins and, consequently, their downstream targets including Nrf2, AP, and FoxOs which attenuate aging and AADs. Alternatively, there are certain drugs, e.g., rapamycin, that can inhibit mTOR1 and allow activation of AMPK-SIRT1-Sestrins pathways. Natural products and chemicals possessing these properties are the subjects of intense investigations, and their mechanisms actions as well as clinical trials are challenging questions that require further research and stringent tests, since they could potentially be adopted to enhance both life and health spans.
AADs: age-associated diseases
AD: Alzheimer disease
AI: acute inflammation
AMP: adenosine monophosphate
AMPK: adenosine monophosphate-activated protein kinase
AP: autophagy
APCs: antigen-presenting cells
ARA: arachidonic acid
ATP: adenosine triphosphate
CAT: catalase
CO: carbon monoxide
COXs: cyclooxygenases
CR: caloric restriction
CTRs: cytokine receptors
CVD: cardiovascular disease
CYP: cytochrome P450
DAMP: damage-associated molecular pattern
ECM: extracellular matrix
EGCG: epigallocatechin 3-gallate
eNOS: endothelial nitric oxide synthase
ER: endoplasmic reticulum
ETC: electron transport chain
FAK: focal adhesion kinase
FoxO: forkhead box O
GPx: glutathione peroxidase
GSH: glutathione
H2O2: hydrogen peroxide
HNE: 4-hydroxy-2-nonenal
HO-1: haem oxygenase-1
IFN: interferon
IIS: insulin/insulin-like growth factor signaling
IkB: inhibitor of nuclear factor kappa B
IKK: inhibitor of nuclear factor kappa B kinase
IL1β: interleukin 1β
iNOS: inducible nitric oxide synthase
JAK: Janus kinase
JNK: c-Jun N-terminal kinase
KEAP1: Kelch-like ECH-associated protein 1
LKB1: liver kinase B1
MAPK: mitogen-activated protein kinase
MDA: malondialdehyde
MSCs: mesenchymal stem cells
mtDNA: mitochondrial DNA
mTOR1: mammalian targets for rapamycin complex 1
mTORC1: mammalian targets for rapamycin complex 1 complex
NAD+: nicotinamide adenine dinucleotide
NADH: reduced nicotinamide adenine dinucleotide
NADPH: nicotinamide adenine dinucleotide phosphate
NAFLD: nonalcoholic fatty liver disease
NF-kB: nuclear factor kappa B
NLRP3: NOD-like receptor family pyrin domain containing 3
NO: nitric oxide
NOXs: nicotinamide adenine dinucleotide phosphate oxidases
Nrf2: nuclear factor erythroid 2-related factor 2
OH•: hydroxyl radical
ONOO•: peroxynitrite
OS: oxidative stress
PAMPs: pathogen-associated molecular patterns
PD: Parkinson disease
PGC-1α: peroxisome proliferator-activated receptor-gamma coactivator 1α
PKB: protein kinase B
PRRs: pattern recognition receptors
Prx: peroxiredoxin
RNS: reactive nitrogen species
ROS: reactive oxygen species
SASPs: senescence-associated secretory phenotypes
SCI: systemic chronic inflammation
SIRT1: silent information regulator 1
SOCS1,3: suppressor of cytokine signaling 1,3
SODs: superoxide dismutases
STATs: signal transducer and activator of transcriptions
TAZ: transcriptional co-activator with PDZ-binding motif
TLRs: toll-like receptors
TNFα: tumor necrosis factor α
Trx: thioredoxin
TrxR: thioredoxin reductase
ULK1,2: Unc-51-like autophagy activating kinase 1,2
UPR: unfolded protein response
UPS: ubiquitin-proteasome system
YAP: Yes-associated protein
PS: Conceptualization, Writing—original draft. GS: Investigation, Visualization, Writing—original draft. SW: Writing—review & editing. All authors have read and agreed to the submitted version of the manuscript.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Peter Martin ... Leonard W. Poon
Renad I. Zhdanov ... Alexey S. Sozinov
Naoko Suga ... Satoru Matsuda
Freda Gonot-Schoupinsky
