
Abstract
DNA paralogs that have a length of at least 1 kilobase (kb) and are duplicated with a sequence identity of over 90% are classified as low copy repeats (LCRs) or segmental duplications (SDs). They constitute 6.6% of the genome and are clustering in specific genomic loci. Due to the high sequence homology between these duplicated regions, they can misalign during meiosis resulting in non-allelic homologous recombination (NAHR) and leading to structural variation such as deletions, duplications, inversions, and translocations. When such rearrangements result in a clinical phenotype, they are categorized as a genomic disorder. The presence of multiple copies of larger genomic segments offers opportunities for evolution. First, the creation of new genes in the human lineage will lead to human-specific traits and adaptation. Second, LCR variation between human populations can give rise to phenotypic variability. Hence, the rearrangement predisposition associated with LCRs should be interpreted in the context of the evolutionary advantages.
Keywords
Genomic disorders, low copy repeats, segmental duplicationsIntroduction
Pathological alterations in the DNA structure or composition result in genetic disorders. Different classes can be distinguished, ranging from aneuploidies, defined by a change of the total number of chromosomes, to single nucleotide variations, in which the disease is caused by the change of one single base pair (bp) [1, 2]. Rearrangement of a segment of the chromosome, typically encompassing 100 bp or more, is classified as structural variation [3]. Genomic disorders are a subclass of genetic disorders in which the disease phenotype is caused by these DNA rearrangements, rather than single nucleotide changes [2].
Technological innovation has driven our ability to detect the structural variation. First, G-banded karyotyping provided indications of larger-scale rearrangements. Second, the introduction of array comparative genomic hybridization and single nucleotide polymorphism assays enabled the scanning of the genome for copy number variations (CNVs) without a priori assumptions. Systematic screening of patients with developmental disorders with chromosomal microarrays resulted in the identification of several hitherto unknown genomic disorders [4]. Third, the structural variation catalogue was rapidly expanding by the use of whole-exome and whole-genome sequencing based on read depth or presence of split reads and conflicting mate pairs in short-read sequencing data [3], but still limited by short-read sequencing associated problems. Nowadays, these problems are solved by long-read sequencing approaches resulting in the gap-free assemblies of whole human chromosomes [5, 6] and even a complete human genome [7]. This started a new era of structural variation detection overload, switching the challenges from detection to documentation, interpretation, and clinical validation of newly observed CNVs. To that aim, the Human Pangenome Reference Consortium was established [8].
Rearrangements via non-allelic homologous recombination
Genomic disorders can be subdivided into the non-recurrent and recurrent rearrangements. Non-recurrent and complex rearrangements are characterized by locus and breakpoint variability, which are not predictable based on the genomic architecture. Molecular mechanisms responsible for these CNVs include non-homologous end-joining (NHEJ), fork stalling and template switching, and microhomology-mediated break-induced replication (MMBIR) [2]. In the recurrent rearrangements, several patients are described with breakpoints clustering in a specific locus. This is caused by the presence of duplication modules in this locus, serving as drivers for the rearrangements [4].
Recurrent genomic disorders are caused by meiotic misalignment of high sequence identity (> 90%) blocks (Figure 1A), resulting in rearrangements of the involved segment, a mechanism known as non-allelic homologous recombination (NAHR) [1]. The NAHR substrates are typically low copy repeats (LCRs) or segmental duplications (SDs), which are flanking the involved locus [1]. LCRs are blocks of DNA with a length of at least 1 kb and duplicated to several inter- and intra-chromosomal loci in the genome [9, 10].
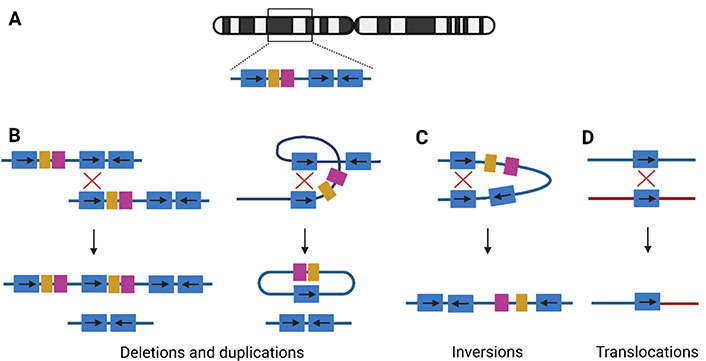
NAHR. (A) Example of LCR structure on a random chromosome. The blue duplicons can be in direct or inverted orientation, indicated by arrows; (B) NAHR between two direct repeats on two homologous chromosomes, interchromosomal NAHR (left), will lead to the creation of a duplication and deletion allele. Intrachromosomal NAHR (right), between two repeats on the same chromosome, leads to deletions and a ring chromosomal segment; (C) the recombination between indirect repeats on the same chromosome will lead to an inversion of the segment; (D) translocations are shaped if the NAHR is between two different chromosomes. →: orientation of the LCR; ↓: recombination event;
If both LCRs are in direct orientation, NAHR between homologous chromosomes will result in a chromatid with a deletion and another with the reciprocal duplication (Figure 1B). These are considered CNVs since they are associated with the gain or loss of DNA [3]. Intrachromosomal NAHR only can create deletions and a ring-shaped chromosomal segment (Figure 1B). Therefore, deletions are more frequently observed compared to duplications. Both deletions and duplications can lead to a clinical phenotype via a diversity of mechanisms: gene-dosage effect [11], expression of a new gene via gene fusion [12], interaction with regulatory elements [13], and interruption of chromatin structure [14]. Over 250 micro-deletion and -duplication syndromes are known and a limited overview is provided in Table S1 [2, 15–18]. In general, duplication syndromes have a milder phenotype compared to the deletion syndromes, since gene deficiencies overall have more phenotypic consequences [4]. Therefore, duplication carriers are less represented in clinical cohorts and the population incidence was long underestimated.
NAHR between LCRs in opposite orientation will lead to inversions (Figure 1C). These are copy-neutral events, since no gain or loss is associated with the rearrangement [3]. If heterochromatic sequence is inverted, the inversion will be harmless. If the inversion directly affects a gene, this can lead to disease, either by disrupting the gene or by alteration of the expression level [19]. For example, the majority of severe hemophilia A cases are caused by an inversion disrupting the coagulation factor VIII (F8) gene on chromosome X [Mendelian Inheritance in Man (MIM) 306700], mediated by two LCRs [20]. Another recurrent inversion involves the iduronate 2-sulfatase (IDS) gene on chromosome X, encoding the IDS enzyme, responsible for breakdown of glycosaminoglycans. The inversion causes IDS gene disruption, leading to mucopolysaccharidosis type II (MIM 309900), a lysosomal storage disorder [21]. Although not causing disease, some human inversion polymorphisms are associated with an abnormal phenotype [19]. The largest known human inversion polymorphism is located in the 8p23.1 locus, spanning a length of 4.5 megabases (Mb). These 8p23.1 inversion carriers have a lower risk of developing systemic lupus erythematosus and rheumatoid arthrosis compared to individuals carrying the reference allele [22].
Translocations are created by crossovers between elements on different chromosomes (Figure 1D). Examples of recurrent constitutional translocations are t(11;22)(q23;q11), t(8;22)(q24.13;q11.21), and t(4;8)(p16;p23). Carriers of the balanced translocation are phenotypically normal in most cases, but their offspring are at risk of inheriting a derivative chromosome, leading to genomic imbalance [23]. For example, in the unbalanced der(4)t(4;11)(p16.2;p15.4) translocation, the 4p16.2→pter monosomy expresses as Wolf-Hirschhorn syndrome (MIM 194190), and the imprinted 11p15.4→pter manifests as Silver-Russell syndrome (MIM 180860) or Beckwith-Wiedemann syndrome (MIM 130650), when maternally or paternally inherited, respectively [23].
Hence, the NAHR mechanism is responsible for a range of rearrangements, involving several, but LCR-specific parts of the genome. If this rearrangement manifests as an abnormal clinical phenotype, it can be classified as a genomic disorder.
Genomic predisposition for deletion/duplication syndromes
Parental inversion polymorphisms between LCRs predispose the region to NAHR, resulting in offspring with genomic disorders [24]. Population-embedded inversion polymorphisms are drivers of many genomic disorders. For example, the 1.5 Mb inversion on 7q11.23 is a driver of the deletion causing Williams-Beuren syndrome (MIM 194050) [25]. The inversion has a frequency of 12.4% in deletion-transmitting parents, although only present in 2.9% of the control population [19]. Other examples are the inversions leading to Angelman syndrome (15q11.2, MIM 105830, inversions in 33% of the mothers), Sotos syndrome (5q35.5, MIM117550), 8p23.1 microdeletion, and 15q23 or 15q24 microdeletion syndrome [19]. In addition, in some cases, these disease-predisposing inversion polymorphisms can be linked to phenotypic consequences as well. For example, the two haplotypes of the 1.5 Mb inversion polymorphism in the 17q21.31 locus have different characteristics: the direct H1 haplotype carries mutations linked to Parkinson’s disease and other neurodegenerative diseases, the inverted H2 haplotype is associated with an increased risk for 17q21.31 rearrangements and positive selection in the human population [26, 27]. Thus, although not directly related to disease, inversion polymorphisms between LCRs are an important driving cause of genomic disorders.
Distribution, origin, and evolution of LCRs
SDs or LCRs play an important role in the origin of genomic disorders. They constitute 6.6% of the human genome [7]. The LCR fragments span a length larger than the average sequencing read length. Therefore, these fragments will have multiple mapping options and are therefore frequently misassigned, creating errors and gaps in reference assemblies. As a consequence, they are frequently removed from standard analysis pipelines. Specialized approaches such as long-read sequencing and alternative bioinformatic approaches are necessary to investigate their importance in genome stability and evolution [28].
LCRs are not randomly distributed across the genome, but are primarily clustering in pericentromeric, subtelomeric, and interstitial loci. In these regions, there is up to 10-fold enrichment for LCRs with chromosome-specific differences: chromosome 3 has a low LCR density, while chromosomes 22 and Y harbor the largest LCR proportions [9]. They are composed of regular genomic architectural features such as genes, repeat elements, and regulatory sequences, but differ from the standard unique sequence in that they are copied to inter- and intra-chromosomal loci. However, compared to other repeat elements, their copy number is limited and ranges between 2 and 50 [29].
Pericentromeric, subtelomeric, and interstitial LCRs differ in characteristics and mechanism of origin (Figure 2A). The duplication content of pericentromeric LCRs originated mainly from interchromosomal duplication events, as proposed in the two-step model (Figure 2B). In a first “initial seeding event”, LCR sequence from different genomic loci is juxtaposed in a duplicon block. Afterwards, these duplicon blocks are copied to other pericentromeric sites, creating a mosaic structure [28]. Interchromosomal duplications are enriched in subtelomeric LCRs via serial translocations (Figure 2C): consecutive events of double-strand breakage and repair in these subtelomeric regions created a mosaic pattern of LCR-containing sequences [28, 30]. The largest LCRs in the human genome, are located in interstitial regions and are enriched for intrachromosomal duplications. Those interstitial LCR paralogues have the highest similarity. The complex patterns are formed by serial duplication, using the LCRs themselves as homology substrates in consecutive rounds (Figure 2D) [28, 30]. Alu repeats are frequently observed at or in the vicinity of the boundaries of LCRs, suggesting involvement of both NAHR and replication-based mechanisms (NHEJ, MMBIR) in the creation of these complex structures [28–31].
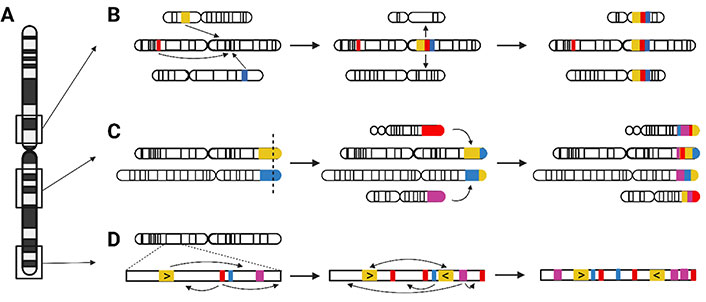
Origin of pericentromeric, interstitial, and subtelomeric LCRs. (A) Relative chromosomal location of pericentromeric, interstitial, and subtelomeric LCRs; (B) two consecutive events created the pericentromeric LCRs. First, during the initial seeding event, LCR sequences from different chromosomes are merged into an LCR block. Second, blocks are duplicated to other pericentromeric loci; (C) mosaic patterns of the interstitial LCRs were formed by several rounds of serial duplication; (D) serial translocation, by double-strand breakage and repair, is responsible for the subtelomeric LCR structure. →: consecutive rearrangement events
The proportion of LCR sequence varies substantially between the genomes of different species. The number of LCRs is very low in fly and worm in comparison to the human genome [28]. Although first thought that the duplication content was lower in mammalian genomes (mouse, dog, cow) as well [28], genome sequencing revealed similar levels of recent duplications [32]. However, the architecture of these duplications differs radically from the human mosaic LCR structure, since they show a tandem organization [32].
The LCRs evolved into their current organization starting in primates and are therefore of relatively recent origin. The genome of the marmoset, a New World monkey, has lower LCR levels than hominids (great apes: orangutan, gorilla, chimpanzee, bonobo; and humans), suggesting an expansion of LCRs after the divergence of Old and New World monkeys, 35 million years ago [32, 33]. This is consistent with the LCR duplication burst during human evolution [34]. Investigation and comparison of great ape and human genomes revealed more genetic variation due to LCR structure than single nucleotide variants in other genomic loci [35]. Due to the presence of Alu elements at the boundaries and breakpoints of the LCRs, the expansion is thought to be caused by Alu-Alu mediated rearrangements. This hypothesis is concordant with the burst of Alu elements 35 million years ago [32, 35]. Hence, LCRs are considered as fundamental components in the shaping process of primate genomes.
Transcriptomic innovation and phenotypic effects on evolution
Incorporation of one or more extra copies in the genome is associated with disease and evolutionary consequences. On the one hand, two identical copies on different chromosomal locations can act as NAHR substrates, leading to genetic instability, rearrangements, and eventually disease. On the other hand, natural evolutionary processes can act on the copied segment itself, creating gene segments and transcripts with a completely new function (neofunctionalization) or altered function (subfunctionalization), compared to the ancestral gene [32, 36]. Hence, due to their duplication potential and nature, LCRs are ideal substrates to influence gene evolution. If these duplications are specific to the human lineage, they can contribute to important adaptive traits.
One mechanism leading to a new gene product is incomplete duplication of an ancestral gene. For example, the SLIT-ROBO Rho guanosine-triphosphate hydrolase (GTPase) activating protein 2A (SRGAP2A) gene (chromosome1q32.1) is important in neuronal migration in mammals and is partially duplicated in the human lineage (SRGAP2B, chromosome1q21.1; SRGAP2C, chromosome1p11.2). The SRGAP2C paralogue is the most recent one and dimerizes with SRGAP2A in the human embryonic cortex. The function of SRGAP2C is antagonistic to the ancestral SRGAP2A, since it is a cortical development gene involved in dendritic spine maturation [37, 38]. Another example is the human-specific Rho GTPase activating protein 11B (ARHGAP11B) gene (chromosome15q13.2), the product of incomplete duplication of ARHGAP11A (chromosome15q13.3). It exerts a completely new function, by influencing progenitor cells of the radial glia neurons, leading to cortical layer expansion of the developing brain [39]. The Notch homolog 2 N-terminal-like (NOTCH2NL) gene has three human-specific paralogues: NOTCH2NLA (chromosome1q21.1), NOTCH2NLB (chromosome1q21.2), and NOTCH2NLC (chromosome1q21.2). They are expressed in radial glia and are important in the Notch signaling pathway. In that way, they influenced human-specific neuronal differentiation and alterations in the size and complexity of the human neocortex [40]. Hence, the emergence of human-specific genes due to incomplete duplications has contributed to critical adaptive traits regulating brain size and function.
If the duplication juxtaposes two partial genic fragments and the necessary regulatory elements, a fusion gene with a new function can be created. Indeed, there is evidence of an enrichment of gene fusions in human LCR regions [41]. The HYDIN gene (chromosome16q22.2), involved in cilia motility, was duplicated in the human lineage, although the promotor and polyadenylation site were missing. However, the partial duplication was juxtaposed with active regulatory elements in the locus, leading to the transcription of the HYDIN2 gene (chromosome1q21.1). Interestingly, whereas the ancestral HYDIN gene is mainly expressed in ciliated tissues, the human-specific HYDIN2 transcripts are specific for neuronal tissues [42]. The incomplete duplication and consecutive fusion between FAM7A and cholinergic receptor nicotinic alpha 7 (CHRNA7) gene created the CHRFAM7A fusion gene (chromosome15q13.2). Although research is hampered due to the large sequence identity between the ancestral and fusion gene, the gene product is involved in ion channel function [35, 43]. So, in addition to incomplete gene duplication, gene fusion is another mechanism contributing to gene evolution.
To conclude, the presence of genes in loci subjected to duplication has tremendous evolutionary potential. Human-specific genes were identified with important functions in the development and maturation of the brain, differentiating humans from chimpanzees [36]. The next step will be to link these human-specific genetic alterations to complex brain diseases such as schizophrenia, intellectual disability, and developmental delay. Due to their complex genetic architecture, targeted studies are essential and therefore, the extent of the evolutionary and adaptive impact is only starting to be discovered.
Structural variation of LCRs in the human population
The LCR loci are important substrates for the creation of human-specific genes via duplication, but due to their duplication potential, they can also be polymorphic within the human population [44]. These inter-human CNVs can give rise to phenotypic differences between individuals of different populations. The copy number of the amylase gene (AMY1, chromosome1p21.1) differs between 2 and 15 in modern humans. The gene has an essential function in the digestion of starch and copy number is therefore correlated with the amount of starch in the diet between populations [45]. The BolA family member 2 (BOLA2) unit (chromosome16p11.2) is a CNV under positive selection and 3 to 8 copies are reported in the human population. This polymorphism is associated with the maturation of iron-sulfur proteins and iron homeostasis: anemia is described in individuals with lower copy number and deletions of BOLA2, while expansions are protective against iron deficiency [46, 47]. Another interesting CNV is domain of unknown function 1220 (DUF1220, chromosome1q21.1), characterized by neuron-specific expression and positive selection. There is a strong association between head circumference in the normal population and brain-size manifestations in the disease population [48]. Transient receptor potential cation channel subfamily M member 8 (TRPM8) channel-associated factor (TCAF) genes (chromosome7q35) are highly conserved in evolution but show CNV in the human population. They have a valuable effect on regulating TRPM8, the ion channel for thermal sensation in somatosensory neurons. Copy numbers can be linked to demographic differences and implicate an adaptive role in changing conditions [49]. So, human polymorphic LCR loci are common and phenotypic consequences can be expected if genes are involved in the duplication.
Human-specific structural variation is described for several LCR loci. The 15q13.3 locus comprises five possible structural configurations, including CNVs and inversions. The locus, and more specifically the golgin A (GOLGA) repeat, is important in evolution and a hub for the breakpoint loci of Prader-Willi syndrome (MIM 176270)/Angelman syndrome (MIM 105830), 15q24 and 15q25.3 microdeletions [50]. Nine structural variants were observed in the 17q21.31 locus, associated with Koolen-de Vries syndrome (MIM 610443), and including partial duplications of the KAT8 regulatory NSL complex subunit 1 (KANSL1) gene [26]. However, since the reconstruction of the LCR region is challenging using short-read sequencing data, specialized techniques providing long-range structural information were used to map larger haplotypes. For example, Mostovoy et al. [51] leveraged Bionano optical mapping to uncover the variety of haplotypes in the 7q11.23, 15q13.3, and 16p12.2 loci. In addition, an astonishing number of structural variants was identified in the 22q11.2 locus, with more than 25 haplotypes of the first chromosome 22q11 LCR block ranging in size from 200 kb to over 2 Mb [52, 53]. The variability was considered human-specific in comparison to the shorter haplotypes of great apes [54]. The locus is associated with deletions causing the 22q11.2 deletion syndrome (MIM 192430), characterized by a variable phenotypic expression and penetrance. However, although several genes are embedded within the copy number variable subunits of these LCRs (PI4KAP, FAM23O, TMEM191, etc.), the transcriptional effect and phenotypic impact could not be associated (yet).
These studies introduce a paradox: LCRs, which are associated with an increased susceptibility for genomic rearrangements, and therefore are expected to be ‘selected out’ during evolution, are actually expanded during human evolution [44]. An explanation is that the genomic instability created by an increased copy number is neutralized or compensated by an evolutionary advantage, highlighting their extreme evolutionary and adaptive potential [48].
Conclusions
Although LCRs constitute a large portion of the human genome [7], their function and importance have long been unknown. They act as hubs for genomic disorder breakpoints, in which the rearrangement is caused by meiotic misalignment of similar sequences and subsequent NAHR. In addition, duplications of complete or partial genic segments create an opportunity to expand the gene catalogue, by neo- or sub-functionalization or gene fusion. This specific class of genes has proven to be important during human evolution. Hence, we suggest that the increased genomic instability associated with LCRs is a consequence of the evolutionary potential associated with this DNA class.
Several LCR regions remain elusive to short-read and even standard long-read sequencing technologies. Most recent optical mapping and ultra-long sequencing will assist in scaffolding the LCR and the associated structural variants. What will be the impact of specific structural variants at the transcriptome level and how the architecture alters the three-dimensional (3D) organizational structure? How do LCRs and LCR variability influence our phenotypes both in the general population and in the different genomic disorders? Multi-omics and multi-disciplinary investigation of the LCRs will be essential to further unravel their role and effects.
Abbreviations
| CNVs: | copy number variations |
| IDS: | iduronate 2-sulfatase |
| LCRs: | low copy repeats |
| Mb: | megabases |
| MIM: | Mendelian Inheritance in Man |
| NAHR: | non-allelic homologous recombination |
| NOTCH2NL: | Notch homolog 2 N-terminal-like |
| SRGAP2A: | SLIT-ROBO Rho guanosine-triphosphate hydrolase activating protein 2A |
Supplementary materials
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/1001131_sup_1.pdf.
Declarations
Author contributions
LV: Writing—original draft. LV and JRV: Writing—review & editing. Both of the authors read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by Fonds Wetenschappelijk Onderzoek [G0E1117N, G0A2622N] to JRV and by Stichting Marguerite-Marie Delacroix [GV/B-453] to LV. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2023.