 Review
Review

Affiliation:
1Hainan Provincial Key Laboratory of Carcinogenesis and Intervention, Hainan Medical University, Haikou 571199, Hainan, China
†These authors have contributed equally to this work and share the first authorship.
ORCID: https://orcid.org/0009-0000-3808-4471
Affiliation:
1Hainan Provincial Key Laboratory of Carcinogenesis and Intervention, Hainan Medical University, Haikou 571199, Hainan, China
†These authors have contributed equally to this work and share the first authorship.
ORCID: https://orcid.org/0009-0003-9124-6208
Affiliation:
1Hainan Provincial Key Laboratory of Carcinogenesis and Intervention, Hainan Medical University, Haikou 571199, Hainan, China
ORCID: https://orcid.org/0009-0007-1815-826X
Affiliation:
1Hainan Provincial Key Laboratory of Carcinogenesis and Intervention, Hainan Medical University, Haikou 571199, Hainan, China
2Department of Medical Oncology, Second Affiliated Hospital, Hainan Medical University, Haikou 570311, Hainan, China
3Institution of Tumour, First Affiliated Hospital, Hainan Medical University, Haikou 570102, Hainan, China
Email: mengsenli@163.com
ORCID: https://orcid.org/0000-0003-2512-0386
Affiliation:
1Hainan Provincial Key Laboratory of Carcinogenesis and Intervention, Hainan Medical University, Haikou 571199, Hainan, China
Email: mingyuezhu2002@163.com
ORCID: https://orcid.org/0000-0001-5997-4991
Explor Med. 2023;4:541–559 DOI: https://doi.org/10.37349/emed.2023.00160
Received: February 09, 2023 Accepted: April 17, 2023 Published: August 31, 2023
Academic Editor: Paul Zarogoulidis, “General Clinic Euromedica” Private Hospital, Greece
Abnormal energy metabolism is one of the ten hallmarks of tumors, and tumor cell metabolism provides energy and a suitable microenvironment for tumorigenesis and metastasis. Tumor cells can consume large amounts of glucose and produce large amounts of lactate through glycolysis even in the presence of oxygen, a process called aerobic glycolysis, also known as the Warburg effect. Lactate is the end product of the aerobic glycolysis. Lactate dehydrogenase A (LDHA), which is highly expressed in cancer cells, promotes lactate production and transports lactate to the tumor microenvironment and is taken up by surrounding stromal cells under the action of monocarboxylate transporter 1/4 (MCT1/4), which in turn influences the immune response and enhances the invasion and metastasis of cancer cells. Therapeutic strategies targeting lactate metabolism have been intensively investigated, focusing on its metastasis-promoting properties and various target inhibitors; AZD3965, an MCT1 inhibitor, has entered phase I clinical trials, and the LDHA inhibitor N-hydroxyindole (NHI) has shown cancer therapeutic activity in pre-clinical studies. Interventions targeting lactate metabolism are emerging as a promising option for cancer therapy, with chemotherapy or radiotherapy combined with lactate-metabolism-targeted drugs adding to the effectiveness of cancer treatment. Based on current research, this article outlines the role of lactate metabolism in tumor metastasis and the potential value of inhibitors targeting lactate metabolism in cancer therapy.
Cancer is generally considered a process driven by genomic and genetic instability, and its occurrence also involves effects on the tumor microenvironment [1]. Abnormal energy metabolism is one of the ten characteristics of tumors. In the early 20th century, studies carried out by Warburg et al. [2], Cori and Cori [3] showed that tumors consume vast quantities of glucose and secrete lactate. In general, the glycolytic pathway is the way how normal cells produce glucose to pyruvate and then, in oxygen presence, enter mitochondrial for oxidative phosphorylation to provide energy for cells, which is the main way of glycometabolism for energy supply in vivo. However, in oxygen deficiency, pyruvate is reduced to lactate. Interestingly, Warburg found that cancer cells depend primarily on glycolysis to metabolize glucose to lactate even in the presence of oxygen, a phenomenon of aerobic glycolysis identified as the Warburg effect [4]. The majority of cancer cells rely on aerobic glycolysis for lactate production to maintain a high proliferation rate, except for a few cells such as in breast cancer stem cells (CSCs) where reduced lactate production is found and in isocitrate dehydrogenase (IDH) mutant tumor cells where low lactate concentrations and near normal intracellular pH are found [5, 6].
Lactate has an essential function in the progression of cancer, as lactate secreted by the microenvironment acts both as a metabolic fuel for oxidative cells and as a signaling molecule in the tumor microenvironment, promoting cancer cell proliferation, metastasis, and invasion, increasing acidosis of the tumor microenvironment extracellularly, promoting chemoresistance, angiogenesis, and immune escape [7, 8].
Lactate dehydrogenase (LDH) is an oxidoreductase enzyme of the lactate metabolic pathway that reversibly catalyzes lactate oxidation to pyruvate. The subunit of LDH, LDHA, is a key enzyme in the production of lactate from pyruvate and plays an important role in lactate metabolism. When LDHA is deficient, patients have no specific clinical symptoms other than myoglobinuria and muscle stiffness after strenuous exercise [9]. That is, inhibiting the lactate metabolic pathway in which LDHA is involved may have fewer side effects on the body because almost all normal cells in the body are involved in the production of pyruvate from glucose, and inhibiting this process may cause inhibition and damage to normal cells. This is why there is a growing interest in drugs targeting the lactate metabolism pathway, and a new class of drugs targeting lactate metabolism is expected to emerge. The intrinsic metabolic properties and the surrounding environment affects the growth and metastasis of cancer cells [10, 11]. So understanding the metabolic characteristics of cancer cells and their interactions with the surrounding environment may provide new insights into cancer therapy and guide the development of more effective metabolic targeted drugs.
LDHA and/or monocarboxylate transporters (MCTs) are key targets of lactate metabolism, and their overexpression is related to treatment resistance and low prognosis in cancer patients [12–14]. In the past ten years, many molecules that inhibit key targets of lactate metabolism have been investigated. Among these targeted inhibitors, the LDHA inhibitor N-hydroxyindole (NHI) has a high value for pre-clinical studies [15]; in vivo experiments with the MCT1 inhibitor AZD3965 have shown that AZD3965 causes lactate accumulation in the Raji Burkitt lymphoma model and leads to significant tumor growth inhibition, which is a relatively promising compound [16]. This article describes the abnormalities of lactate metabolism and its effects on tumors and related carcinogenic signaling pathways; analyses lactate metabolism function in tumor cell metastasis; and discusses the potential value of inhibitors targeting lactate metabolism, such as LDHA and MCTs inhibitors, in cancer therapy.
In the last few decades, cancer metabolism has received increasing attention as an area of research. In the 1920s, Warburg [17] and colleagues observed tumors taking up huge quantities of glucose compared to the surrounding tissue. And for the first time, they described the metabolic phenomenon of rapid conversion of glucose to lactate in tumor cells even with sufficient oxygen. In later experiments, Warburg [2] used transplanted tumors from mouse sarcomas and found that for tumor cells, lactate production was very high. Heneberg’s [18] analysis of 57 published cases also reveals that systemic lactic acidosis is associated with an extremely poor prognosis in patients with solid tumors [18]. And lactic acidosis in association with hematologic malignancies also carries an extremely poor prognosis [19]. Suggesting that as an end product of the glycolysis, lactate production in cancer cells plays an essential role in tumor development.
Glycolytic pathway begins with the uptake of glucose, which is the primary source of lactate production. Glucose enters the cell mainly through the glucose transporters-1 (GLUT-1) and undergoes the glycolytic pathway to produce pyruvate. Pyruvate enters mitochondria for oxidative decarboxylation to generate acetyl coenzyme A (acetyl CoA), which enters the tricarboxylic acid cycle (TCA cycle) for oxidative phosphorylation; LDH can directly catalyze pyruvate in the cytoplasm to produce lactate, which is a reversible reaction. When cancer occurs, cancer cells provide energy to the cells primarily through a pathway that produces lactate from pyruvate. There are five isozymes of LDH, LDH1-LDH5, consisting of tetramers of LDHA or LDHB subunits in different proportions [20]. LDHA is mainly involved in pyruvate conversion to lactate and nicotinamide adenine dinucleotide (NADH) oxidation to NAD+ to meet the energy requirements of cancer cells.
In contrast, LDHB mainly contributes to the reverse conversion of lactate to pyruvate and NADH [21]. LDHA is regulated by hypoxia-inducible factor-1 (HIF-1) and c-Myc, which synergistically promote the transcription of LDHA in cancer cells (Figure 1) [22]. Post-translational modifications, including phosphorylation, are the way by which LDHA can control the activity of cancer cells [23]. LDHA is also related to cancer cell invasion, metastasis, and drug resistance and is a potential target for chemotherapy [24]. The abnormal LDHA expression has been identified to be closely related to a diversity of malignancies. It is highly expressed in liver cancer [25], cervical cancer [26], breast cancer [27, 28], pituitary adenoma [29], glioma [23], and lung cancer [30]. The above studies suggest that high expression of LDHA promotes the development of tumor cells. One of the possible reasons for this is that it stimulates the generation of huge quantities of lactate in tumor cells, which in turn promotes the growth, invasion, and metastasis of tumor cells, so targeted inhibition of LDHA may suppress the malignant behavior of tumor cells.
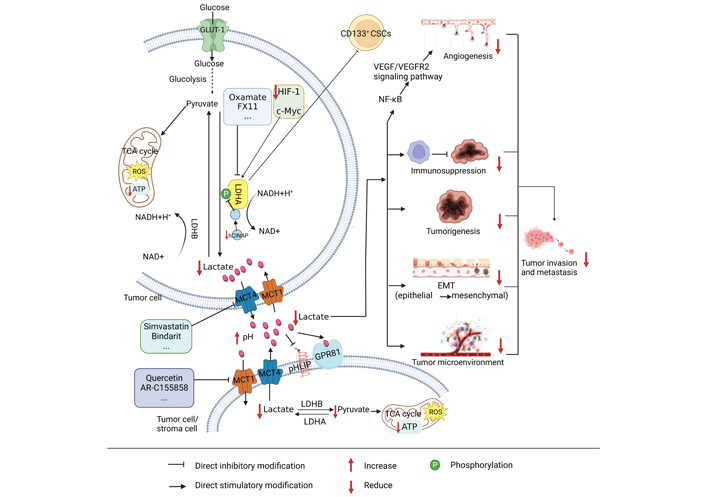
The process of lactate metabolism in tumors. CD: cluster of differentiation; VEGF/VEGFR: vascular endothelial growth factor/vascular endothelial growth factor receptor; ATP: adenosine triphosphate; ROS: reactive oxygen species; pHLIP: pH low insertion peptide; GPR81: G-protein-coupled receptor 81; EMT: epithelial-mesenchymal transformation; hCINAP: human coilin interacting nuclear ATPase protein; NF-κB: nuclear factor-kappa B. It is created by BioRender
Lactate is the product of glycolysis. For a long time, the role of lactate in tumor metabolism has been underestimated and considered a waste of glycolytic metabolism. Recently, an increasing count of investigations has demonstrated that lactate has an essential role in tumor development, mainly in terms of its effects on metabolic regulation, immune escape, tumor growth, angiogenesis, tumor metastasis, and intercellular interactions in the tumor microenvironment (Figure 1) [31–33]. Under normal conditions, cells produce 36 mol of ATP and a small amount of lactate through oxidative phosphorylation, while cancer cells produce 4 mol of ATP and a large amount of lactate through aerobic glycolysis, but because cancer cells metabolize glucose at a much faster rate than normal cells, the total amount of ATP and lactate produced in the same time is much higher than that obtained by oxidative phosphorylation in normal cells, which provides a large amount of energy and a suitable environment for the development and metastasis of cancer cells [34–36]. Lactate transport is dependent on MCT, a proton-linked membrane transporter protein located on the cell membrane, and the solute carrier family 16 (SLC16) is comprised of 14 members of the MCT family. however, only MCTs (1–4) contribute to monocarboxylate transport in human cells. Of these, those responsible for lactate entry and exit from cells are MCT1 and MCT4, respectively [37]. The lactate formed in tumor cells is transported outside the cells with the help of MCT4. Lactate excessive secretion decreases the pH in the microenvironment. Lactate in the microenvironment can induce drug resistance in colon cancer cell lines [38], promote tumor growth [39], and suppress lymphocyte immune function [40]; lactate in the microenvironment enters the surrounding cancer cells under the transport of MCT1 and generates pyruvate under the action of LDHB, making it enter the TCA cycle and provide energy for cancer cells [21, 37]. Hanson et al. [41] have established that inhibition of MCT1, MCT2, and MCT4 alone had a minor effect on tumors. In contrast, combined inhibition led to higher inhibition of lactate export and induced tumor cell death.
GPR81 (HCA1 or HCAR1) acts as the receptor of lactic acid [42] and is mainly expressed in adipose tissue [43] and also in skeletal muscle [44], liver, and pancreas [45]. GPR81 has been proven to play a crucial role in cancer. Roland et al. [46] conducted experiments on colon, breast, lung, hepatocyte, pancreas, and other cancer cell lines and found that almost all cancer cells expressed high levels of GPR81. In contrast, normal cells, such as pancreatic ductal cells, expressed only very low levels of GPR81. The results of Longhitano et al. [47] also showed that GPR81 expression was upregulated in breast cancer. Therefore, targeting key enzymes, receptors, and products of lactate metabolism may provide help in the search for tumor therapy.
Lactate metabolism can promote immunosuppression, EMT, tumor generation and metastasis, and the formation of tumor microenvironment. Oxamate, simvastatin, quercetin, and other small molecule inhibitors can respectively target LDHA, MCT4, and MCT1 to decrease lactate generation, accumulate ROS and deplete ATP, and hinder the development of tumors.
Metastasis results from a multi-step cellular biological process that manifests as the spread of cancer cells to more distant organ sites and their evasion of host defenses and adaptation to a new microenvironment, culminating in progressive growth and metastasis [48]. Cancer cells reach target organs through the circulation of body fluids such as blood, and the microenvironment determines the viability of cancer cells. More than 90% of deaths from solid tumors are attributed to metastases rather than primary tumors [49]. Cancer cell metastasis is characterized by cell shape rearrangements involving the formation of actin-based protrusions and new adhesions to surfaces, as well as cell contraction. For cancer cell metastasis actin and cell contractility-targeting drugs have obtained the expected results in preclinical studies and are a promising cancer chemotherapy drug [50]. In addition, lactate has been found to induce metastasis of tumor cells [51]. And an increasing number of experiments support the involvement of tumor metabolism and metabolic reprogramming in the metastasis of tumor cells, providing them with a large amount of energy, and targeting tumor metabolism may prevent the power provided for cell migration [52]. Therefore, studying the mechanisms involved in lactate metabolism, lactate metabolism-targeted drugs in combination with, for example, actin and contractility-targeting drugs for treatment may provide help in finding ways to inhibit cancer development and metastasis.
Tumor cell metastasis requires overcoming the damage caused by highly evolved human normal tissues [49]. The generation of lactate from glucose by glycolysis provides favorable conditions for tumor metastasis, and important factors of lactate metabolism, such as LDHA, lactate, and MCTs, have a crucial function in tumorigenesis and metastasis. High expression of LDHA correlates with expression of VEGF and can significantly promote angiogenesis by activating the VEGF/VEGFR2 signal pathway (Figure 1). Anti-angiogenic therapy benefits patients with LDHA-high expressing cancers, while LDHA overexpressing tumors are particularly aggressive [53, 54], suggesting that LDHA may affect the tumor microenvironment by promoting angiogenesis and invasion, which in turn promotes tumor growth and metastasis. It has been found that lactate released from tumor cells can promote acidification of the microenvironment and reduce adhesion between tumor cells, thereby promoting their invasion and metastasis [55]. Lactate also triggers the pro-angiogenic pathway via MCT4 output and MCT1 input [56]. Inhibition of the binding of MCTs to its chaperone Basigin inhibits VEGF expression and tumor angiogenesis in tumors of mice with glioblastoma stem cell (GSC)-derived xenografts [57]. Moreover, lactate secreted by human head and neck squamous cell carcinoma (HNSCC) may promote tumor migration and invasion by inhibiting the nuclear factor-kappa B (NF-κB) pathway [58], and NF-κB may also stimulate bile duct cancer metastasis by increasing VEGF (Figure 1) [59]. Taken together, high expression of LDHA may promote lactate production, which activates the NF-κB pathway and subsequently promotes activation of the VEGF/VEGFR2 signaling pathway, ultimately promoting angiogenesis and tumor invasion, and metastasis.
Aerobic glycolysis is used by tumor cells to meet their energy and biosynthetic needs, and lactate is the most significantly elevated metabolite in tumors, which is excreted into the microenvironment via MCT4. Peripheral stromal cells take up secreted lactate via MCT1, thereby meeting their energy requirements and promoting cancer cell invasion and metastasis (Figure 1) [60, 61]. Conversely, stromal cells can also secrete lactate as ‘fuel’ for cancer cells and stimulate their development and metabolism [62]. These studies suggest that the metabolic cooperation between tumor cells and stromal cells can promote tumor development, that lactate is the key link between their cooperation, and that inhibiting lactate production by tumor cells or stromal cells may be able to block malignant behaviors of cancer.
The phenotype of CSCs is related to tumorigenesis and metastasis, as well as lactate metabolism (Figure 1). In low miR-192-5p and CSC-positive hepatocellular carcinomas (HCC), cancer cells generate excessive quantities of lactate, which may further increase the malignant characteristics of HCC by interacting with non-tumor cells in the microenvironment [63]. Similarly, the hCINAP binds to the C-terminal structural domain of LDHA. It relies on its adenylate kinase activity to stimulate LDHA phosphorylation at tyrosine 10 sites and confer a metabolic advantage for colon CSC invasion (Figure 1) [64]. It has been shown that CD133+ hepatocellular CSCs have a more active glycolytic profile than oxidative phosphorylation in comparison with CD133– cells [65]. Inhibition of pyruvate dehydrogenase kinase 4 (PDK4) and LDHA significantly suppressed the stem cell characteristics of CD133+ and overcame resistance to sorafenib. Adding glucose or lactate to CD133– cells promoted the CSC phenotype, as demonstrated by elevated CD133+ cell populations, increased stemness gene expression, and promoted sphere development. It has also been shown that glioblastoma multiforme (GBM)-derived CSCs are more sensitive to glycolysis inhibition than non-CSCs. That CSCs may be more susceptible to the effects of LDHA [66]. It is speculated that inhibition of LDHA could inhibit the oncogenicity of CD133+ tumor stem cells, reducing lactate production and ultimately inhibiting tumorigenesis and metastasis.
EMT is a biological process in which epithelial cells lose epithelial properties, including cell polarity and cell contact, and gain mesenchymal properties, such as increased motility and the ability of quiescent adherent cells to migrate. Increased lactate secretion in oral squamous cell carcinoma and lung cancer cells has stimulated cell proliferation and migration and induced EMT to work (Figure 1) [67, 68]. lactate secretion by tumor cells may promote EMT, which in turn promotes lactate secretion and tumor development, invasion, and metastasis. High LDHA expression is related to invasion and low prognosis and can promote the transcription of genes contributing to the EMT process [69]. Another study showed that alterations in metabolic characteristics occurred in the EMT experimental model, including increased glucose uptake, lactate secretion, and lack of respiration, and 13C-glucose tracing data confirm that lactate secretion originates from glucose, suggesting that EMT can promote lactate production [70]. Therefore, the research and discovery of inhibitors targeting lactate metabolism may facilitate tumor treatment.
Due to the importance of lactate metabolism in tumor metabolic reprogramming, the development of anticancer drugs targeting lactate metabolism has gradually attracted the interest of scientists. Many small molecule inhibitors targeting lactate metabolism have been identified, but most of these molecules are still in the pre-clinical stage or clinical research stage. To date, the main targets include lactate production mediated via LDHA and lactate transport mediated via MCTs, while inhibitors of the GPR81 have not yet been reported. Therefore, the critical research on LDHA and MCTs inhibitors and the growth, invasion, metastasis, and metabolism of cancer cells were described in detail.
LDHA catalyzes the conversion of pyruvate to lactate. It was found that overexpression of LDHA decreased phosphorylation of the metabolic regulator adenosine 5’-monophosphate (AMP)-dependent protein kinase AMP-activated protein kinase (AMPK) and stimulated phosphorylation of downstream mammalian target of rapamycin (mTOR) in pancreatic adenocarcinoma (PAAD) cells. mTOR blockage inhibited LDHA-induced aggressiveness of PAAD cells. Cancer cells prefer to produce ATP by generating lactate through the glycolysis, and LDHA is an essential component of this pathway and is considered one of the potential targets for cancer therapy [12]. And LDHA-deficient individuals do not exhibit any specific clinical symptoms except muscle stiffness and myoglobinuria after strenuous exercise [9]. Several inhibitors developed for LDHA and their effects on cancer treatment are revealed in Table 1 and Figure 1.
List of information on LDHA inhibitors
| LDHA inhibitors | Competitive sites | LDHA (Ki value) | Shortcomings | References |
|---|---|---|---|---|
| Oxamate | Pyruvate | 136.3 μM | High polarity, low exocytosis across cell membranes, high dosing concentration | [71–75] |
| (R)-(–)-gossypol | NADH | 1.9 μM | Non-specific toxicity | [76–78, 84, 85] |
| FX11 | NADH | 8 μM | Not yet used in clinical trials | [76, 78] |
| Quinoline 3-sulfonamides | NADH | 4–6 nM | Low clearance in vivo | [88] |
| GF | Pyruvate | 5.46 μM | Possible inhibition of RNA synthesis rather than blockage of glycolysis | [89, 92] |
| NADH | 56 μM | |||
| NHI | Pyruvate, NADH | 8.9 μM | Novel inhibitors to be studied | [15, 93] |
| GNE-140 | NADH | - | Novel inhibitors to be studied | [96] |
GF: galloflavin. Ki: inhibition constant; -: no literature has demonstrated it
Oxamate, an isotope of pyruvate, has been shown that non-selectively inhibit LDHA (Ki = 136.3 μM) by competing with pyruvate [71]. The study also found that the half-inhibitory concentration (IC50) values were 74.6, 32.4 and 17.8 mM and 62.3, 44.5, 31.6 mM at 24, 48 and 72 h in the nasopharyngeal carcinoma cancer cells CNE-1 and CNE-2 [72]. Although some studies indicated that inhibition of LDHA by oxamate significantly increased human non-small cell lung cancer A549 and H1975 cells radiosensitivity and promoted ionizing apoptosis and autophagy induced by radiation in cells, associated with altered distribution of cell cycle. In addition, LDHA inhibition triggered a tumorigenic cascade response, inducing ROS buildup and cellular ATP lessening, which may elevate DNA damage and impede the activity of DNA repair (Figure 1) [30]. However, comparative studies have also found that oxamate inhibits glycolysis, proliferation and motility in medulloblastoma cell lines, but not small interfering RNA (siRNA) of LDHA, so oxamate may not act exclusively on LDHA [73]. In terms of medication, oxamate has very low therapeutic toxicity but is highly polar [74]. Its application is limited due to low ectopicity across cell membranes and high dosing concentrations, and if used in the clinic, consider eliminating its limiting factors first [75].
(R)-(–)-gossypol is a potent LDH inhibitor and is a natural compound [76]. It inhibits LDHA non-selectively by competing with NADH [77] and has a Ki value of 1.9 [78]. Gossypol was found to suppress the proliferation of human breast cancer MCF-7 cells [79]. Certain concentrations also inhibited the development of human melanoma cell lines, colon cancer, fibrosarcoma, promyelocytic leukemia, breast adenocarcinoma, T-lymphocytoma, and other cancer cells [80]. The (R)-(–)-enantiomer of gossypol is more cytotoxic than the (S)-(+)-enantiomer. Studies have shown that (R)-(–)-gossypol triggers cell shrinkage, loss of cell adhesion, membrane vesicles, and DNA breakage [81] and also has good in vitro cytotoxicity in human glioma cell lines and adrenocortical carcinomas [82, 83]. However, tests of gossypol in humans have led to non-specific toxicity, mainly with side effects such as hypokalaemia, fatigue and muscle weakness, kidney damage, and even paralysis [84, 85]. The above results suggest that gossypol has limited clinical application due to its non-specificity in treatment.
The small molecule inhibitor FX11 is a derivative of the natural product gossypol [78]. It inhibits LDH by competing for NADH binding sites and is an LDHA competitive inhibitor (Ki = 8 uM) [76]. FX11 inhibits cell proliferation, metastasis, and invasion and promotes apoptosis in human prostatic cancer PC-3 and DU145 cells by inhibiting LDHA activity. Mechanistically, in PC-3 and DU145 cells, LDHA knock-down or FX11 treatment was found to reduce the Warburg effect, as evidenced by decreased glucose depletion and lactate release and downregulated expression of matrix metalloproteinase-9 (MMP-9), plasminogen activator urokinase (PLAU), and histone B [86]. FX11 has pre-clinical efficacy in lymphoma, prostate cancer, and neuroblastoma [86, 87]. It is a potentially useful inhibitor for cancer therapy but has not been applied in clinical trial studies.
Quinoline 3-sulfonamides inhibit LDH by competing for NADH binding sites, and their selectivity to LDHA is 10–80 times than LDHB. Treatment with quinoline 3-sulfonamides increased oxygen consumption in HCC SNU398 cells, inhibited lactate production and cell proliferation, and promoted apoptosis. Unfortunately, in vivo, the use of quinolone 3-sulfonamides is unacceptable due to their pharmacokinetic properties, low clearance rate in vivo, and incompatibility with oral bioavailability [88], thus inhibiting their clinical application.
GF inhibits LDHA by competing with pyruvate (Ki value of 5.46 μM) and with NADH (Ki value of 56 μM) [89]. GF effectively inhibited cell growth in endometrial cancer cell lines and primary cultures of human endometrial cancer that regulate metabolism, cell cycle, apoptosis, cell stress, and metastasis. The IC50 values for the endometrial cancer ECC-1 and Ishikawa cells were 25 μM and 43 μM after 72 hours of treatment with GF [90]. It downregulated essential protein expression in cellular glycolysis and caused a decrease in glucose uptake and lactate generation [91]. However, GF was found to prevent LDHA from binding to single-stranded DNA and to inhibit RNA synthesis and not lactate formation in human colon cancer SW620 cells cultured in glucose-free conditions after a short exposure, which excludes the impact of LDHA inhibition. Therefore, the anticancer effect of GF on tumor cells may block not only glycolysis but also RNA synthesis, and the inhibition of RNA synthesis has nothing to do with the effect of glycolysis [92]. The inhibition of GF may not be a specific LDHA inhibitor, and its particular mechanism of tumor inhibition remains to be investigated.
NHI inhibitors are a new class of LDHA inhibitors with a central indole scaffold. They are competitive inhibitors of NADH and pyruvate and have Ki values in the low micromolar range, anti-proliferative activity, and the ability to inhibit lactate formation [15]. In a 2013 study, Granchi et al. [93] identified an NHI-like methyl ester [methyl 1-hydroxy-6-phenyl-4-(trifluoromethyl)-1H-indole-2-carboxylate] with cell membrane permeability, reduced formation of cellular lactate and anti-proliferative action against cancer cells, which may be suitable for creating LDHA-targeted cancer treatments. Di Bussolo et al [94], in 2015, developed a glucose-fixed N-hydroxyindolyl. They found that α-D-mannose derivative 2 exhibited optimal performance in cellular assays, demonstrating potent anti-glycolytic and anti-proliferative activity in cancer cells. In a 2020 study, EI Hassouni et al. [95] designed NHI-glucose conjugates-2 (NHI-Glc-2) for dual targeting of cancer cells, exhibiting inhibition of LDHA enzyme activity, human pancreatic cancer PANC-1 cell growth and disrupting the integrity of the 3D sphere model, which refers to cells cultured into spheres via a matrix or scaffold that more accurately represents the natural environment experienced by cells in an organism, and which allows for cellular interactions with more realistic biochemical and physiological responses. Exploration of NHI derivatives may assist in the search for more optimal LDHA inhibitors.
GNE-140 is a novel inhibitor of LDHA and LDHB [96]. Interestingly, Mazzio et al. [97] blocked lactate production and attenuated cell division but unimpaired cell viability after acting on triple-negative breast cancer MDA-MB-231 cells with GNE-140, and the addition of exogenous (+) lactate did not reverse the cytostatic effect, and how precisely LDH activity controls the cell cycle remains to be investigated. GNE-140 mechanism of action in vivo is unknown and needs to be further investigated.
In conclusion, although small molecule inhibitors of LDHA have been presented to have cancer therapeutic activity in pre-clinical trials, none have yet reached the clinical trial stage. There is a need to optimize current inhibitors, such as eliminating high polarity, reducing non-specificity, increasing in vivo clearance or increasing compatibility with oral biological formulations, or developing new effective LDHA inhibitors with low toxicity.
Lactate is transported across membranes via the MCT family. Lactate can promote tumorigenesis and metastasis of tumors. It enters endothelial cells through MCT1, triggering phosphorylation/degradation of inhibitor kappa B alpha (IκBα), which activates the autocrine NF-κB/IL-8 (CXCL8) pathway to drive cell migration and angiogenesis, and lactate secretion from tumor cells via the MCT4 transporter, and stimulates IL-8-dependent angiogenesis and tumor development. [39]. MCTs are highly expressed in cancer cells, and lactate export, live cell counts, and intracellular pH can be partially reduced by a single knock-down of MCT1, MCT2, or MCT4. However, simultaneous inhibition of all three molecules is the most efficient [41]. However, pancreatic ductal adenocarcinoma (PDAC) cell migration is non-significantly affected by MCT1/MCT2 suppression or MCT1 depletion but by MCT4 knock-down [98]. Studies have also found that the prognosis of triple-negative breast cancer is negatively correlated with MCT4 levels and that MCT4 may be used as a target for drug therapy [99]. Interestingly, MCT1 is also a novel biomarker for prognosis, and inhibition of MCT1 enhanced the response of human glioblastoma to temozolomide [14]. MCTs are considered an essential target for cancer therapy, and several inhibitors against MCTs and their effects on cancer are listed below (Figure 1 and Table 2).
List of information on MCTs inhibitors
| MCTs inhibitors | Site of action | Shortcomings | References |
|---|---|---|---|
| Lonidamine | MCT1, MCT2, MCT4 | Less effective | [102] |
| Quercetin | MCT1 | Does not affect cell migration | [103, 106] |
| Simvastatin | MCT4 | Does not affect cell migration | [104, 106] |
| DIDS | MCT Family | Does not affect cell migration | [105, 106] |
| AR-C155858 | MCT1 (Ki = 2.3 nM), MCT2 | Novel inhibitors to be studied | [107] |
| AZD3965 | MCT1 (Ki = 1.6 nM), MCT2 (Ki = 20 nM) | Novel inhibitor in Phase I clinical trials | [16, 111, 113] |
| BAY-8002 | MCT1, MCT2 (MCT1 is 5 times more selective than MCT2) | Novel inhibitor, not in clinical trials | [114] |
| Bindarit | MCT4 | Newly identified inhibitors to be studied | [115] |
| VB124 | MCT4 | Novel inhibitors to be studied | [117] |
DIDS: 4,4’-diisothiocyanatostilbene-2,2’-disulfonic acid
Lonidamine is an antitumor agent that selectively sensitizes tumors to chemotherapy, thermotherapy, and radiotherapy. Investigations into the molecular mechanisms of the anti-cancer effects of lonidamine have revealed that the drug is an inhibitor of the glycolytic enzyme hexokinase [100]. Lonidamine also inhibits mitochondrial pyruvate carrier (MPC) and respiratory chain complex I/II activity [101]. Because of the inhibition effect on MCT, it can synergistically inhibit MCT1, MCT2, and MCT4 but has low potency, limiting its use as a targeted MCTs inhibitor due to K0.5 of 36–40 µM [102].
Quercetin is an MCT1 inhibitor [103]; simvastatin is an MCT4 inhibitor [104], and DIDS is an anion transporter protein inhibitor that inhibits the functional activity of all proteins in the MCT family [105]. However, when quercetin, simvastatin, and DIDS were used to act on human lung cancer A110L cells, cell invasion was inhibited, but migration activity and cellular MCT1 and MCT4 expression were unaffected [106]. The above MCTs inhibitors have limited their clinical application because of their modest inhibitory effect on tumor cells.
AR-C155858, a novel MCTs inhibitor, has been demonstrated to be a non-competitive inhibitor of MCT1 with almost 2.3 nM Ki value for MCT1 in rat erythrocytes, and AR-C155858 also inhibits MCT2 but not MCT4. AR-C155858 may be much more sensitive to the inhibition of MCT1-mediated lactate transport than MCT2-mediated lactate transport [107]. However, in vivo treatment with AR-C155858 is inefficient in a mouse 4T1 xenograft mammary tumor model [108], and the application of this inhibitor remains to be investigated.
AZD3965 is an analog of AR-C155858 and an oral biologically effective inhibitor of MCT1. It has high affinity and selectivity for MCT1 (Ki value is 1.6 nM), has little effect on MCT2 (Ki value is 20 nM), and has no activity on MCT4 [16]. Study shows effect of AZD3965 in lymphoma xenograft model; AZD3965 inhibited the growth of 99% of Burkitt lymphoma CA46 in mice after 24 days of continuous oral administration [109]. However, one study found little overall impact on tumor growth and increased lung metastasis following the action of AZD3965 in a mouse 4T1 cells tumor model of triple-negative breast cancer [110]. AZD3965 has been evaluated in advanced solid tumors during phase I clinical trials and lymphomas in the UK. In phase I clinical study of the drug for the treatment of patients with advanced solid tumors reported in 2017, side effects included nausea and fatigue, and elevated cardiac troponin and reversible retinal function changes were detected at 20 mg oral dose but were generally within the range of pre-clinical effectiveness [111]. However, in the 2020 clinical trial, a patient of 47 years old with metastatic melanoma developed refractory hyperlactatemia after the first dose of AZD3965, and analysis of this event may have been drug-related. Patients with elevated plasma lactate were excluded from screening in subsequent clinical trials [112]. A clinical trial of 11 patients with lymphoma reported in 2021 also showed the effects of urinary lactate and urinary ketones in one patient following oral administration of the recommended phase 2 dose of AZD3965 at 10 mg, but well tolerated by the drug. However, one study found that AZD3965 resistance was associated with the overexpression of MCT4 [113]. In summary, the inhibition of MCT1 by AZD3965 may block the exchange of lactate and thus inhibit tumor growth, a promising anticancer drug. However, treatment should also consider excluding factors such as hyperlactatemia and high MCT4 expression that affect AZD3965.
BAY-8002 is a novel and potent inhibitor of MCT1. Although structurally different from AZD3965, dual MCT1 and MCT2 inhibitors are five times more selective than MCT2 and unselective for MCT4. Treatment of human Burkitt lymphoma-bearing mice with BAY-8002 resulted in the aggregation of lactate in the tumor [114]. This potential tumor suppressor has not been tested in clinical trials.
There are not many selective inhibitors of MCT4. Recently, bindarit (2-[(1-benzyl-1H-indazol-3-yl) methoxy]-2-methylpropanoic acid), the primary MCT4 inhibitor to be proposed, was identified as a highly potent, highly selective, and non-competitive inhibitor [115]. When bindarit was applied to the breast cancer MCF-7 and T47D cells, the uptake of 14C-L-lactate was significantly inhibited, and bindarit significantly elevated HIF-1α levels in MCF-7 cells [116]. However, additional clinical validation of this MCT4 inhibitor has yet to be performed.
Recently, the small molecule compound VB124 was produced and used as an effective, safe, and selective MCT4 inhibitor that explicitly inhibits lactate efflux [117]. When treating a mouse model of liver cancer C57BL/6 cells with VB124, the reduction in MCT4 activated the immune system of the mice, which in turn reduced tumor growth [13]. More and more in-depth validation of the new inhibitor needs to be done to determine the effect of the inhibitor.
GPR81 is a G-protein-coupled receptor for lactate. its expression is mainly in adipocytes, skeletal muscle, and the brain. The presence and function of GPR81 in the regulation of lactate sensitivity in cancer cell metabolism were first demonstrated by Roland et al. [46]. GPR81 is present in cancer cell lines of the colon, breast, hepatocyte, and pancreas, and 94% of pancreatic cancer patients express high levels of GPR81. In the lack of glucose and lactate, GPR81 is essential for cancer cell survival. Knocking out GPR81 reduces the proliferation and metastasis rates of cancer cells in vivo. Therefore, GPR81 is a very novel target for cancer drug therapy. With no confirmed reports of inhibitors or antagonists of GPR81 to date, it is crucial to find inhibitors or antagonist drugs of GPR81 and to study their effects on tumors.
pHLIP is a peptide carrier that can target the extracellular micro acidic environment, and this carrier can selectively transport the load to the diseased cells in a low pH-dependent manner. Under normal pH conditions, pHLIP has a high affinity to cell membranes, but under low pH conditions, pHLIP can fold and insert into cell membranes, which brings new opportunities for tumor diagnosis and targeted therapy. Compared with normal cells, the vast majority of tumor cells can produce large amounts of lactate through aerobic glycolysis, which lowers the extracellular pH. pHLIP has been shown to target cancer tumor acidity, which can bring hope for targeted tumor therapy if it carries targeted drugs [118]. However, how to apply pHLIP more effectively in tumor therapy and how to carry targeted drugs to target tumor tissues precisely and unambiguously is the problem to be solved at present.
Although many drugs targeting cellular metabolism have been studied to enable them to exert antitumor effects, targeting metabolism alone is insufficient for cancer treatment. In terms of combining targeted therapy and immunotherapy, pHLIP combined with the development of a novel STING agonist (STINGa) achieved the desired effect. It was found that in a tumor mouse model, in the acidic microenvironment of the tumor, the use of pHLIP-targeted STINGa eradicated CT26 tumors and developed anti-cancer immunity in mice, with no reappearance of tumors against re-injected cancer cells, while in nude mice lacking T cells, the therapeutic effect against CT26 tumors was negligible [119]. Targeting tumor metabolism in combination with chemotherapeutic agents, immune blockers, or radiotherapy may be the best therapeutic strategy. In terms of combination chemotherapeutic agents, the LDHA inhibitor NHI derivative NHI-Glc-2 inhibited cell growth and disrupted the integrity of 3D spheroid model when administered alone, whereas the anticancer drug gemcitabine had no effect in this model; however, the overall effect was more pronounced when NHI-Glc-2 was combined with gemcitabine compared to monotherapy [95]. In terms of combined immune blockers, targeting MCT4 (VB124) in conjunction with anti-programmed cell death protein 1 (anti-PD-1) therapy was found to be more efficacious than treatment with VB124 or anti-PD-1 alone in a mouse model of HCC; It may be because inhibition of MCT4 enhances antitumor immune response in HCC and targeting MCT4 enhanced anti-PD-1 treatment in mice [13], which may be a promising method of treatment. In combination radiotherapy, when mice with small cell lung cancer were treated with radiotherapy combined with the MCT1 inhibitor AZD3965, tumor growth was significantly reduced compared to either AZD3965 inhibitor or radiation therapy alone. It may be that AZD3965 is particularly effective in killing tumors in the hypoxic area. The effectiveness of radiotherapy is limited by cellular resistance in the hypoxic zone of tumor. When radiotherapy is combined with AZD3965, AZD3965 inhibits MCT1 and may enhance radiosensitivity by reducing lactate transport, thereby reducing tumor growth [120]. In terms of combined oxidative phosphorylation inhibitors, Burkitt lymphoma CA46 cells were exposed to AZD3965 for seven weeks in vitro, resulting in higher dependence on oxidative phosphorylation. When AZD3965 was combined with a mitochondrial complex I inhibitor (acting on oxidative phosphorylation), this combination reduced CA46 disease burden in mice and significantly induced lymphoma cell death in vitro [109]. The compensatory metabolic changes observed following AZD3965 administration support an adaptive metabolic response leading to a dependence on oxidative phosphorylation. This is when drugs that inhibit the glycolysis and target oxidative phosphorylation are needed to ensure efficacy against the tumor. The above results suggest that targeted lactate metabolism, combined with other therapeutic approaches, has great potential for cancer treatment.
Tumor is a complex, multifactorial disease. Although the mechanisms of carcinogenesis are uniquely influenced by various risk factors such as environment, nutrition, and lifestyle, the primary premise of precision medicine is that individuals with similar molecular pathology may have similar etiologic mechanisms and similar responses to drugs and other interventions [121]. While the focus of tumor pathology research has been devoted to tumor cells and the surrounding tumor microenvironment, the influence of other exogenous and endogenous factors (including but not limited to environmental, microbial, nutritional, lifestyle, and socioeconomic) on the pathogenic process should also be investigated to improve our understanding of tumors [122, 123]. It has been found that the intra-tumor microbiota is highly organized in particles that can promote cancer progression [124]. Poor diet can also have an impact on microbial composition and thus potential impact on cancer prevention and treatment [125]. Studies have also suggested that reduced production of short-chain fatty acids (SCFA) may disrupt the normal bacterial ecology of intestinal microbes, leading to malnutrition and increased glycolysis, increased lactate metabolism, and ultimately the development of colorectal cancer (CRC) [126]. Molecular pathology epidemiology (MPE) will play a very important role in the study of these factors and can be used not only to assess definitive clinical outcomes but also to predict intermediate biomarkers for future full-blown disease [127, 128]. This also provides a reliable research paradigm for targeting lactate metabolic pathways for tumor therapy.
Metabolic reprogramming is how cancer cells metabolize to obtain more energy faster and more often, especially when proliferating and metastasizing. Metabolic inhibition is a therapeutic approach to control the energy uptake and metastasis of cancer cells. Cancer cells undergo aerobic glycolysis, which in turn leads to an exuberant lactate metabolic pathway; therefore, targeted small molecule inhibitors of the lactate metabolic pathway are increasingly being investigated and explored, but only a small number of inhibitors have entered clinical trials and are not yet formally used widely in patients. Analysis of lactate metabolisms in cancer cells, such as the interaction between the tumor microenvironment and benign or malignant tumor cells, the metabolic profile of source tissue associated closely with the cancer cells, the metabolic changes between primary and metastatic tumors, and the impact on immune molecules can all help to guide drug development and research. MPE may also provide a new research paradigm for targeting lactate metabolic pathways for the treatment of tumors. LDHA, MCTs, or both are ideal targets for cancer treatment. However, it may be impractical to treat cancer with a single drug, this is because lactate metabolism-driven cancer cells, although important, are only a subpopulation and do not represent the whole spectrum of cancer cells, and chemotherapy or radiotherapy combined with lactate metabolism-targeting drugs would be a promising therapeutic method.
anti-PD-1: anti-programmed cell death protein 1
ATP: adenosine triphosphate
CD: cluster of differentiation
CSCs: cancer stem cells
DIDS: 4,4’-diisothiocyanatostilbene-2,2’-disulfonic acid
EMT: epithelial-mesenchymal transformation
GF: galloflavin
GPR81: G-protein-coupled receptor
HCC: hepatocellular carcinomas
hCINAP: human coilin interacting nuclear ATPase protein
HIF-1: hypoxia-inducible factor-1
LDH: lactate dehydrogenase
LDHA: lactate dehydrogenase A
MCT: monocarboxylate transporter
NADH: nicotinamide adenine dinucleotide
NF-κB: nuclear factor-kappa B
NHI: N-hydroxyindole
NHI-Glc-2: N-hydroxyindole-glucose conjugates-2
pHLIP: pH low insertion peptide
ROS: reactive oxygen species
TCA cycle: tricarboxylic acid cycle
VEGF/VEGFR: vascular endothelial growth factor/vascular endothelial growth factor receptor
WX, XL, and YZ: Writing—original draft. MZ and ML: Writing—review & editing, Supervision.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work is supported by the
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

