 Review
Review

Affiliation:
PanTherapeutics, CH1095 Lutry, Switzerland
Email: lundstromkenneth@gmail.com
ORCID: https://orcid.org/0000-0002-0580-5209
Explor Med. 2023;4:670–687 DOI: https://doi.org/10.37349/emed.2023.00168
Received: March 16, 2023 Accepted: April 19, 2023 Published: October 10, 2023
Academic Editor: Ragusa Marco, Università degli Studi di Catania, Italy
The article belongs to the special issue RNA World in Health and Disease
Self-replicating RNA viruses such as alphaviruses, flaviviruses, paramyxoviruses, and rhabdoviruses have been engineered as expression vectors for vaccine development. The prominent feature of self-replicating RNA viruses is their RNA-dependent RNA polymerase activity, which generates massive self-amplification of RNA in the cytoplasm of infected host cells leading to extreme levels of transgene expression. Infectious diseases have been targeted by overexpression of surface proteins of pathogens as antigens for vaccine development. Moreover, overexpression of tumor-associated antigens and immunostimulatory genes has been the basis for cancer vaccines. Proof-of-concept of robust antigen-specific immune responses and protection against challenges with lethal doses of infectious agents have been demonstrated. Likewise, vaccine development against various cancers has elicited strong immune responses and resulted in tumor regression and eradication, cure, and prolonged survival in animal tumor models. Good safety and immune responses have been achieved in clinical trials. The ERVEBO® vaccine, based on the vesicular stomatitis virus, has been approved for immunization against the Ebola virus disease.
The classic approach for vaccine development against viral infections has relied on the application of killed or live-attenuated whole viruses [1]. The progress in recombinant protein expression in the 1980s and 1990s facilitated the development of protein subunit and peptide vaccines [2]. It also opened the possibility of utilization of viral and non-viral vectors for efficient delivery and overexpression of antigens suitable for eliciting robust antibody responses for vaccine development against infectious diseases and cancers [3]. The recent coronavirus disease 2019 (COVID-19) pandemic has taken vaccine development to unprecedented levels related to both viral vectors and nucleic acid-based vaccines [4]. Moreover, the application of viral vectors for the expression of tumor-associated antigens (TAAs) and immunostimulatory genes has been an attractive alternative for cancer vaccine development [5–7]. Additionally, therapeutic and prophylactic efficacy has been obtained by viral vector-based delivery of anti-tumor, and suicide genes [8].
A variety of viral vectors have been utilized for vaccine development against infectious diseases and cancers as described before [9]. For this reason, some key findings are briefly presented below. For example, adenovirus (Ad)-based vaccines such as ChAdOx1 nCoV-19 [10], Ad26.COV2.S [11], and Ad5S-nb2 [12] have demonstrated robust immune responses and good vaccine efficacy against severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) as well as being granted emergency use authorization (EUA) for mass vaccinations in different countries [13–15]. In the case of cancer therapy, Ad vectors demonstrated complete regression of pancreatic neuroendocrine tumors (PNETs) in mice [16]. The administration of the herpes simplex virus (HSV)-HF10 resulted in substantial breast tumor regression and prolonged survival in mice [17] and in partial response (PR) in three patients, stable disease (SD) in four patients and in progressive disease (PD) in two patients with pancreatic cancer in a phase I trial [18]. In the context of retroviruses, retroviral replicating vector (RRV) Toca 511 was administered to patients with recurrent or progressive high-grade glioma (HGG) in a phase I study, showing statistically prolonged survival [19]. However, in a phase II/III trial in HGG patients, no prolongation of overall survival was detected [20]. In a phase II/III trial, the administration of HSV talimogene laherparepvec (T-VEC) vector expressing the granular macrophage-colony stimulating factor (GM-CSF) demonstrated good tolerance and promising therapeutic efficacy in melanoma patients [21], which has contributed to the approval of HSV T-VEC for the treatment of advanced melanoma in the US, Europe, and Australia [22].
In this review, the focus is solely on vaccine development by applying self-replicating RNA viruses. As detailed reviews have recently been published on both vaccine development against infectious diseases and cancers [23, 24], the goal here is to discuss the different types of viral vectors used and through the presentation of limited examples describe findings from both preclinical studies in animal models and evaluation in human volunteers and in cancer patients in clinical trials.
Among self-replicating RNA viruses, alphaviruses, flaviviruses, paramyxoviruses, and rhabdoviruses have been engineered as expression vector systems. However, while the single-stranded RNA (ssRNA) genomes of alphaviruses [25] and flaviviruses [26] are of positive polarity, paramyxoviruses [27] and rhabdoviruses [28] possess negative sense ssRNA genomes. The polarity plays an essential role in the engineering and utilization of self-replicating RNA virus vectors as described below and illustrated in Figure 1. The positive sense ssRNA is directly translated in the cytoplasm, which is substantially enhanced by the formation of the replicase complex (RC) after processing of the non-structural polyprotein expressed from the non-structural protein 1–4 (nsP1–4) genes resulting in an estimated 106 copies of subgenomic RNA per cell [29].
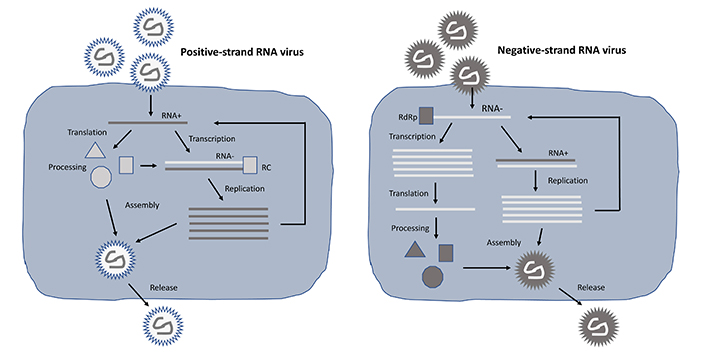
Lifecycle of positive- and negative-stranded self-replicating RNA viruses. In the case of positive-stranded RNA viruses (left), translation from the RNA plus strand can occur directly, whereas for negative-stranded RNA viruses (right) a plus strand copy needs to be synthesized first. In both cases, massive RNA replication takes place, RNA genomes are packaged into nucleocapsids, and viral particles are released from cells by budding. RdRp: RNA-dependent RNA polymerase
Alphavirus expression systems have been engineered to allow the utilization of recombinant viral particles, RNA replicons, and DNA replicons for the Semliki Forest virus (SFV) [30], Sindbis virus (SIN) [31], and Venezuelan equine encephalitis virus (VEE) (Figure 2) [32]. In the case of recombinant viral particles, both replication-deficient and replication-proficient systems have been developed. Briefly, the generation of replication-deficient particles takes place from co-electroporation or co-transfection of an alphavirus vector and a helper vector. The expression vector carries the nsP1–4 genes responsible for RC formation for massive viral RNA self-replication. The gene of interest (GoI) is introduced downstream of the highly efficient SFV subgenomic promoter (26S) located at the 3’ end of the nsP4 gene. As the alphavirus structural genes are deleted from the expression vector, they are provided in trans from the helper vector. RNA molecules from both expression and helper vectors are replicated in transfected host cells, but as the packaging signal is located in the nsP2 gene for SFV [33] or in the nsP1 gene for SIN and VEE [34], only RNA from the expression vector will be packaged into recombinant viral particles. These so-called suicide particles show the same broad host range as wild-type alphaviruses, but due to the absence of any viral structural genes, no further viral progeny will be produced. However, alphavirus suicide particles generate extreme levels of transgene expression. As no virus progeny is produced and the viral ssRNA is quickly degraded, active antigen production lasts for approximately 5–7 days in vivo, which is actually ideal for immunization with no risk of long-term extrachromosomal presence or chromosomal integration. In the case of replication-proficient alphavirus particles, transgene expression can be achieved by the introduction of the GoI either downstream of the non-structural genes or alternatively after the structural genes driven by a second 26S. Transfection of in vitro transcribed full-length alphavirus RNA and the GoI into mammalian host cells will generate fully replication-proficient particles. Importantly, as the generated alphavirus particles are both infectious and capable of generating viral progeny, the biosafety requirements are tighter compared to replication-deficient particles.
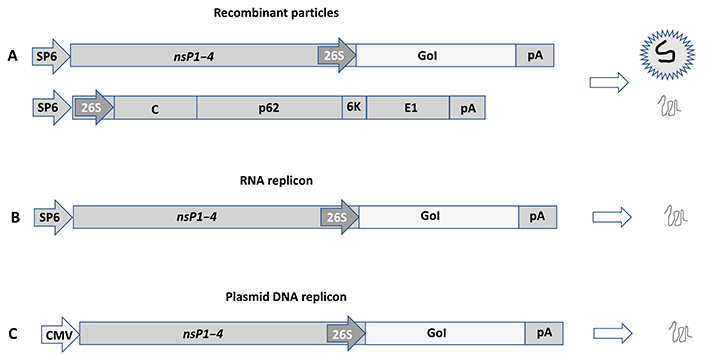
Alphavirus expression systems illustrated by SFV vectors. (A) Recombinant viral particles. Co-electroporation/transfection of in vitro transcribed RNA from the expression vector carrying the nsP1–4 genes and the GoI and the helper vector providing the SFV E genes (C-p62-6K-E1) into mammalian host cells generates replication-deficient recombinant viral particles and expression of the GoI; (B) RNA replicons. In vitro transcribed RNA from the expression vector can be transfected to host cells or injected in vivo for expression of the GoI; (C) plasmid DNA replicons. Transfection of the plasmid DNA replicon vector, where the SP6 RNA polymerase promoter is replaced by a CMV promoter generated expression of the GoI. SP6: SP6 RNA polymerase promoter; pA: polyadenylation signal; C: capsid; p62: precursor of E2 and E2 envelope proteins; 6K: 6K protein; E1: E1 envelope protein; CMV: cytomegalovirus
In the context of RNA-based delivery, in vitro transcribed RNA from the alphavirus expression vector containing the GoI can be directly administered to cell cultures or in vivo. However, the sensitivity of ssRNA to degradation [35] has triggered the encapsulation of RNA molecules in nanoparticles to improve their delivery, stability, and extended circulation [36]. Alternatively, DNA-based vectors can be used. In this context, the SP6 or T7 RNA polymerase promoter (T7) in alphavirus expression vectors has been replaced by a mammalian host cell compatible eukaryotic RNA polymerase type II promoter such as CMV [37]. The attractive features of DNA-based administration include their easy handling and rapid and inexpensive manufacturing. However, the application of these DNA replicons is dependent on efficient transfection methods, which are inferior to viral delivery. Another issue relates to the need for plasmid DNA to translocate to the nucleus for mRNA transcription followed by its transport to the cytoplasm for translation, which has substantially reduced transgene expression [38]. Different methods such as electroporation [39], gene gun [40], liposome [41], and polymer-based [42], nanoparticle formulation have been developed as well as the application of nuclear localization signals for optimized DNA [43].
In the case of flaviviruses, the Kunjin virus (KUN) has been subjected to the engineering of an expression system [44] (Figure 3). Generally, the GoI is introduced between the first 60 nucleotides of the C20 core protein and the last 22 codons of the E2 envelope protein [44]. As the GoI is expressed as part of a larger polyprotein, an FMDV2A protease sequence was engineered into the KUN vector for removing any foreign flanking regions [45]. Moreover, the generation of a packaging cell line has facilitated KUN particle production [46]. Other flaviviruses such as West Nile virus (WNV) [47], yellow fever virus (YFV) [48], Dengue virus (DENV) [49], tick-borne encephalitis virus (TBEV) [50], and Zika virus (ZIKV) [51] have been engineered. Additionally, expression vector systems have been constructed for the bovine viral diarrhea virus (BVDV) by insertion of the green fluorescent protein (GFP) reporter gene between the N(pro) and C genes in the non-cytopathic type-1 BVDV strain SD1 [52]. The classical swine fever virus (CSFV) has also been engineered as a bicistronic vector containing the GFP gene [53].

Flavivirus expression vectors exemplified by KUN. (A) RNA replicon vector. The GoI is introduced between the first 60 nucleotides of the C20 core protein and the last 22 codons of the E2 envelope protein in the KUN genome; (B) DNA replicon vector. The SP6 RNA polymerase promoter has been replaced by a CMV promoter. UTR: untranslated region; U: ubiquitin; F: foot-and-mouth disease virus 2A protease; ns1–5: non-structural protein genes 1–5; HDVr: hepatitis D virus ribosome
In contrast to alphaviruses and flaviviruses, the negative-stranded ssRNA viruses such as rhabdoviruses the RNA self-replication is based on the large protein (L) gene coding for the RdRp and its cofactor in the phosphoprotein (P) gene [28]. Expression vectors for vesicular stomatitis virus (VSV) have been engineered by insertion of the GoI between the glycoprotein (G) and the L genes for the generation of recombinant VSV particles or alternatively by replacing the VSV G by the GoI for pseudovirus production (Figure 4A) [54, 55]. Pseudovirus VSV particles can be produced from plasmid DNA in transfected mammalian cells followed by infection with the VSV G-complemented VSVΔG, which can be used for infection of mammalian host cells leading to transgene expression but no generation of viral progeny [54]. Fully infectious VSV particles can be produced by the infection of mammalian producer cells with VSV G-complemented recombinant virus replacing the VSV G [55]. Reverse genetics has been applied for VSV based on vaccinia virus vectors [56]. Moreover, a vaccinia virus-free system has been engineered to allow the production of infectious VSV particles from DNA [57]. In the context of large-scale production, an inducible VSV L cell line has been engineered for the packaging of recombinant VSV particles [58]. In the context of rabies virus (RABV), expression vectors have been engineered, where the GoI is introduced between the nucleoprotein (N) and P genes, or alternatively, between the G and L genes (Figure 4B) [59]. A reverse genetics system has also been constructed for RABV based on vaccinia virus-free vectors [60].
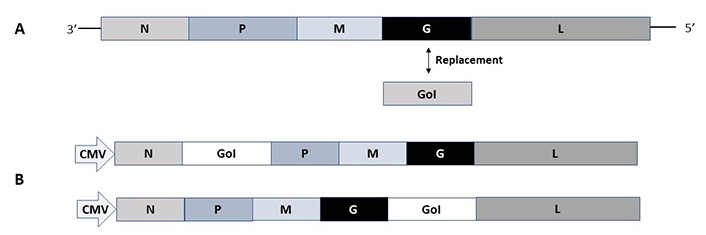
Rhabdovirus expression vectors. (A) VSV pseudovirus vector. The VSV-G protein gene is replaced by the GoI; (B) RABV vectors. The GoI can be inserted in the RABV vector either between the N and P genes or the G and L genes. M: matrix protein
Among the negative strand ssRNA paramyxoviruses, measles virus (MV) vectors have been engineered for the insertion of the GoI between the P and the M genes or alternatively between the hemagglutinin (HA) gene and the L genes (Figure 5) [27]. Packaging systems and helper cell lines have been engineered for MV, which will allow the rescue of replicating MV from plasmid DNA vectors [61, 62].
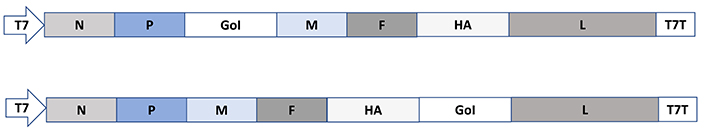
Paramyxovirus expression vectors exemplified by MV. In MV vectors, the GoI is introduced either between the P and M genes or the HA and L genes. F: fusion protein; T7T: T7 terminator
Self-replicating RNA viruses have been used for a large number of studies in preclinical animal models and a limited number of clinical trials. This is also reflected in the very low number of approved vaccines, so far. In this section, examples of applications of self-replicating RNA viruses from preclinical studies to clinical trials are described and summarized in Table 1 and Table 2.
Examples of preclinical studies and clinical trials on vaccines developed for infectious diseases
| Disease | Vector | Findings |
|---|---|---|
| CHIKV | MV-CHIKV | Protection against CHIKV challenges in macaques [63] |
| MV-CHIKV | Seroconversion in 100% of volunteers in phase I [64] | |
| MV-CHIKV | Good safety, and strong immune responses in phase II [65] | |
| EVD | VSV-EBOV-GP | Protection against two EBOV strains in macaques [66, 67] |
| VSV-EBOV-GP | Good safety and tolerability in phase I volunteers [68] | |
| VSV-ZEBOV | High vaccine efficacy, and protection in phase III [69] | |
| VSV-ZEBOV | High vaccine efficacy in phase III volunteers [70] | |
| VSV-ZEBOV | Approval of ERVEBO® by the FDA and EMA [71] | |
| ZIKV | MV-ZIKV-E | ZIKV-specific Abs, protection against ZIKV of mice fetus [72] |
| MV-ZIKV-E | Despite the completion of phase I, no results are available [73] | |
| MV-ZIKV-RSP-E | Phase I study in progress [75] | |
| MV (TMV-083) | Th1-biased Ab and T cell responses in mice [76] | |
| MV (TMV-083) | Weak immune responses, phase I trial discontinued [77, 78] | |
| COVID-19 | VSV-SARS-CoV-2 S | Protection against SARS-CoV-2 challenges in mice [79] |
| VSV (V590) | Weak immune responses, phase I trial discontinued [80, 81] | |
| VSVΔG-SARS-CoV-2 S | Protection against SARS-CoV-2 in hamsters [82] | |
| VSVΔG-SARS-CoV-2 S | Phase I/II study in progress [83] | |
| LNP-nCoVsaRNA | Strong SARS-CoV-2 specific Ab responses in mice [86] | |
| LNP-nCoVsaRNA | Safe but not 100% seroconversion in phase I [87] |
Ab: antibody; CHIKV: chikungunya virus; EBOV-GP: Ebola virus-glycoprotein; EMA: European Medicines Agency; EVD: Ebola virus disease; FDA: US Food and Drug Administration; LNP: lipid nanoparticle; LNP-nCoVsaRNA: LNP-VEE-SARS-CoV-2 S RNA; SARS-CoV-2 S: SARS-CoV-2 spike; ZEBOV: EBOV-Zaire strain
Examples of preclinical studies, clinical trials, and approved cancer vaccines
| Disease | Vector | Findings |
|---|---|---|
| Breast | VEE-HER-2 | Prevention of tumor growth and death in mice [89] |
| VEE-HER-2 | Good tolerance, PR in 1 patient, SD in 2 patients in phase I [90] | |
| VEE-HER2 | Combination with pembrolizumab (ICB) in phase II [88] | |
| Cervical | SFV-E6/E7 | Tumor regression or complete eradication in mice [91] |
| SFV-E6/E7 (Vvax001) | Good tolerability, HPV-specific responses in phase I [92] | |
| Colon | VEE-CEA | Anti-tumor activity in mice with MC38-CEA tumors [93] |
| VEE-CEA | Immune responses, prolonged survival in phase I patients [94] | |
| Ovarian | MV | Anti-tumor activity in mice with ovarian tumor xenografts [95] |
| MV | SD in 67% of RROC patients in phase I [96] | |
| Prostate | VEE-PSMA | Robust PSMA-specific immune responses in mice [97] |
| VEE-PSMA | No clinical changes in CRPC patients in phase I [98] | |
| Pancreatic | VEE-CEA | CEA-specific T-cell responses in transgenic mice [99] |
| VEE-CEA | Ab and T-cell responses, prolonged survival in phase I [99] | |
| Various | ChAd + VEE neoAg | Clinical benefits in cancer patients in phase I/II [100] |
CEA: carcinoembryonic antigen; ChAd: chimpanzee Ad; CRPC: castrate-resistant prostate cancer; E6/E7: human papillomavirus envelope proteins 6 and 7; HER-2: human epidermal growth factor receptor 2; HPV: human papillomavirus; ICB: immune checkpoint blockade; MC38: mouse colon cancer cell; neoAg: neo-antigen; PSMA: prostate-specific membrane antigen; RROC: refractory recurrent ovarian cancer
In the case of infectious diseases, MV-based particles were applied for the expression of the CHIKV C and envelope (E) protein genes, which elicited strong immunogenicity and provided protection in immunized macaques [63]. Furthermore, the MV-CHIKV particles were subjected to a phase I trial in healthy volunteers [64]. After a single dose of MV-CHIKV, the seroconversion rate was 44–92%, and after a second dose, it reached 100% in all vaccinated volunteers. Moreover, in a phase II follow-up study, strong neutralizing antibodies were obtained without any serious adverse events registered [65].
Related to EVD, VSV particles have been used for the expression of the EBOV-GP gene, which protected immunized macaques against challenges with the EBOV-Makona [66] and ZEBOV [67] strains. In the context of clinical evaluation, VSV-EBOV-GP showed good safety and tolerability in a phase I study in healthy volunteers [68]. Moreover, a good safety profile and excellent vaccine efficacy were demonstrated for the VSV-ZEBOV vaccine in 7,651 vaccinated individuals in a phase III trial in Guinea [69]. The VSV-ZEBOV vaccine efficacy was further confirmed in another phase III study in Guinea and Sierra Leone [70]. Approval of VSV-ZEBOV against EVD under the brand name ERVEBO® was granted in 2020 [71].
In the case of ZIKV vaccine development, the ZIKV precursor membrane (prM) protein and the soluble form of the envelope protein (ZIKV-sE) were expressed from MV particles, which elicited E-protein specific and neutralizing antibodies in mice [72]. Moreover, challenges with ZIKV resulted in reduced virus load and prevented fetus infection in an allogenic mouse pregnancy model. Two clinical studies on MV-ZIKV have so far been conducted. Although a dose-finding phase I trial has been carried out in 48 volunteers with the MV-ZIKV (V186-001) vaccine candidate, no results have been published yet [73]. Additionally, the safety and immunogenicity of the MV-ZIKV-RSP (V187-001) vaccine candidate, similar to MV-ZIKV-E but with a different insert position [74], is under investigation in phase I study in volunteers in Austria [75].
Due to the COVID-19 pandemic, massive efforts have been dedicated to the development of vaccines against SARS-CoV-2 resulting in EUA of vaccines based on Ad vectors and nucleic acids [13, 14]. Self-replicating RNA viruses have also been utilized. For example, MV-based expression of the SARS-CoV-2 S protein elicited strong Th1-biased antibody and T-cell responses in mice [76]. However, the MV-SARS-CoV-2 S (TMV-083) vaccine candidate induced disappointingly weak immune responses in volunteers in a phase I clinical study [77], which resulted in the termination of the trial [78]. The SARS-CoV-2 S gene has also been expressed from VSV vectors resulting in neutralizing antibody responses and protection against SARS-CoV-2 challenges in mice [79]. Moreover, the VSV-SARS-CoV-2 S (V590) vaccine candidate has been evaluated in 252 volunteers in a phase I trial [80]. Although good safety and tolerability profiles were achieved, the immune responses were weaker than those seen in COVID-19 patients, causing the discontinuation of the trial [81]. The replication-competent VSVΔG vector, where the VSV G protein was replaced by the SARS-CoV-2 S protein gene, was engineered [82]. Immunization of Syrian golden hamsters with VSVΔG-SARS-CoV-2 S induced strong neutralizing antibody responses and provided protection against challenges with SARS-CoV-2. Furthermore, the VSVΔG-SARS-CoV-2 S vaccine candidate has been subjected to a phase I/II trial where the safety and vaccine efficacy will be monitored for 12 months [83].
Another application of self-replicating RNA viruses comprises RNA-based delivery. Due to the sensitivity of degradation of ssRNA, the VEE-SARS-CoV-2 S RNA has been formulated in LNPs. However, LNP formulations of self-replicating RNA molecules have proven more difficult than the smaller-size synthetic mRNA [84]. The application of the three FDA-approved ionizable lipids (6Z,9Z,28Z,31Z)-heptatriacont-6,9,28,31-tetrqene-19-yl 4-(dimethylamino) butanoate (MC3), 6-((2-hexyldecanoyl)oxy)hexyl-N-(4-hydroxybutyl)hexan-1-aminium (ALC-0315), and 9-heptadecanyl 8-{(2-hydroxyethyl)[6-oxo-6-(undecyloxy)hexyl]amino}octanoate (SM-102) for LNP formulations contributed to achieving maximal transgene expression. Another aspect to consider for LNP formulations is its effect on the acute systemic cytokine responses to self-replicating RNA [85]. For example, LNP formulations that induced stronger cytokine responses also elicited enhanced antibody responses showing an impact on the adaptive immune response after vaccinations with LNP-formulated self-replicating RNA. Intramuscular immunization of BALB/c mice with LNP-nCoVsaRNA induced strong dose-dependent SARS-CoV-2 specific antibody responses and neutralization of both pseudovirus and wild-type virus [86]. The LNP-nCoVsaRNA vaccine candidate has been subjected to the first-in-human dose-ranging phase I trial [87]. Compared to conventional mRNA-based vaccines, substantially lower doses of 0.1 to 10 μg of LNP-nCoVsaRNA were administered to 192 healthy volunteers. The immunization was safe with no serious adverse events detected. However, although seroconversion was obtained, it did not reach 100% and modifications are required to optimize humoral responses.
In the context of cancer vaccines, examples from studies in animal tumor models and clinical trials [88] are presented and summarized in Table 2. For example, VEE particles expressing the HER-2/neuroblastoma (neu) gene significantly inhibited or prevented the growth of HER-2/neu-expressing mouse breast tumor cells in mice after injection into mammary tissue or after intravenous administration [89]. Moreover, tumor formation and death in mouse mammary tumor virus (MMTV)-C-neu transgenic mice were completely prevented. VEE particles expressing the extracellular domain (ECD) and transmembrane (TM) domains of HER-2 have been subjected to a phase I study in patients with stage IV HER-2 overexpressing breast cancer, which showed good tolerance and resulted in PR in one patient and continued SD in two other patients [90]. A phase II trial in combination with ICB has been initiated with pembrolizumab in women with advanced HER-2+ breast cancer [88]. Three patients received combination therapy, which was well tolerated and did not cause any dose-related toxicity. Moreover, enhanced lymphocyte infiltration in tumors and increased infiltration of both CD4+ and CD8+ T-cells and B-cells were observed.
In the case of cervix cancer caused by the HPV, vaccines have been developed against the HPV oncoproteins E6 and E7. For example, expression of the E6/E7 fusion protein from SFV vectors generated tumor regression and complete eradication of established tumors in immunized C57BL/6 mice [91]. Moreover, the Vvax001 vaccine candidate, based on an SFV vector containing the translation enhancement signal from the SFV C gene expressing the HPV E6/E7 fusion protein, has been subjected to a phase I study in 12 cervical intraepithelial neoplasia patients [92]. High safety and tolerability were seen, and HPV-specific immune responses were obtained in all 12 patients in the study.
Related to colon cancer, VEE particles expressing the CEA elicited immune responses and generated anti-tumor activity in mice implanted with MC38-CEA-2 colon tumors, which could be further enhanced by co-immunization with VEE-CEA and VEE particles expressing interleukin-12 (VEE-IL-12) [93]. In a clinical setting, VEE-CEA particles were evaluated in stage IV colorectal cancer in a phase I trial [94]. Moreover, stage III colorectal cancer patients were included in the study. Immunization elicited antigen-specific immune responses and also resulted in prolonged overall survival.
In the context of ovarian cancer, MV vectors were applied for the expression of CEA and the sodium iodide symporter (NIS) in mice with implanted SKOV3ip.1 xenografts [95]. The combination of MV-CEA and MV-NIS showed superior anti-tumor activity compared to treatment with either MV-CEA or MV-NIS alone. In a phase I trial, the MV-CEA vector was evaluated in patients with taxol and RROC [96]. Intraperitoneal MV-CEA administration was safe and well tolerated. The dose-dependent biological activity resulted in SD in 14 out of 21 RROC patients.
VEE particles have been applied for the expression of the PSMA as a vaccine candidate for prostate cancer [97]. Immunization of BALB/c and C57BL/6 mice elicited strong PSMA-specific immune responses. Furthermore, patients with progressive CRPC have been subjected to VEE-PSMA immunizations in the first-in-human phase I trial [98]. The immunization was well tolerated, but no significant clinical changes such as those measured by PSA or circulating tumor cells were detected.
In the context of pancreatic cancer, VEE particles expressing the CEA gene were administered to transgenic C57BL6-CEA mice, which elicited a high magnitude of CEA-specific T-cell responses [99]. A phase I clinical trial has been conducted with VEE-CEA particles in pancreatic cancer patients [99]. Clinically relevant antibody and T-cell responses were seen in patients, which were intramuscularly injected with VEE-CEA particles. The overall survival was also improved.
Finally, self-replicating RNA has been used in a prime-boost regimen, where the primary immunization has taken place with ChAd vectors followed by self-replicating VEE RNA expressing neoepitopes from autologous tumors [88]. Different types of cancers such as colorectal cancer, gastroesophageal adenocarcinoma, and non-small cell lung cancer have been evaluated in a phase I/II clinical trial according to the ChAd-VEE RNA prime-boost immunization strategy [100]. No dose-related toxicity was detected in any of the 26 patients enrolled and only minor adverse events such as fever and reactions at the injection site were recorded. After prime immunization, neoantigen-specific T-cell responses were recorded, which further increased after the booster vaccination with self-replicating RNA. Clinically, a complete response (CR) was confirmed in one gastroesophageal cancer patient, SD in five patients, PD in eleven patients, and no measurable disease in two individuals.
The common factors for all vaccine development for both infectious diseases and cancers are the application of self-replicating RNA viral vectors and the overexpression of recombinant proteins used for antigen production. Moreover, the route of administration is of importance, where intramuscular injection is the most common approach. Below are summarized the findings from preclinical studies and clinical trials.
Preclinical studies have provided proof-of-concept for the vaccine establishment to demonstrate on a small scale as cost-effectively as possible that robust immune responses can be induced and in the case of infectious diseases protection against challenges with lethal doses of infectious agents can be achieved. In the case of cancer vaccines, demonstration of tumor growth inhibition, tumor eradication, and complete cure in animal models have been indications that expensive clinical trials can be initiated. Moreover, cancer vaccines are expected to provide protection against challenges with tumor cells in animal models. As described above and summarized in Table 1 and Table 2, these criteria have been reached both for infectious diseases and various cancers, which has allowed moving forward to clinical evaluation.
In the case of clinical assessment, relatively few studies have been conducted, so far. In quite a few cases of infectious diseases, relatively modest immune responses have been obtained in phase I and II trials in healthy volunteers, which is a clear indication of the less straightforward transition from animal studies to humans. For example, clinical evaluation of both MV- and VSV-based vaccine candidates against SARS-CoV-2 was discontinued in phase I despite promising results in animal models [78, 81]. In contrast, VSV-based vaccine development against EDV was highly successful, showing excellent vaccine efficacy in phase III studies [69, 70], leading to vaccine approval by both the FDA and the EMA [71].
The trend for cancer vaccines is similar. Several studies have demonstrated tumor inhibition [89], tumor eradication [93], and even long-term tumor-free survival [91] in different animal models. However, the transition to humans has resulted in less efficient tumor eradication, although PR, SD, and CR have been obtained in individual cases [90, 97].
Although plenty of engineering has been dedicated to the development of more efficient self-replicating RNA virus vectors, there is still room for improvement (Table 3). Related to vector development, several efforts have been made to specifically generate less cytotoxic alphavirus vectors by introducing point mutations in the non-structural protein genes [101, 102]. In the case of cancer vaccines and especially targeting existing tumors, this approach is not necessarily of advantage. Instead, the application of naturally occurring or engineered oncolytic viruses, which specifically replicate in tumor cells leading to their killing [103] might be a better approach. Recently, trans-amplifying SFV vectors, a split-vector derivative of self-replicating RNA vectors, have been engineered for the amplification of transreplicon RNA coding for antigens, which requires minimal amounts of RNA for vaccine development [104].
Potential vaccine improvement strategies and expectations
| Strategy | Approach | Expectations |
|---|---|---|
| Vector engineering | Mutations | Less cytotoxic vectors, prolonged transgene expression Tumor targeting Targeting APCs for improved immunogenicity |
| Antigens | taRNA | Use of minimal vectors, easier handling, and readiness |
| Oncolytic viruses | Tumor targeting and killing | |
| Pseudoviruses | Introduction of tumor targeting epitopes Introduction of immunogenic epitopes | |
| Protein and epitope optimization | Improved immune responses Combination of epitopes for a larger spectrum of immunogenicity | |
| Epitope selection | Identification of tumor-specific epitopes on cancer cells More potent personalized immune responses | |
| RNA delivery | Stabilization | Improved resistant to degradation, longer RNA half-life |
| Encapsulation | Improved stability, delivery, prolonged circulation in vivo | |
| Combination therapy | Prime-boost | Prime-boost vaccinations with Ad and other viral vectors |
| ICB, other drugs | Superior to self-replicating vector alone | |
| Process optimization | Vaccine dose | Optimization of applied dose |
| Route | Evaluation of administration route |
APCs: antibody-producing cells; taRNA: trans-amplifying RNA
In the context of antigen targets, the choice of regions or epitopes generating the best possible immune responses is important. In the case of infectious viruses, surface proteins easily recognized by the host cell immune system and/or structures involved in viral replication are desirable. For example, the replacement of the VSV-G protein with target surface proteins such as the SARS-CoV-2 S protein [82]. As epitopes have proven good as antigens, the combination of various epitopes can provide a larger spectrum against infectious viruses or their variants providing enhanced vaccine efficacy. Related to cancer vaccines, desirable antigen targets are those that are commonly expressed or uniquely overexpressed in cancer cells. More recently, personalized antigen approaches have become feasible, where neoepitopes derived from autologous tumors can potentially elicit more potent and personalized immune responses [100].
The delivery of viral vectors has been an area of optimization too. The introduction of targeting sequences by generating pseudoviruses or using strains or mutants capable of selectively targeting antigen-presenting cells (APCs) can improve and target delivery. Related to delivery, the route of administration and dose optimization are key factors for the improvement and optimization of immune responses and vaccine efficacy.
In the case of using RNA replicons instead of recombinant viral particles, prevention of ssRNA degradation has been addressed by stabilization of RNA by chemical modifications [35] and encapsulation of RNA replicons in nanoparticles to protect against degradation, enhance delivery, and prolong circulation of RNA in vivo [36].
Combination therapy of self-replicating RNA viruses has also been attractive. In this context, prime-boost vaccinations with other viral-based vaccines [100], other antiviral and anti-cancer drugs [105], and ICB drugs [88] have been tested.
In summary, self-replicating RNA viruses have been evaluated in preclinical animal models and clinical trials for vaccine development against infectious diseases and various cancers. Both viruses with ssRNA genomes of positive and negative polarity have been used. Proof-of-concept for vaccine candidates against both infectious diseases and cancers has been established in animal models. However, although good safety and tolerability profiles have been seen in clinical trials, similar levels of vaccine efficacy as seen in animal models have been difficult to reproduce in humans. Despite many years of experience, recent developments such as the approval of the VSV-based vaccine against EDV have encouraged further vaccine development efforts. Moreover, the recent success achieved for SARS-CoV-2 vaccines, not the least mRNA-based vaccines, has presented excellent opportunities for the development of novel vaccines based on self-replicating RNA viruses.
26S: Semliki Forest virus subgenomic promoter
Ab: antibody
Ad: adenovirus
CEA: carcinoembryonic antigen
ChAd: chimpanzee adenovirus
CHIKV: chikungunya virus
CMV: cytomegalovirus
COVID-19: coronavirus disease 2019
E6/E7: human papillomavirus envelope proteins 6 and 7
EBOV-GP: Ebola virus-glycoprotein
EVD: Ebola virus disease
FDA: US Food and Drug Administration
G: glycoprotein
GoI: gene of interest
HA: hemagglutinin
HER-2: human epidermal growth factor receptor 2
HPV: human papillomavirus
HSV: herpes simplex virus
ICB: immune checkpoint blockade
KUN: Kunjin virus
L: large protein
LNP: lipid nanoparticle
LNP-nCoVsaRNA: lipid nanoparticle-Venezuelan equine encephalitis virus-severe acute respiratory syndrome-coronavirus-2 spike RNA
M: matrix protein
MV: measles virus
N: nucleoprotein
neu: neuroblastoma
NIS: sodium iodide symporter
nsP1–4: non-structural protein 1–4
P: phosphoprotein
PR: partial response
PSMA: prostate-specific membrane antigen
RABV: rabies virus
RC: replicase complex
RROC: refractory recurrent ovarian cancer
SARS-CoV-2 S: severe acute respiratory syndrome-coronavirus-2 spike
SARS-CoV-2: severe acute respiratory syndrome-coronavirus-2
SD: stable disease
SFV: Semliki Forest virus
SP6: SP6 RNA polymerase promoter
ssRNA: single-stranded RNA
T7: T7 RNA polymerase promoter
VEE: Venezuelan equine encephalitis virus
VSV: vesicular stomatitis virus
ZEBOV: Ebola virus-Zaire strain
ZIKV: Zika virus
KL is the sole author of the manuscript and contributed to: Conceptualization, Writing—original draft, Writing—review & editing.
The author declares that he has no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 7055
Download: 63
Times Cited: 0
Hiromichi Sato ... Hideshi Ishii
Cristina Barbagallo ... Marco Ragusa
Azizul Haque, Anudeep B. Pant
