Review
Review

Affiliation:
1MM College of Pharmacy, Maharishi Markandeshwar (Deemed to be University), Mullana-Ambala 133207, Haryana, India
ORCID: https://orcid.org/0000-0001-5702-0932
Affiliation:
1MM College of Pharmacy, Maharishi Markandeshwar (Deemed to be University), Mullana-Ambala 133207, Haryana, India
ORCID: https://orcid.org/0000-0002-4784-6383
Affiliation:
2Department of Pharmaceutical Sciences, Maharishi Dayanand University, Rohtak 124001, Haryana, India
Affiliation:
3Department of Pharmacology, Indore Institute of Pharmacy, Indore 453331, Madhya Pradesh, India
Email: rupeshgautammmu@gmail.com
Affiliation:
4Department of Biosciences, Saveetha School of Engineering, Saveetha Institute of Medical and Technical Sciences, Chennai 602105, Tamil Nadu, India
ORCID: https://orcid.org/0000-0001-8867-7603
Affiliation:
1MM College of Pharmacy, Maharishi Markandeshwar (Deemed to be University), Mullana-Ambala 133207, Haryana, India
ORCID: https://orcid.org/0000-0003-2980-7791
Affiliation:
4Department of Biosciences, Saveetha School of Engineering, Saveetha Institute of Medical and Technical Sciences, Chennai 602105, Tamil Nadu, India
5Institutes for Systems Genetics, Frontiers Science Center for Disease-related Molecular Network, West China Hospital, Sichuan University, Chengdu 610065, Sichuan, China
6King Fahd Medical Research Center, King Abdulaziz University, Jeddah 21589, Saudi Arabia
7Department of Pharmacy, Faculty of Allied Health Sciences, Daffodil International University, Dhaka 1207, Bangladesh
8Enzymoics, 7 Peterlee Place, Novel Global Community Educational Foundation, Hebersham 2770, Australia
ORCID: https://orcid.org/0000-0003-0088-0565
Explor Med. 2024;5:89–100 DOI: https://doi.org/10.37349/emed.2024.00208
Received: October 14, 2023 Accepted: November 08, 2023 Published: February 28, 2024
Academic Editor: Hua Su, University of California, USA
Nerve cell death is the central aspect of human neurodegenerative disorders. Neuronal death in results leads to the onset of various human neurological disorders such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, and stroke. In developing neurons, apoptosis is assumed to provide a counterbalance to overexuberant cell replication. Numerous signals may induce apoptosis in neurons, such as the absence of neurotrophic factor support, increased levels of metabolic and oxidative stress, and overstimulation of glutamate receptors (leading to the calcium influx). Cell death and neurological disorders have been related to oxidative stress, which creates an imbalance between antioxidant defenses and free radical production. In this paper, a summary of the engrossment of oxidative stress, neuronal apoptosis, and mitochondrial dysfunction in neurodegenerative disorders has been discussed. Antioxidant therapy’s potential assistance for neurodegenerative illnesses in human beings is still up for dispute, despite encouraging pre-clinical research findings. One elucidation for this disparity could be the non-existence of an accurate way to assess oxidative stress in the brain. The explosion in research on apoptosis in neurodegeneration has stemmed from the conception that persuading neuronal apoptotic death may be crucial to the progression of a disease and that anti-apoptotic approaches may be useful in the prevention of neurodegenerative processes. A deeper understanding of the role that apoptosis plays in neurodegenerative processes will serve as the foundation for future research into the development of focused, effective treatment modalities.
Apoptosis is a phenomenon of programmed cell death, in which cells die sequentially at the end. The central nervous system is at fatal risk during both acute and chronic neurodegenerative disorders (NDs) [1]. The onset and prevalence of NDs such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and stroke lead to an alarming rate of increase in the number of people suffering from it. These disorders cause the degeneration of neurons oxidative stress and excessive cell-mediated proteolysis [2].
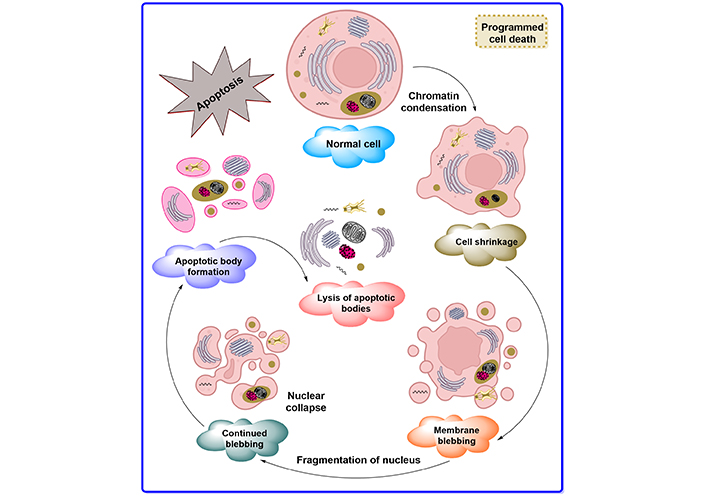
Degenerating neurons exhibit oxidative stress and excessive calcium-mediated proteolysis at the cellular level, which eventually results in apoptosis. In apoptosis, the cell and nucleus shrink and form blebbing, causing chromatin fragmentation, loss of adhesion, and rapid engulfment by phagocytosis [3]. Apoptosis also causes several biochemical changes along with morphological alterations such as DNA fragmentation, plasma membrane depolarization, lysosome permeability, and enhanced formation of reactive oxygen species (ROS) [4]. Induction of apoptosis is achieved by many methods depending on their pathways such as internal and external stimuli. Internal stimuli are triggered by activating intrinsic pathways such as abnormalities in the DNA and external stimuli are activated by cytokines such as extrinsic signaling pathways [5]. The description of normal apoptotic pathways by Guerin et al. [6] is depicted in Figure 1.
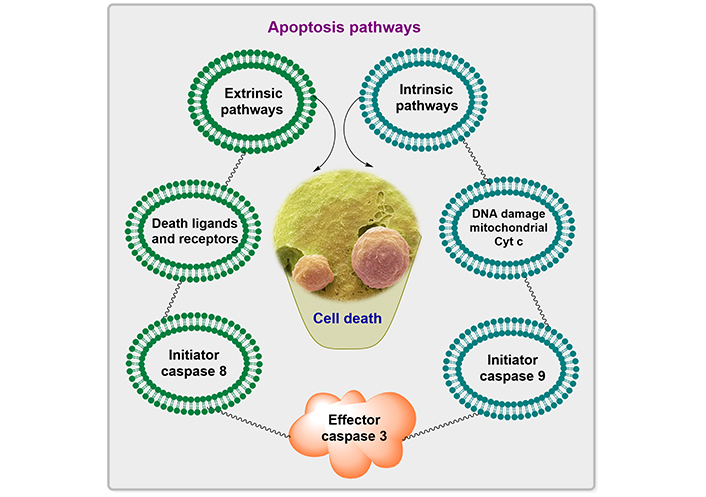
The apoptosis pathways elaborated in Figure 1 show different phases such as shrinking of the cell, membrane blebbing, the collapse of the nucleus, normal apoptotic body, and at the end leads to lysis of the apoptotic body. The descriptive pathways include two different methods of apoptosis such as intrinsic and extrinsic pathways for cascades, as depicted in Figure 2.
The non-receptor-mediated intracellular signals that are part of the intrinsic signaling pathway for programmed cell death include the numerous mitochondrial processes that start apoptosis [7]. The intrinsic pathway for programmed cell death can also be activated by damage to cellular DNA. Pro-apoptotic proteins set off caspases, which then regulate several cell death pathways. These proteins also enter the cellular nucleus where they trigger apoptosis and DNA damage. The removal of growth factor supplements from culture media, exposure to ultraviolet (UV) light, or cell stressors are examples of situations that can start the intrinsic pathway (for instance osmotic or metabolic). In the end, these circumstances result in DNA cell damage. When exposed to UV radiation, thymine base pairs adsorb UVB photon, which then joins together to generate pyrimidine dimers. ROS production may rise in response to growth factor withdrawal or other stressful circumstances. ROS can harm DNA through several mechanisms, including base modification [8].
The literature has reported that several diseases, including hypoxia, irreversible DNA damage, oxidative stress, and viral infections are triggered by intrinsic pathways, which will cause the alteration in the release of cytochrome from mitochondria, and form the complexes with the apoptotic protease activating factor-1 (Apaf-1) and procaspase [9].
Transmembrane death receptors, which are the products of the tumor necrosis factor (TNF) receptor gene, are a component of the extrinsic signaling cascade that results in apoptosis [10]. These receptors interact with extrinsic ligands to bind and translate intracellular signals that lead to cell death. It works on the active receptor and subsequently l binds procaspase-8 and TNF R1 (receptor type 1). Procaspase-8 is activated by this multiprotein complex, which starts apoptosis, or the process of cell death. Several caspases, which are proteases with specific biological targets, are involved in the signal transduction of the extrinsic pathway. The activation of caspases affects a variety of cellular processes and plays a significant part in the process that results in cell death [11].
A high level of ROS such as hydrogen peroxide radicals (•H2O2), hydroxyl radicals (•OH), and superoxide anion and an imbalance of antioxidant enzymes contribute to oxidative stress, which is a detrimental factor in memory dysfunctions [12]. Oxidative stress may be associated with cell membrane mutilation from lipid peroxidation, vicissitudes in protein structure and function, and structural damage to DNA. As the brain is one of the most metabolically active organs in the body, it is particularly susceptible to oxidative stress for the reasons listed below: First, its high oxygen demand (20% of the body’s oxygen intake). Second, the brain is full of redox-active metals like copper and iron, which constantly accelerate ROS production. Third, the membranes of brain cells contain substantial amounts of polyunsaturated fatty acids, that act as substrates for lipid peroxidation. Fourth, the brain has comparatively low levels of GSH, which functions as an endogenous antioxidant that helps in eliminating ROS [13].
It was evident from a research study that showed a prompt increase in oxidative stress levels as compared to neurologically impaired patients with aging factors. From the immunohistological examination, it was evident the increased protein oxidation, and lipid peroxidation in brain areas having neurofibrillary tangles and plaques. In AD, the cerebrospinal fluid had increased lipid peroxidation levels such as 4-hydroxynonenal [14]. Variations in the antioxidant enzymes catalase, Cu/Zn-superoxide dismutase (SOD), and Mn-SOD are caused by the increased oxidative stress in AD. These enzymes present in the neurons in the membrane cause lipid peroxidation with an increase in the secretion of toxic aldehydes such as 4-hydroxynonenal or malondi-aldehyde. These aldehydes disturb cellular Ca2+ homeostasis by interfering with the activity of the adenosine triphosphatases (ATPases), glucose, and glutamate transporters. Oxidative stress accumulation leads to the generation of ROS, which causes mitochondrial dysfunctions [15].
Nitric oxide (NO), which functions as a second messenger and can combine with superoxide anion to generate peroxynitrite [ONOO(-)], produces an interesting concept concerning oxidative stress in neurons. It has been demonstrated that ONOO(-) is neurotoxic and induces apoptosis in leukemic cells [16]. Several substances including vitamin E, estrogens, uric acid, and glutathione, are utilized to alleviate oxidative stress. They have been reported to prevent cellular injury, and membrane lipid or protein damage. Through the tumor protein (p53), poly(ADP-ribose) polymerase (PARP), or the ataxia telangiectasia mutant (ATM), ROS can cause DNA damage, which triggers lethal apoptotic signaling [17]. It was evident that oxidative stress in AD has scavenging free radicals which are used as a neuroprotective therapy for the disease. Moreover, prophylactic treatment with free radical scavengers such as vitamin E may reduce the risk of AD.
The alteration and dysfunction in the Ca2+ regulation have been reported in various clinical studies on patients with AD. The neurofibrillary changes are exhibited by calpain-II which gets activated by the activation of Ca2+-dependent protease [18]. The oxidative stress or greater intracellular calcium levels can activate cysteine proteases called caspases, such as calpain linked to neuronal cell death. Some conditions, such as cerebral ischemia, epilepsy, and traumatic brain injury, affect the calcium homeostasis of neurons and overstimulate capillaries [19].
To maintain cellular ion homeostasis and cell viability, calpain depends on a variety of substrates, including membrane receptors, kinases, phosphatases, cytoskeletal proteins, and transporters. Increased levels of calpains or the endogenous calpain inhibitor, calpastatin, have been connected to neuron loss in several neurodegenerative illnesses, including amyotrophic lateral sclerosis (ALS), PD, and AD [20]. Calpain inhibitors inhibited neuronal cell death in a variety of experimental scenarios, supporting the idea that calpains are crucial in the regulation of apoptosis and necrosis. The existence of higher Ca2+ binding proteins such as calretinin and calbindin, which are resistant to neurofibrillary degeneration, has been found to promote neurodegeneration in AD illness [21].
It is widely known that the cytotoxic buildup of intracellular calcium plays a crucial role in neuronal cell death as well as other stress conditions like toxicity. The cytotoxic intracellular Ca2+ is mediated via the stimulation of glutamate receptors. This strengthens the voltage-dependent Ca2+ channels, metabotropic glutamate receptors, and Ca2+ permeable α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors [22]. The lethal accumulation of Ca2+ in apoptotic cells is caused due to the over-activation of glutamate receptors. This fatal result arises due to the cleavage of ion pumps due to variability in the physiological environment which supports the pumping out of Ca2+ to maintain a stable state of cytosolic calcium [23]. On exposure to apoptotic damage, the caspases or calpains become activated and cleave the ion pumps. This lets the Ca2+ ion pump remove huge amounts of calcium that have accumulated in the cytosol. Because of this, calcium homeostasis is upset, which ultimately causes apoptotic signaling to change to necrosis.
It reveals that the signaling pathways of apoptosis are crucial for the development and control of illness. There are still a large number of proteins that are unknown and are involved in various signaling pathways. The purpose of that unidentified protein would be as a linker protein, which would be able to treat or stop the aberrant loss of neural cells in neurodegenerative illnesses. One of the key goals of current science is to understand the mechanism of proteins in the signaling pathways of apoptosis and anti-apoptosis [24].
Proteopathies are diseases in which some proteins develop aberrant structural characteristics, which are aggregated at various cellular levels and in tissues causing the interruption of their activities [25]. The proteopathies comprise several NDs in which peculiarly accumulated proteins seem to play a vital role [26]. When the misfolded or atypical proteins accumulate in the nuclear, cytoplasmic, and extracellular enclosures, it leads to disruption of organelle and synaptic dysfunction in the nervous system. Neuronal apoptosis in neurodegenerative illnesses can be induced via oxidative stress, disruption of calcium homeostasis, and metabolic impairment, at the cellular level. The anti-apoptotic or neuroprotective signaling pathways comprising neurotrophic factors, cytokines, and conditioning responses may offset the aging effects and hereditary predisposition in the experimental models of NDs [27].
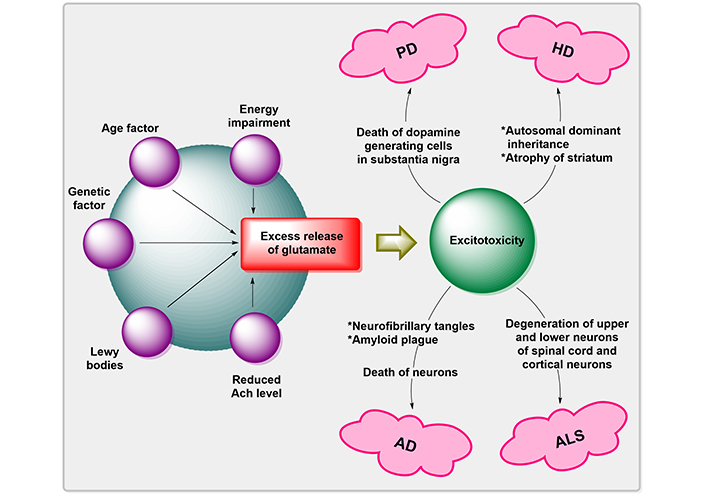
Free radical production, activation of the pathway for NO synthesis, disruption of cellular calcium homeostasis, and programmed cell death are all factors that contribute to the progression of neurodegenerative illnesses associated with glutamatergic dysfunction [28, 29]. These methods may induce mutilation of proteins, nucleic acids, and lipids, as well as open the pores of mitochondrial permeability transition, which may increase the ROS generation, energy failure degeneration, and proclamation of proapoptotic substances like Cyt c in the cytoplasm [30]. The overproduction of ROS and downregulation of antioxidant defense cause the death of NDs’ neuronal cells [31]. A variety of neurodegenerative illnesses are linked to inappropriate regulation of Wnt/β-catenin (WβC) signaling [32]. Degenerative nerve diseases are thought to contribute to the deterioration of various vital bodily functions, including memory, breathing, balance, speech, movement, and heartbeat [29]. The factors for the pathogenesis of neurogenerative disorders by Mehta et al. [33] are illustrated in Figure 3.
Apoptosis is a fundamental physiologic process that contributes to cellular homeostasis. Apoptosis (programmed cell death) is a type of physiological cellular suicide that occurs in several biological processes, including immune response, synaptogenesis, embryogenesis, and normal tissue and organ involvement. Normal brain development depends on programmed cell death. It affects the quantity and type of cells in the growing brain and spinal cord and is essential for the development of a functional neural network. In pathological circumstances, it is also partly accountable for physiological aging and the death of neurons linked to NDs [27]. Apoptotic cell death is well characterized via a sequence of morphological variations that comprise attenuation of nuclear and cytoplasmic compartments, condensation of chromatin, DNA degradation into fragments of oligonucleosomes, and an assortment of nuclear material into the vesicular apoptotic bodies [34].
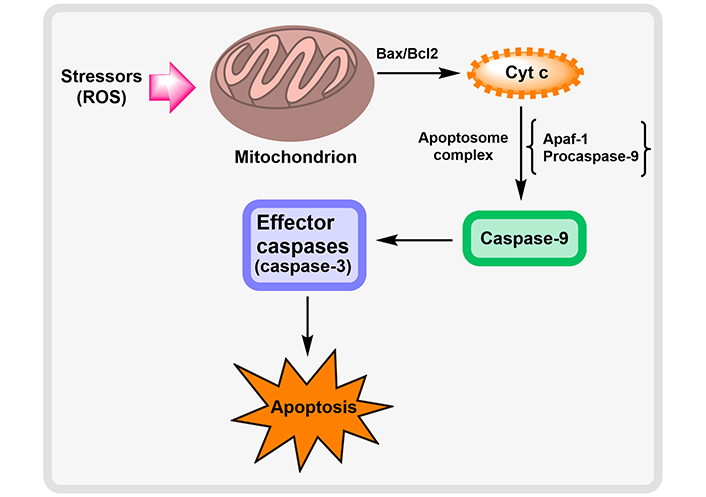
Neurodegenerative disorders cause neuronal death at the cellular level as a result of oxidative stress, DNA damage as a result of endoplasmic reticulum (ER) stress, and disruption of cellular calcium homeostasis. The apparent upsurge in the lifespan of individuals and the normal age of the population is tactlessly allied to a liberal intensification in the vast number of persons having NDs [35]. Apoptosis induction via pathogenic proteins of NDs by Yang et al. [36] is described in Figure 4.
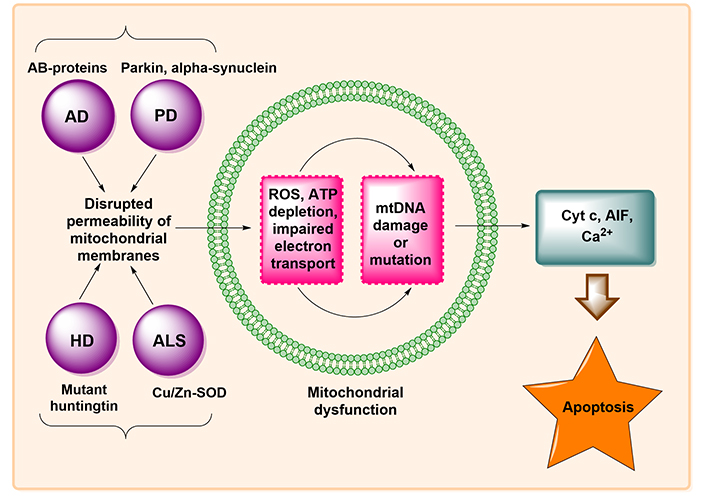
Induction of apoptosis by pathogenic proteins of NDs. ATP: adenosine triphosphate; mtDNA: mitochondrial DNA; AIF: apoptosis-inducing factor
Mitochondrial dysfunction plays a crucial role in the etiology of NDs. Numerous fragments of data suggest that NDs have profoundly impaired mitochondrial activity. The mitochondria are highly vulnerable to oxidative stress as it has the primary organelles for the production of ATP and are the main source of ROS generation [37]. The release of Cyt c, which occupies center stage as the starting point and mediator of the apoptosis cascade, is the major paradigm in mitochondria-mediated apoptotic signaling. Cyt c acts as a molecular adaptor that couples the procaspase-9 and Apaf-1 to produce the apoptosome complex during the activation of the death protease, caspase-3. Cyt c is released from the mitochondria into the cytosol either by non-specifically expanding and rupturing the mitochondria or by opening a particular channel or permeability transition pore in the mitochondrial membrane. Members of the pro- and anti-apoptotic subfamilies of B-cell lymphoma 2 (Bcl-2) are the guardians of channel-mediated Cyt c release within mitochondria. This ROS mechanism of apoptosis signaling is probably going to be crucial in reperfusion-induced neuronal cell death since post-ischemic mitochondria can produce more ROS [34]. This mitochondrial pathway of cellular apoptosis is illustrated in Figure 5.
The apoptosis induction in tumor cells affects the anti-tumor efficacy of radiotherapy and chemotherapy. However, the significant adverse impact of those pro-apoptotic therapies has prompted researchers to seek out other ways to promote tumor apoptosis [38]. Antioxidants or medications that block glutamate receptors or calcium channels have traditionally been used to reduce neuronal death in NDs. Anti-inflammatory medicines and caspase inhibitors are also being utilized. The biology of neural stem cells and the “anti-aging” enzyme telomerase have also been utilized to create two distinct strategies for avoiding apoptosis and replacing damaged or dead neurons. The six-base DNA repeat sequence (TTAGGG) is added to chromosomal ends by the telomerase reverse transcriptase (TERT) enzyme, which has recently attracted a lot of attention due to its purported “anti-aging” capabilities. TERT may assist an anti-apoptotic activity in neurons by suppressing the TERT expressions or activity, which makes them more vulnerable to oxidative and excitotoxic stimuli. Conversely, overexpression of TERT protects the neurons from apoptosis [39].
Acetylcholine (Ach) is a parasympathetic neurotransmitter that also enhances synaptogenesis in living cells to influence memory and learning in people. With three approved medications (donepezil, galantamine, and rivastigmine), acetylcholinesterase inhibitors (AChEIs) are the most well-researched medications and the first-choice treatment for NDs globally. Nevertheless, the AChEIs are less valuable in the advanced stages as they necessitate a higher dose which also upsurges the risk of adverse effects. Over the years, many potential candidates like metrifonate and tesofensine came under clinical trial as AChEIs but failed at certain stages mainly due to their safety profile. Altogether, this evidence indicates an awful and unmet necessity for the development of novel AChEIs [40].
Some potent apoptotic medications used for the treatment of NDs are described in Table 1.
Some potent apoptotic medications used for the treatment of NDs
| Sr. No. | Drugs | Mechanism of action | Model used | References |
|---|---|---|---|---|
| 1. | Minocycline | Inhibition of the generation of free radicals and the neurotoxicity caused by 6-OHDA in rat CGN. | Cultured rat CGN | [41, 42] |
| 2. | Rasagiline | This causes MAO-B inhibition, which may decrease oxidation and the buildup of free radicals while simultaneously increasing the amount of monoamines in the brain through the suppression of their catabolism. | Aged male C57Bl/6J mice | [43, 44] |
| 3. | Resveratrol | Present in red wine, grapes, and other fruits that inhibit oxidative stress via attenuation of 6-OHDA-induced oxidative damage and dopamine depletion. | 6-OHDA-induced PD rat model | [45, 46] |
| 4. | Coenzyme Q10 | By reducing oxidative stress and activating the P13K pathway, amyloid beta-induced neuronal cell death can be prevented. | Amyloid beta25–35-induced rat cortical neurons | [47] |
| 5. | Metformin | By lowering the transcription of Nrf2 and neurotrophic factors, the brain is protected against neurodegeneration without suffering cognitive impairment. | Aged male C57Bl/6J mice | [48] |
| 9. | Mucuna pruriens | Its treatment significantly reduced nitrite levels, lipid peroxidation, and iNOS expression, indicating that it lowers NO in PD. | PQ-induced PD mouse model | [49] |
| 10. | Ursolic acid | Reduction of oxidative stress in nigrostriatal tissue and enhancement of neurobehavioral functioning in PD patients. | MPTP-induced Parkinsonian mouse model | [50] |
Sr. No.: serial number; 6-OHDA: 6-hydroxydopamine; CGN: cerebellar granule neurons; MAO-B: monoamine oxidase B; P13K: phosphoinositide 3-kinase; C57Bl/6J: parentral substrain of mice; Nrf2: nuclear factor erythroid 2–related factor 2; PQ: paraquat; iNOS: inducible NO synthase; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
When subjected to excessive oxidative stress, neurons may respond adaptively to overcome the stress, or they may activate a programmed cell death pathway called apoptosis. Alterations in the ER and mitochondria, as well as the activation of cysteine proteases known as caspases, are the characteristics of apoptosis. There is mounting evidence that neurodegenerative diseases cause malfunction and neuronal death due to apoptotic biochemical pathways. Indeed, the notion that neuronal apoptotic death induction plays a crucial role in the progression of the disease, the recent advances in basic apoptosis research, and the recognition that apoptosis has a broad range of clinical applications, have prompted much of the research into investigations of intervention tactics directed at the apoptosis control or apoptosis induction in the neuronal apoptotic death program. Potential remedies can be inferred from the hypothesis that oxidative stress is shared by multiple illnesses involving increasing cell death. Among these, using neurotrophic growth factors and antioxidant molecules could be a common strategy to shield neurons from oxidative damage.
The field of apoptosis research has recently gained exciting additional insights into the mechanics of cell death programs that may perhaps contribute to the degeneration and death of neurons after acute brain trauma and the evolution of neurodegenerative diseases. The focus of this review paper will be on contemporary ideas that are important for comprehending the apoptotic death program, the mediators and regulators of cellular apoptosis, and the connection between aberrant apoptosis and the emergence of neurodegenerative illnesses. A deeper comprehension of the mechanisms underlying apoptosis may open the door to more advanced therapeutic approaches and treatment modalities that aim to regulate the neuronal apoptotic death pathway. To date, caspases and the Bcl-2 protein family are two members of the apoptotic machinery that have been the subject of much investigation. Future treatment modalities that emerge from research to more precisely identify the redox-sensitive molecular components of the neuronal apoptotic death machinery and their regulation in the various acute and chronic neurodegenerative pathologies should be challenged by the successes of these current approaches. More apoptotic inhibitors will probably be added to physicians’ daily toolkits in the coming years for treating neurologic conditions involving caspase-mediated cell malfunction and cell death.
6-OHDA: 6-hydroxydopamine
AChEIs: acetylcholinesterase inhibitors
AD: Alzheimer’s disease
ALS: amyotrophic lateral sclerosis
Apaf-1: apoptotic protease activating factor-1
Bcl-2: B-cell lymphoma 2
Cyt c: cytochrome c
DNA: deoxyribonucleic acid
NDs: neurodegenerative disorders
NO: nitric oxide
PD: Parkinson’s disease
ROS: reactive oxygen species
SOD: superoxide dismutase
TERT: telomerase reverse transcriptase
UV: ultraviolet
Authors are thankful to their organizations for their support.
RG and KW: Writing—original draft, Writing—review & editing, Visualization. AS and HC: Supervision, Investigation, Conceptualization, Writing—review & editing. RKG and SG: Formal analysis, Resources, Visualization. MAK: Writing—review & editing, Supervision, Methodology. All the authors have equally contributed to conceiving this paper and participated in its revisions. All authors read and approved the final manuscript.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.