 Review
Review

Affiliation:
UMR PNCA, AgroParisTech, INRAE, Université Paris-Saclay, 91120 Palaiseau, France
Affiliation:
UMR PNCA, AgroParisTech, INRAE, Université Paris-Saclay, 91120 Palaiseau, France
Email: francoismichel.blachier@gmail.com
ORCID: https://orcid.org/0000-0002-8501-0990
Explor Med. 2024;5:257–279 DOI: https://doi.org/10.37349/emed.2024.00220
Received: November 25, 2023 Accepted: February 07, 2024 Published: April 24, 2024
Academic Editor: Amedeo Lonardo, Azienda Ospedaliero-Universitaria di Modena, Italy
In this review, we present the main luminal fuels that are responsible for energy production in colonocytes, namely the bacterial metabolites short-chain fatty acids and lactate, which are produced from undigestible polysaccharides and proteins, and hydrogen sulfide that is mainly produced from undigested proteins. In addition to these luminal fuels, colonocytes can use glutamine, and to a lower extent glucose, as energy substrates provided by arterial capillaries. The effects of excessive concentrations of bacterial metabolites within the colonic luminal fluid (including butyrate, hydrogen sulfide, p-cresol, indole derivatives, ammonia, 4-hydroxyphenylacetic acid, and acetaldehyde) on the mitochondrial energy metabolism in colonic epithelial cells and the consequences of altered ATP production on the colonic epithelium renewal and barrier function are detailed, as well as consequences for water and electrolyte absorption. The relationships between modifications of these latter processes and development of colitis are then discussed. Finally, several mechanisms that are considered as adaptive against deleterious effects of bacterial metabolites on colonic epithelial cell energy metabolism are presented.
The colonic epithelium is made of absorptive colonocytes, mucus-secreting goblet cells, enteroendocrine cells, and tuft cells. The metabolic pathways which are operative in the colonic epithelial cells for energy production have been almost exclusively studied in the absorptive colonocytes [1]. Absorptive colonocytes display high energy demand, corresponding notably to the Na/K ATPase activity which regulates sodium concentration in the colonocytes in the context of sodium absorption [2], and to the renewal within 3–4 days of the colonic epithelial layer [3]. Such renewal requires ATP-dependent anabolic metabolism [4]. This is illustrated by the fact that although the gastrointestinal tract represents approximately 5% of the body weight, this tract is responsible for around 20% of the whole-body oxygen consumption [5].
In the present review, we will examine the main substrates that are used by colonic epithelial cells for energy production. The association of defective energy metabolism and epithelial barrier dysfunction, as well as the implication of such dysfunction in the etiology of intestinal inflammation will be presented. We will then focus on the bacterial metabolites recovered in the feces or in the colonic luminal fluid which, when present in excess, have been shown to affect energy production in the intestinal epithelial cells. Some recent data regarding the mechanisms involved in such alterations will be then detailed. In addition, we will examine the consequences of alterations of energy metabolism in the intestinal epithelium, and thus of ATP production, for different ATP-dependent processes. These processes include epithelium renewal and maintenance of the intestinal barrier function. Finally, we will present some of the adaptive mechanisms that are involved in the capacity of the colonic epithelium to cope with marked changes in the concentrations of some alimentary-derived bacterial metabolites in the colonic luminal fluid.
Absorptive cells, which are the most abundant cells within the differentiated colonic epithelial cells, are polarized cells which are present mainly on the surface epithelium. These cells are responsible for water absorption, and for both absorption and secretion of electrolytes, with an overall net movement toward electrolyte absorption from the luminal fluid to blood capillaries [6]. To make a long story short, it can be useful to consider that overall, water follows Na+ during its transcellular journey from the lumen to the blood. This function allows the progressive dehydration of the luminal fluid within the large intestine. This fluid is viscous in the proximal colon while becoming more solid in the rectal part of the large intestine. The brush-border membranes situated at the luminal side of the colonocytes and the baso-lateral membranes situated near to blood capillaries are equipped with a battery of transporters and channels for different minerals, including sodium, potassium, and chloride [7].
The respective parts of total respiratory rate devoid of the Na/K ATPase activity and macromolecule synthesis have not been determined in colonocytes. However, from experiments with other mammalian cells, it can be presumed that these processes correspond to at least approximately half of the total cellular respiratory rate [8], thus pointing out the high energy requirement for these processes in mammalian cells in general, and in colonocytes in particular.
Such ATP production can be obtained through the oxidation of energy substrates which can be supplied to colonocytes from both the apical and the baso-lateral sides. Regarding substrates supplied from the arterial capillaries through the baso-lateral membranes, absorptive colonocytes use mainly glutamine [9]. This amino acid is oxidized in the mitochondria of colonocytes allowing ATP synthesis. Blood glucose is also a contributor to ATP synthesis in colonocytes, but since colonocytes use this hexose mainly in the glycolytic pathway, the contribution of glucose to ATP production is believed to be minor when compared with glutamine (Figure 1). Regarding luminal fuels originating from the metabolic activity of the intestinal microbiota, they are supplied to colonocytes through the apical side of the cells. Indeed, since in usual dietary and physiological situations, glucose is absorbed in the small intestine [10], almost no dietary glucose is available from the luminal fluid for colonic absorption. Regarding amino acids, they also do not represent major luminal fuels available from the luminal side since the colonocytes do not absorb these compounds to any significant extent [11]. The main luminal fuels for ATP production are short-chain fatty acids (SCFAs), lactate, and hydrogen sulfide (H2S) which are mainly produced from dietary and endogenous undigested polysaccharides and proteins [1] (Figure 1).
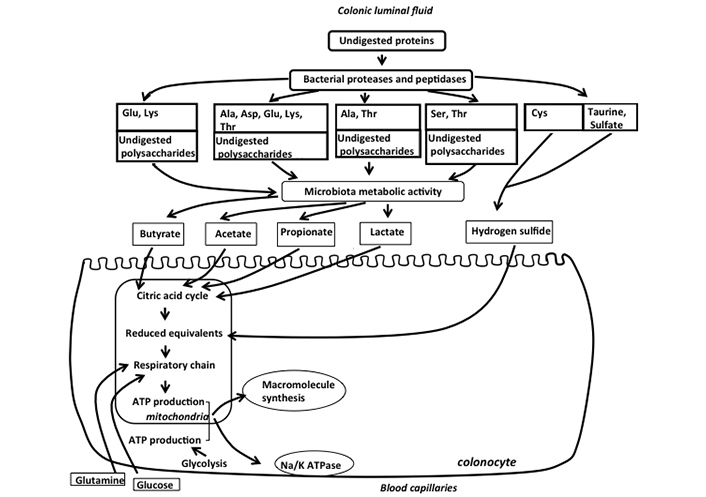
Schematic representation of the main fuels available from the luminal and baso-lateral sides of colonic absorptive cells for mitochondrial synthesis of ATP in colonocytes. Undigested polysaccharides and amino acids released by bacterial proteases and peptidases are metabolized by the colonic microbiota which produces SCFAs, lactate, and H2S. These bacterial metabolites are used as fuels by colonocytes for mitochondrial ATP production, such ATP being mainly used for macromolecule synthesis (notably proteins) and for the functioning of the Na/K ATPase. Glutamine, and to a lesser extent glucose from arterial blood, are also used as oxidative fuels by colonocytes, thus participating in ATP production. Please note that the ports of entry of the luminal and blood energy substrates in the mitochondrial system of ATP synthesis can be different and are not specified in the figure. Glu: glutamate, Lys: lysine, Ala: alanine, Asp: aspartate, Thr: threonine, Ser: serine, Cys: cysteine
Although it is predictable that mucous-secreting goblet cells depend on high ATP synthesis rate for mucin synthesis (especially in the colon where the goblet cells represent as much as approximately 20% of all epithelial cells [12]), the ways by which energy substrates are utilized by these cells remain poorly understood [13]. Regarding enteroendocrine cells, here again there is a paucity of data regarding the energy substrates they use for ATP production (see the section H2S below).
The SCFAs are butyrate, acetate, and propionate. In the human colon, acetate is the most abundant SCFA, averaging 54 mmol, while propionate and butyrate represent approximately one third of acetate concentration, thus averaging 18 mmol [14, 15]. Although the main substrates for SCFA production by the intestinal microbiota are dietary fibers and resistant starch [16], several amino acids may contribute as substrates for such production by the intestinal bacteria [17] (Figure 1). These amino acids are released by the bacterial proteases and peptidases from both alimentary and endogenous proteins that have not been fully digested in the small intestine [18]. Of note, in a rodent model, the consumption of a diet containing high amounts of proteins, results in a marked increase of SCFAs in the colonic luminal fluid [19], reinforcing the view that amino acids are significant contributors for microbial SCFA production.
These compounds are transported from the luminal fluid to the intracellular medium of colonocytes by several processes. Specific transporters, notably monocarboxylate transporter 1 (MCT1) which are members of the proton-coupled monocarboxylate electroneutral transporters, and sodium-coupled MCT1 (SMCT1) which are members of sodium-linked electrogenic transporters, represent the predominant transporters for monocarboxylates (SCFAs and lactate) from the luminal fluid to the intestinal epithelial cells [20]. However, butyrate can also enter within the colonocytes, although to a lesser extent, by using the SCFA/HCO3– exchanger [21]. In addition, a minor amount of SCFAs in undissociated form can enter colonocytes by diffusion [22]. Butyrate, acetate, and propionate, after their entry inside colonocytes are efficiently oxidized in the mitochondria of colonocytes, thus serving as fuels for ATP synthesis [23]. This explains the micromolar concentrations of these compounds in the portal blood as compared to millimolar concentrations in the colonic luminal fluid [24] (Figure 2). Importantly, the oxidation of butyrate within colonocytes has been proposed as a way to regulate its intracellular concentration, and thus presumably its effect on the level of histone acetylation and consequently on nuclear gene expression [25, 26] (Figure 2). In fact, butyrate has been shown to dose-dependently inhibit histone deacetylase activity in models of epithelial cells originating from human colonic adeno-carcinoma. Such inhibition increases histone acetylation, resulting in alteration of cell cycle regulatory protein expression, and finally reducing markedly cell growth [27]. Of note, among SCFAs, propionate represents also an efficient inhibitor of histone deacetylase activity [28]. In contrast, acetate shows no effects on histone deacetylase activity, and incidentally exerts no effect of colonic epithelial cell growth [26, 29].
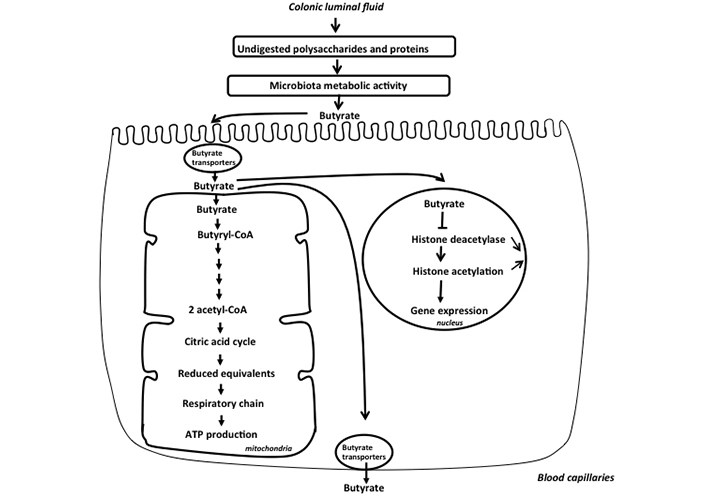
Schematic representation of the metabolism of butyrate in the colonic epithelial cells. Butyrate is produced by the colonic microbiota from undigested polysaccharides and proteins. This SCFA, after entering inside colonocytes is metabolized in the mitochondria leading to acetyl-CoA production that enters the citric acid cycle, allowing synthesis of reduced equivalents that allows ATP production in the respiratory chain. Butyrate that has not been metabolized in the mitochondria enters nucleus and inhibits histone deacetylase activity, thus increasing the level of histone acetylation, such epigenetic event modulating the expression of several genes in the nucleus. Finally, the minor part of unmetabolized butyrate is released in the portal vein
The concept that butyrate metabolism within colonocytes regulates its intracellular concentration is indirectly supported by the pioneering work of Roediger [30]. In this work, it was observed that although well oxidized in the mitochondria of colonocytes, butyrate very moderately increases oxygen consumption in these cells. This can be explained by the fact that butyrate suppresses the oxidation of endogenous energy substrates in colonocytes. In other words, when butyrate is available to colonocytes, these cells use preferentially butyrate for ATP synthesis, and thus use less endogenous substrates to maintain energy homeostasis, allowing regulation of its concentration within colonocytes. Of note, and in a similar line of thinking, it has been shown that oxidation of butyrate by the differentiated absorptive colonocytes in the surface epithelium and the upper part of the colonic crypts regulates the concentration of butyrate in vicinity of healthy proliferating epithelial stem/progenitor cells in the lower part of the crypts, thus protecting them from the antiproliferative effects of butyrate at excessive concentrations [31].
Regarding lactate which is generally produced by the intestinal microbiota from amino acids and undigestible saccharide [19, 32, 33], this organic acid is found in the human feces at much variable concentrations among individuals ranging from undetectable amount up to millimolar concentrations [34]. In human colon, lactate concentration averages approximately 3.5 mmol [35]. The absorptive colonocytes have been shown to be characterized by high capacity for L-lactate oxidation [36] (Figure 1).
More recently, H2S produced by the intestinal microbiota has been shown to represent a mineral fuel for ATP production in colonocytes. H2S is produced by different bacterial species from different alimentary and endogenous compounds including cysteine, taurine, and sulfite, and by sulfate-reducing bacteria [37–39] (Figure 1). The H2S-producing bacterial species include Fusobacterium, Clostridium, Escherichia coli, Salmonella, Klebsiella, Streptococcus, Desulfovibrio, and Enterobacter [1]. H2S which is not bound to luminal components, thus either as a gas pocket or as a gas dissolved in the liquid phase of the colonic luminal fluid, represents the chemical form that diffuses through the mucus layer, and then into the colonocytes [1]. Several dietary compounds like zinc and proanthocyanidins (contained in plants) bind H2S, thus reducing its concentration in free form [40, 41]. Interestingly, in germ-free mice, H2S concentrations in its free form are reduced in plasma and intestinal tissues when compared with normal animals equipped with an intestinal microbiota [42]. These data suggest that the microbiota is likely a purveyor of this compound for the host. The concentration of total H2S, in its free and bound forms, measured in the mammalian colonic luminal fluid is rather divergent, according notably to the specificity of techniques used for measurement, thus ranging from micromolar to low millimolar concentrations [43]. In isolated colonocytes, micromolar concentrations of H2S (5–65 µmol) increase instantaneously oxygen consumption by entering the mitochondrial respiratory chain [44]. This increased consumption is concomitant with an inner mitochondrial energization and synthesis of ATP [41], thus establishing H2S as the first mineral energy substrate in human cells [44]. This discovery has challenged the previous concept which considers that mammalian cells are mostly dependent on carbon-based fuels for ATP production. The mitochondrial ATP synthesis through H2S oxidation in colonocytes is made possible by the catalytic activity of the sulfide oxidation unit which drives the conversion of H2S into thiosulfate [45] (Figure 3).
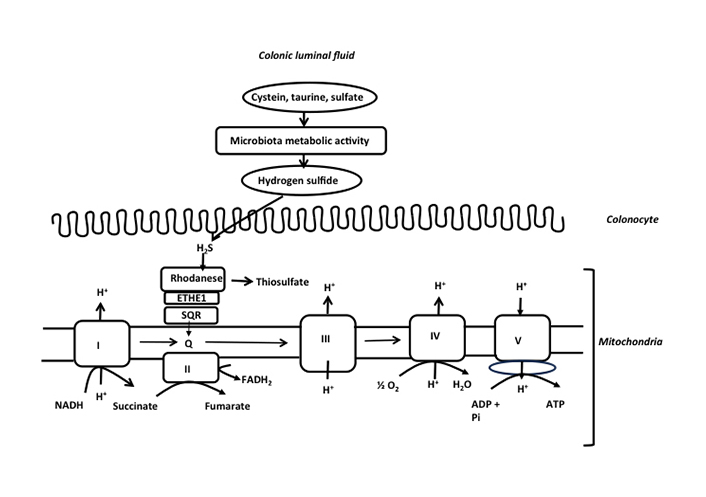
Schematic representation of the detoxification of H2S and production of ATP in the colonic epithelial cells. H2S is mainly produced by the colonic microbiota from cysteine, taurine, and inorganic sulfate. H2S at low micromolar concentration is metabolized by the sulfide oxidation unit which is made of sulfide quinone reductase (SQR), persulfide dioxygenase (ETHE1), and rhodanese. SQR is an electron donor to the mitochondrial respiratory chain through quinone, allowing mitochondrial ATP production. Please note the transfer of electrons by SQR into the mitochondrial respiratory chain through quinone as indicated in the figure. This electron transfer is followed by transfer to complex III cytochrome c, and finally to complex IV (cytochrome oxidase). NADH: nicotinamide adenine dinucleotide; FADH2: flavin adenine dinucleotide; Q: quinone; Pi: orthophosphate
Regarding the colonic enteroendocrine cells, a recent study has shown that these cells are equipped with the 3 enzymes constituting the sulfide-oxidizing unit, and that low amounts of the sulfide donor NaHS increase oxygen consumption in the enteroendocrine cell model NCI-h716, thus suggesting that H2S is a mineral fuel allowing ATP synthesis in these cells [46].
Inflammatory bowel diseases (IBDs) are associated with marked alterations of the intestinal mucosa associated with exacerbated immune functions [47]. Briefly, IBDs, which include mainly ulcerative colitis and Crohn’s disease, are characterized by chronic inflammation of the intestinal mucosa resulting from inappropriate mucosal immune responses against luminal components in genetically predisposed individuals, with alternating relapse and remission episodes [48–50]. Numerous experimental and clinical evidence indicate that defects of the “barrier function” of the intestinal epithelium might represent one of the initial defects that would be involved in the etiology of IBDs [51–53]. Although it is still debated whether epithelial permeability defects are the primary event that leads to intestinal inflammation development, or if such defects are secondary and triggered by inflammation itself, it looks plausible that defects of barrier function and initial inflammation produce together a vicious circle producing eventually inflammatory flare in the intestine.
The postulate that ulcerative colitis is a colonic energy deficiency disease was postulated more than 40 years ago [54]. Early studies showed mitochondrial morphological anomalies in rectal and colonic biopsies obtained from both ulcerative colitis and Crohn’s disease patients [55, 56]. In both clinical and experimental studies, intestinal mucosal inflammation is associated with lower levels of mucosal ATP content when compared with healthy mucosa, thus pointing out the potential importance of altered mitochondrial function in the etiology of IBD. Indeed, in the inflamed human colon and experimental animal models, decreased mitochondrial metabolic activity together with decreased ATP content have been reported [57]. The concentration of ATP in cells is usually stable, but this concentration decreases in situation of decreased production or/and increased utilization [58]. In support of a causal link between decreased ATP concentration in the inflamed colon mucosa and progression of the disease, an increased mucosal level of ATP was found to protect mice from colitis [59]. In addition, proteomic analysis of colon mucosa obtained from patients with active ulcerative colitis revealed decreased expression of three mitochondrial enzymes involved in mitochondrial energy production when compared with colon mucosa obtained from healthy patients [60], suggesting here again the possible implication of altered energy metabolism in the pathogenesis of IBDs. These results were corroborated by a study showing that the activities of three complexes of the mitochondrial respiratory chain are markedly decreased in the colonic mucosa of patients with ulcerative colitis when compared with the activities measured in the normal colonic mucosa [61]. Similarly, in biopsies obtained from patients afflicted by ulcerative colitis, reduction of the mitochondrial respiratory chain complex II was measured when compared with controls [62]. In newly diagnosed pediatric ulcerative colitis patients, a marked down-regulation of both mitochondrial- and nuclear-encoded genes involved in mitochondrial function was observed [63, 64]. Of note, all the 13-mitochondria-encoded genes were found to be related to ATP production. Interestingly, using colonic biopsies from ulcerative colitis patients, Sünderhauf and collaborators [65] demonstrated that mucosal energy deficiency is linked to impaired goblet cell differentiation, thus pointing out an impaired production and secretion of mucin-producing goblet cells in ulcerative colitis.
When murine intestinal epithelial cells are treated with dextran sodium sulfate, a chemical that is used to provoke experimental colitis when given to rodents in drinking water, marked alterations of mitochondrial bioenergetics are recorded [66], suggesting that experimental colitis affects ATP production within intestinal epithelial cells. In vitro model of monolayers of colonocytes, mitochondrial ATP depletion disrupts the integrity of the monolayers [67]. Mice invalidated for the gene coding for one of the main intestinal mucins (mucin-2 knockout mice) are predisposed to colitis [68] as well as to mitochondrial damage, impaired oxygen consumption, and ATP reduction in the descending colon [69]. In this latter study, using the wild-type mice, it was shown that chemically-induced mitochondrial uncoupling (which decreases the efficiency of mitochondria for ATP synthesis) mimics intestinal barrier disruption in vivo, thus strongly suggesting that decreased capacity of mitochondria for ATP synthesis contributes to the leaky gut syndrome in these models.
The predictable consequences of altered mitochondrial ATP production in regard to ATP requirement in absorptive colonocytes are defects in the process of colonic water absorption. Indeed, it can be predicted that a shortcoming of ATP production within colonocytes would result in a defect of Na/K ATPase activity in baso-lateral membranes, then participating in decreased water absorption and thus diarrhea. Diarrhea is in fact hallmark symptom associated with IBDs, and this symptom is observed in almost 80% of IBD patients [70]. Such energy deficiency in IBDs would be one of the parameters associated with IBDs. Briefly, these parameters are excessive production of inflammatory mediators, damage to the mucosal architecture, disturbed epithelial barrier function, increased access of luminal compounds to the lamina propria, and altered functional entities in colonocyte membranes, such entities including ion transporters and channels, as well as junction complexes [71, 72]. In other words, the cascade of metabolic and functional alterations during IBD development is likely to explain the inadequate water and electrolyte transport leading to the IBD-associated diarrhea.
The fecal and urinary concentrations of several bacterial metabolites may change according to changes in the alimentation, notably in the case of the modified dietary supply of polysaccharides and proteins [73–76]. Accumulation of bacterial metabolites in morning spot urine is observed when these compounds are absorbed without being fully metabolized in colonocytes and peripheral tissues before excretion. These results indicate that an increased supply of substrates from alimentary origin may increase the production of metabolites by the intestinal microbiota [1]. Below, we will examine how inappropriate changes in the amounts of alimentary-derived bacterial metabolites within the large intestine luminal fluid may have an impact on energy metabolism within the colonic epithelial cells.
It is of major interest to consider that in IBD, marked lower fecal SCFA concentrations compared to control have been repeatedly measured [77–82]. Although, as presented above, excessive butyrate (in relation to the metabolic capacity of differentiated colonocytes to oxidize them) may affect proliferating epithelial stem/progenitor cells [31], and thus presumably interfering with the normal process of epithelial renewal, it is conceivable that, conversely, insufficient butyrate availability in relationship with the colonocyte requirement may affect energy homeostasis in these cells (Figure 4). In addition to being luminal fuels for absorptive colonocytes, SCFAs are known to regulate electrolyte and water movement across the colonocytes. Indeed, butyrate, and to a minor extent acetate and propionate, have been shown to play a role in the regulation of electrolyte and water absorption [83, 84]. Then, it is tempting to postulate that a shortcoming of butyrate production by the intestinal microbiota, and thus a defect in the supply of one of the major luminal fuels, would diminish the capacity of the colonic epithelium for water absorption, thus participating notably in the IBD-associated diarrheal process. However, despite encouraging results obtained from experimental studies suggesting beneficial effects of butyrate on the diarrheal process [85, 86], the efficiency of butyrate for treating diarrhea in clinical studies remains controversial. Addition of resistant starch to oral rehydration solution has been reported to decrease duration of diarrhea in adults and children hospitalized for acute diarrhea [87, 88]. However, in a randomized, double-blind, placebo-controlled multicenter study, a mixture of non-digestible carbohydrates was found ineffective as an adjunct to oral rehydration solution for reducing the treatment of acute infectious diarrhea in children [89].
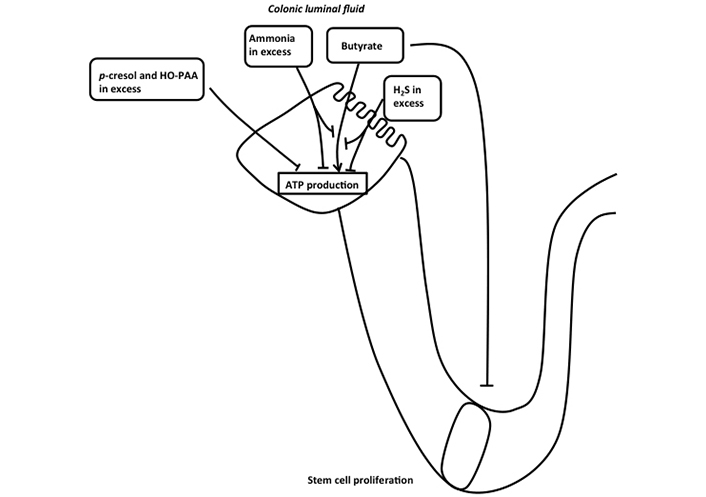
Schematic representation of the effects of different bacterial metabolites on ATP production in colonic absorptive cells. Butyrate is metabolized in colonocytes allowing ATP production in mitochondria. H2S and ammonia in excess inhibit both mitochondrial oxygen consumption and butyrate oxidation, thus affecting ATP production in colonocytes. p-cresol and 4-hydroxyphenylacetic acid (HO-PAA) in excess also affect ATP production in colonocytes. Butyrate that has not been metabolized by differentiated colonocytes affects stem cell proliferation when present at excessive concentration
Several controlled studies have been performed to evaluate the effects of exogenously administered butyrate on several clinical parameters in patients with IBDs. In ulcerative colitis patients treated with butyrate enemas, the results obtained are much heterogeneous. One study found that clinical improvement was proportionally less frequent in the butyrate-treated group than in the placebo group [90]. Two studies found no therapeutic value of SCFA enemas because of no significant clinical improvements [91, 92], and four studies found some modest improvements of inflammatory parameters after enema administration with butyrate or other SCFAs [93–96]. These discrepancies are likely due to differences in the clinical status among volunteers included in each study, in the number of patients recruited, in the dose of SCFA administered, in the duration of treatment, and finally in the parameters measured. By recapitulating the data obtained in randomized controlled trials, it was concluded that the results obtained do not overall support the application of butyrate enemas for the treatment of ulcerative colitis [97, 98]. Regarding Crohn’s disease, there are no reliable data in support of butyrate enemas for the treatment of this disease [97]. These latter conclusions suggest that treatment with butyrate is not able by itself to reduce the inflammatory flare to any valuable extent in ulcerative colitis. These disappointing results are likely due, in part at least, to a reduction of butyrate uptake by the inflamed mucosa because of downregulation of the MCT1, thus limiting its action on the colonic mucosa [99]. Of note, significant alteration in the expression of the MCT1 has also been detected in experimental colitis model [100].
In deep contrast with the demonstration that luminal sulfide at low micromolecular concentrations serves as a mineral fuel in colonocytes, higher concentrations of H2S (above 65 µmol) inhibit colonocyte oxygen consumption by inhibiting the mitochondrial cytochrome c oxidase activity (complex IV of the mitochondrial respiratory chain) after binding to this complex with high affinity [101, 102]. Thus, at concentration above the capacity of colonocytes to oxidize this bacterial metabolite, H2S decreases the capacity of mitochondria within colonocytes to synthesize ATP (Figure 4). In addition, H2S at concentration above 1 mmol dose-dependently inhibits the oxidation of glutamine and butyrate, thus depriving colonocytes from the utilization of their two main fuels [102, 103]. Of note, the oxidation of H2S in colonocytes takes precedence over the oxidation of other carbon-based energy substrates [104]. This fact strongly suggests that the oxidation of H2S by the sulfide-oxidizing unit, likely because of its deleterious effect on mitochondrial ATP synthesis at excessive concentrations, represents a priority. The demonstration that colonocytes appear among the most efficient cells for H2S disposal is not surprising given that these cells are facing the highest sulfide concentrations in the body [105]. Lastly, when the colonocytes undergo either spontaneous or butyrate-induced differentiation, their capacity to metabolize H2S is significantly increased [41], raising the view that differentiated colonocytes on the epithelial surface and on the upper part of the colonic crypts may, like in the situation observed with butyrate [31], protect the proliferating epithelial stem/progenitor cells situated at the bottom of crypts from the deleterious effect of H2S.
As presented above, H2S, when present in excess, severely inhibits oxygen consumption in colonocytes, thus affecting ATP production in these cells. Consistent results have clearly shown that at concentrations where H2S inhibits oxygen consumption in colonic epithelial cells, a marked decrease in cell proliferation has been measured. Indeed, the slow-releasing sulfide donor GYY4137, when used at 3 mmol concentration, inhibits markedly the proliferation of HCT116 cancer-derived colonic cells [106]. In the HT-29 adeno-carcinoma colonic epithelial cell model, the rapid H2S donor NaHS at 0.3 mmol concentration inhibits cell respiration, an effect that coincides with a marked reduction of cell proliferative capacity [102, 107]. Since the colonocytes maintain their ATP cellular content in the presence of NaHS, despite inhibition of mitochondrial oxygen consumption, it has been proposed that by reducing cell growth, and thus ATP-consuming anabolic metabolism, the cells maintain their ATP content and thus their viability under severe restriction of mitochondrial ATP production [102].
The proposition that excessive H2S concentrations may participate in the pathogenesis of IBDs was proposed more than two decades ago [108]. To further test this hypothesis, a study on patients with new-onset pediatric Crohn’s disease demonstrated that the intestinal microbiota composition of these patients was characterized by higher relative abundance of Atopobium, Fusobacterium, Veillonella, Prevotella, Streptococcus, and Leptotrichia [109]. Several members of those genera are known to be H2S producers. In this cohort of children, the abundance of H2S producers from cysteine was correlated with the severity of mucosal inflammation. To identify possible causal links between H2S production and intestinal inflammation, the authors colonized mice invalidated for the production of the regulatory cytokine interleukin-10 (IL-10) (model of mice susceptible to colitis), with the H2S producer Atopobium parvulum, and measured a worsening of colonic inflammation [109]. Since H2S is obviously not the only bacterial metabolite produced by Atopobium parvulum, they used the H2S scavenger bismuth and found that this latter compound was able to attenuate the worsening of inflammation provoked by Atopobium parvulum. Of major interest, the authors of this latter study found that the colonic mucosal biopsies obtained from the young Crohn’s disease patients displayed decreased expression of the mitochondrial enzymes involved in sulfide detoxification. Overall, the results of this study indicate that diminished capacity for H2S disposal in the colonic mucosa of Crohn’s disease patients amplifies the pro-inflammatory effect of H2S overproduction by the intestinal mucosa. Three studies reinforce this latter proposition. In the first one, it has been shown that gene expression and activity of one of the enzymes involved in H2S detoxification, namely thiosulfur transferase, is downregulated in the mucosa of ulcerative patients [110], while in the second and third studies, decreased capacity for H2S detoxification was measured in the mucosa of patients with respectively Crohn’s disease [111] and ulcerative colitis [112].
Results from in vivo studies in rats indicate that intra-colonic instillation with phosphate-buffered saline solution containing NaHS at 1.5 mmol concentration increased in colonocytes expression of the gene coding for the hypoxia inducible factor 1 α (Hif-1α) and of the mucosal inflammation-related genes, including inducible nitric oxide synthase (iNOS) and IL-6 [101]. Also, a study has shown that high concentrations of H2S destabilize the protective mucous layer that covers the intestinal epithelium by reducing disulfide bounds linking the mucin-2 network. This latter process would possibly increase the interactions between bacteria and the epithelium [113]. Then, H2S in excess may favour mucosal inflammation by inhibiting mitochondrial ATP synthesis, thus affecting proliferation of undifferentiated colonic epithelial cells, and in addition by altering the structure of mucus that protects the colonic epithelium. Interestingly, the study by Jowett and collaborators [114] indirectly suggests a pro-inflammatory effect of excessive H2S production by the intestinal microbiota. In this study, following for one year patients with ulcerative colitis in remission, it was found that patients with the highest level of consumption of dietary proteins display a threefold increase in the risk of relapse when compared with patients with the lowest protein intake. In addition, high sulfate dietary intake was also associated with a higher risk of relapse in volunteers. Although H2S concentration in the intestinal fluid was not measured in this study, the fact that high dietary protein consumption is associated with increased fecal H2S concentration [115], and the fact that sulfate is a precursor for H2S synthesis by the intestinal microbiota [116], raises the credible possibility that increased H2S would play a role in the increased risk of relapse in patients in remission. A study in a mouse model deficient in the regulatory cytokine IL-10 shows that a diet with high content in saturated fat increases, in a process modulated by taurine-containing bile acids, the release of H2S produced by the intestinal microbiota, such event appearing to play a major role in the intestinal inflammation observed in this experimental model [117]. Recently, in a Crohn’s disease case-control study, Marcelino and collaborators [118] identify a lack of bacterial species with the capacity to consume H2S as the main distinguishing characteristics of the intestinal microbiota in the context of this inflammatory disease. These results reinforce further the view that failure of the intestinal microbiota to metabolize H2S may contribute to the excessive luminal content of this bacterial metabolite, and thus accordingly to the etiology of Crohn’s disease in predisposed individuals.
However, from experiments performed in animal models in which colitis is induced chemically, and in studies where pharmacological inhibitors of H2S synthesis or H2S-releasing compounds are used, it appears that a low minimal amount of H2S produced endogenously or supplied from the colonic luminal fluid is likely necessary to limit the extent of colonic mucosal inflammation [119–122], thus suggesting that an almost complete depletion of H2S supplied to colonocytes is counter-productive in situation of intestinal inflammation [123]. In addition to the demonstration that, as explained above, low micromolar concentrations of H2S are involved in mitochondrial ATP synthesis in colonocytes [124], H2S have been shown to exert some antioxidant activity, notably by persulfidation of cysteine residues that protects these residues from oxidative damages [45].
Finally, in view of the effects of H2S on the colonic mucosa, the concept that this compound is a double-edged sword for the intestinal epithelium has been proposed in 2019 [43]. In that concept, the effects of the diffusible bacterial metabolite H2S on the inflammatory processes in the colonic mucosa depend on both the low endogenous production by the host colonic mucosa, that is protective, and on the exogenous bacterial production, which is deleterious when produced in excess.
The phenolic compound p-cresol (4-methylphenol) is produced from the amino acid tyrosine by the anaerobic bacteria of the large intestine [125]. The p-cresol concentrations measured in the human colonic fluid are in the low millimolar range [126, 127]. The bacterial metabolite p-cresol can markedly affect colonocyte energy metabolism when present in excess in the colonic fluid. Indeed, 0.8 mmol p-cresol diminishes mitochondrial oxygen consumption in human colonocytes when tested in vitro, and increases anion superoxide production, thus indicating alterations of the mitochondrial functions in these cells [128]. This effect is associated with a reduction of the capacity of colonic epithelial cells to proliferate. At 0.8 mmol, p-cresol increases after 1 and 2 days of treatment the proportion of cells in the S-cell cycle phase, and decreases the proportion of cells in the G0/G1 phase. However, the relative number of cells in the subdiploid G1 peak, that is characteristics of cells undergoing nuclear fragmentation, was not modified by the treatment with 0.8 mmol p-cresol [128]. In this latter study, pretreatment of colonocytes for three days with increasing concentrations of p-cresol resulted in dose-dependent decrease of the intracellular ATP content, thus indicating energy deficiency in these cells (Figure 4). Using monolayers of human colonocytes, p-cresol at millimolar concentrations dose-dependently increases the paracellular transport between colonocytes [129], suggesting that this bacterial metabolite at excessive concentrations can alter intestinal barrier function. The capacity of p-cresol in excess to increase anion superoxide production within colonocytes is of interest since reactive oxygen species have been shown to be produced in experimental situations of inhibition of mitochondrial complexes I, II, and III [130–132]. Excessive production of reactive oxygen species (and notably anion superoxide), in regards to the capacity of colonic epithelial cells to eliminate them, is considered as one parameter that is involved in the development of IBDs [133–136].
Indole and indole-related compounds are synthesized from various intestinal bacterial species [137]. The measurement of indole concentration in feces obtained from volunteers has shown huge inter-individual differences ranging from 0.30 mmol to 6.64 mmol [1]. Although these compounds have been shown in numerous experimental studies to exert protective effects against intestinal mucosal inflammation, notably through binding to aryl hydrocarbon receptor [138], indole at excessive concentration (2.5 mmol) affects the respiration of colonocytes by diminishing mitochondrial oxygen consumption [139], and thus mitochondrial ATP production. Such inhibitory effect was associated with a transient oxidative stress in colonocytes as well as an increased secretion of the pro-inflammatory IL-8 and tumor necrosis factor-α. Indoxyl sulfate, the main indole co-metabolite synthesized by host tissues, displayed similar effects as indole on colonocyte respiration and secretion of pro-inflammatory compounds when tested at 2.5 mmol.
Ammonia (considered as the sum of ammonium NH4+ and NH3) is present at millimolar concentrations mainly in the form of NH4+ in the mammalian large intestinal fluids [140, 141]. Ammonia can be both utilized and produced by the intestinal microbiota. Ammonia provides one of the main sources of nitrogen for bacterial amino acid synthesis, and this compound is notably formed by intestinal microbiota from the conversion of urea to ammonia by the bacterial urease activities and by catabolism of several amino acids [142]. Ammonia used at concentration between 10 mmol and 50 mmol inhibits dose-dependently the mitochondrial oxygen consumption in colonocytes [143], thus affecting colonocyte respiration, and thus energy production, in these cells. In addition, ammonia at 10 mmol concentration inhibits SCFA oxidation in colonic epithelial cells [144] (Figure 4). Of note, at concentrations where ammonia inhibits mitochondrial oxygen consumption in colonocytes, this bacterial metabolite inhibits almost totally the capacity of the human colonic HT-29 cells to proliferate [145]. These latter results suggest that ammonia-dependent alteration of the capacity of colonocyte to produce ATP plays a role in the anti-proliferative effect of this compound.
However, colonocytes are equipped with enzymatic activities that allow to detoxify ammonia, up to a threshold concentration, in the mitochondria of colonocytes. This can be done by converting ammonia into citrulline in two metabolic steps [140], thus limiting the inhibitory effect of excessive ammonia concentration on colonocyte respiration. Interestingly, excessive ammonia concentrations in regards with the capacity of colonocytes to detoxify it, increases the paracellular permeability between colonocytes [146], and ammonia treatment of human colonocytes induces mitochondrial dysfunctions and impairs epithelial barrier integrity as well as the structure of the tight junction proteins [147].
HO-PAA is a bacterial metabolite produced by the intestinal microbiota from tyrosine [148]. This compound when used at concentration equal to 1 mmol decreases in human colonocytes the activity of the complex I of the mitochondrial respiratory chain and the mitochondrial oxygen consumption, thus presumably affecting ATP mitochondria production in these cells [149] (Figure 4). Such effect was paralleled by an increased production of reactive oxygen species in colonocytes.
The intestinal microbiota metabolizes ethanol, either originating from alcohol consumption, or, to a much minor extent, from amino acids, to acetaldehyde [150, 151]. In rodents, by increasing the number of proteins in the diet, the ethanol content in the colonic luminal fluid was increased several folds [19], suggesting capacity of the intestinal microbiota to produce ethanol during amino acid catabolism. Acetaldehyde concentration was increased in the colon of rodents after alcohol administration, and these concentrations were lower in germ-free animals when compared with animal carrying an intestinal microbiota [152], pointing out the role of microbiota for acetaldehyde production from ethanol. Acetaldehyde injected in the lumen of the pig colon is partly metabolized to acetate [153]. Thus, acetaldehyde concentration in the colon appears to depend both on the synthesis from ethanol and on the conversion of acetaldehyde to acetate by the colonic flora.
Acetaldehyde when used between 25 μmol and 100 μmol concentration has been shown to exert deleterious effects on human intestinal goblet cells, inducing cell necrosis. This effect coincided with decreased mitochondrial activity, decreased ATP cellular concentration, and increased production of reactive oxygen species [154]. These findings indicate that acetaldehyde may adversely affect one main component of the intestinal barrier function. In the human colonic mucosa, acetaldehyde in excess reduces the expression of several proteins known to be necessary for normal tight junction and adherent junction functions [155].
Only few studies have examined the adaptive mechanisms that allow to limit the effects of modified concentrations of bacterial metabolites in the colonic luminal fluid. For instance, in the in vitro model of hypoxia-induced loss of colonic epithelium integrity, high millimolar concentration of butyrate can markedly ameliorate such integrity [156], suggesting that this SCFA may favor the colonic epithelial barrier function in situation when this parameter is compromised. Using human colonic cells from cancerous origin, it has been shown that butyrate- and propionate-induced autophagy may serve as an adaptive strategy to delay mitochondria-mediated apoptotic cell death [157]. Whether this situation can be extrapolated to healthy colonic epithelial cells remains however to be demonstrated.
When human colonocytes from cancerous origin are exposed to 1 mmol sulfide concentration, they increase four times their capacity to utilize glucose in the glycolytic pathway [102], but this metabolic pathway is well known to be much less efficient in terms of ATP production than the mitochondrial oxidation of energy substrates. As expected, 1 mmol sulfide severely decreased the capacity of colonocytes to proliferate [102]. Thus, increased capacity for ATP production in the glycolytic pathway may represent an adaptive process aiming at partially compensate for decreased mitochondrial ATP production. Interestingly, using colonic epithelial cells originating from human colon carcinoma, it has been shown that treatment of cells with the H2S donor Na2S used at 0.1 mmol concentration, stimulates the reductive carboxylation of glutamine-derived α-ketoglutarate to drive citrate synthesis [107]. Such process presumably corresponds to metabolic adaptation against deleterious effect of excessive H2S on colonocytes. Also, in rats fed with a high-protein diet for which the amount of H2S increase in the colonic luminal fluid, the expression of the gene corresponding to SQR, the first and rate-limiting enzyme for H2S detoxification, increases in colonocytes [101]. In such situation of high-protein consumption, the water content in the colonic luminal fluid increases markedly as well [143]. Although the mechanisms that allow such water retention are not clear yet, such retention allows to limit the increase of H2S concentration that faces the colonic epithelium, thus limiting its deleterious effects on colonocyte energy metabolism.
Regarding p-cresol, it has been demonstrated that pretreatment of colonocytes for one day with 0.8 mmol p-cresol resulted in an increase of the state 3 oxygen consumption and of the cell maximal respiratory capacity [128]. The increase of the basal and maximal respiratory rate may correspond to an attempt of the cells to counteract the effect of p-cresol on cell respiration. Indole and indoxyl sulfate when used at 2.5 mmol concentration induces an oxidative stress in colonic epithelial cells from cancerous origin. Such oxidative stress is followed by an increased expression of antioxidant enzymes, thus presumably representing an adaptive process against the deleterious effects of indole exposure at excessive concentration [139].
Lastly, in a rodent model, high-protein diet consumption for two days results in increased ammonia concentrations in the intestinal luminal fluid and the colonic vein blood when compared with animals fed with a normal protein diet. Such increases coincided with an increased capacity of colonocytes to convert arginine to ornithine and urea, therefore providing ornithine as substrate for the detoxification of ammonia in excess in two metabolic steps [140].
Further works on the adaptive processes that allow colonic epithelial cells to cope with increased content of deleterious bacterial metabolites are needed.
As presented in the present review, the renewal of the colonic epithelium within few days and the absorption of water and electrolytes are dependent on the continual mitochondrial ATP production within colonic epithelial cells. When this ATP production is, for any reason, compromised, thus failing to meet ATP requirement for anabolism in the colonic epithelium and for physiological functions of this structure, it can be predicted that ATP-dependent processes within the colonic epithelium will be affected. In this paper, we recapitulate data that show what is observed in numerous clinical and experimental situations of mucosal inflammatory flare, and these data suggest that energy deficiency within the colonic epithelium might represent one of the initial events implicated in the etiology of IBDs. Indeed, decreased expression of several proteins involved in mitochondrial ATP production is measured in situations of colitis, and in a rodent model, when mucosal ATP level is increased within the inflamed mucosa, colitis is less severe [59], suggesting a causal link between alteration of mitochondrial ATP production in the colon mucosa and signs of inflammation.
Theoretically, there are several alterations of energy metabolism within the colonic epithelium that would lead to decreased ATP production in this structure. Firstly, any severe deficiency in the supply of luminal and/or blood energy substrates would result in defective mitochondrial ATP production. Secondly, increased concentrations of bacterial metabolites which affect basal mitochondrial ATP production and/or which inhibit the capacity of colonocytes to produce ATP from energy substrates will also result in a decreased ATP production. We have proposed to include these bacterial metabolites in the category of “metabolic troublemakers” [105]. H2S can be included for instance in this latter category of compounds when present in excess. Indeed, as presented above, H2S at excessive concentrations inhibit both basal oxygen consumption and butyrate and glutamine oxidation in colonocytes [102] thus affecting ATP production. These effects are associated with increased expression of pro-inflammatory genes in the colonic epithelial cells [101]. Also, H2S in excess, probably because it alters the capacity of colonocytes to produce ATP, severely decreases proliferative capacity of colonocytes. Regarding, the bacterial metabolite p-cresol, it is of interest to consider its capacity to decrease mitochondrial oxygen consumption in colonocytes, an effect that is associated, here again, with its antiproliferative effect on human colonocytes, and in addition with its deleterious effect on the colonic epithelial integrity [128, 129]. Ammonia is another illustrative example of bacterial metabolite that inhibits at high concentrations both basal mitochondrial oxygen consumption and butyrate oxidation in colonocytes, such inhibition presumably playing a role in the antiproliferative effect of ammonia on colonocytes. Since these effects are associated with severe perturbations of the colonic epithelial barrier function, the role played by excessive ammonia concentration in the colonic luminal fluid for inflammation development requires further experimental works.
The deleterious effects of high concentration of acetaldehyde on goblet cell energy metabolism is of particular interest. Indeed, excessive acetaldehyde decreases the mitochondrial activity in goblet cells, then decreasing the ATP cellular concentration, and finally increasing goblet cell necrosis [154]. Since mice invalidated for one of the main intestinal mucins are predisposed to colitis [68], any bacterial metabolite that alters one component of the colonic barrier function (including mucin production and secretion in that case) is worth to be taken into consideration. Although goblet cells are presumably characterized by high energy requirement for abundant mucin synthesis, very few data are unfortunately available regarding their main energy substrates provided either from the luminal and/or baso-lateral sides.
Attention should be also paid in future works to the fact that in experimental works, bacterial metabolites are often tested individually, even if it is well known that the luminal fluid contain a myriad of bacterial metabolites that may exert additive, synergistic, or opposite effects on energy metabolism in the colonic epithelial cells, and then on the different ATP-dependent epithelial functions. This makes extrapolation from experimental works to “real life situation” difficult. With this reservation in mind, schematically, it is possible to distinguish the beneficial and deleterious bacterial metabolites for the colonic epithelium, keeping in mind that one particular metabolite (like H2S) may prove to be beneficial at low concentrations, while being deleterious at excessive concentrations. The progress in the definition, in individuals at risk for IBDs, of the optimal bacterial metabolite profiles in the colonic and rectal luminal fluids for optimal ATP production in the colonic epithelia (and thus optimal ATP-related functions) is dependent on a constant flow of additional data originating from both experimental and clinical studies.
Most importantly, attention should be paid to the modifications of dietary conditions that decrease concentrations of bacterial metabolites deleterious for the colonic and rectal epithelium energy metabolism in individuals predisposed to IBD, while conversely increasing up to an optimal concentration the bacterial metabolites beneficial for the large intestine epithelium energy metabolism.
H2S: hydrogen sulfide
HO-PAA: 4-hydroxyphenylacetic acid
IBDs: inflammatory bowel diseases
IL-10: interleukin-10
MCT1: monocarboxylate transporter 1
SCFAs: short-chain fatty acids
SQR: sulfide quinone reductase
The authors wish to thank Université Paris-Saclay, AgroParisTech, and INRAe for their constant support regarding our research on the effects of bacterial metabolites on colonic epithelium.
MA: Conceptualization, Writing—review & editing. FB: Conceptualization, Writing—original draft.
François Blachier who is a Guest Editor of Exploration of Medicine had no involvement in the decision-making or the review process of this manuscript.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

