 Original Article
Original Article

Affiliation:
1Team Research of Virology, Oncology, and Biotechnologies, Laboratory of Virology, Oncology, Biosciences, Environment and New Energies, Faculty of Sciences and Techniques Mohammedia, University Hassan II of Casablanca, Casablanca 28806, Morocco
ORCID: https://orcid.org/0000-0002-8151-1595
Affiliation:
2Sequencing Unit, Center of Virology, Infectious and Tropical Disease, Mohamed V Military Teaching Hospital, Rabat 10045, Morocco
ORCID: https://orcid.org/0000-0002-1375-6021
Affiliation:
2Sequencing Unit, Center of Virology, Infectious and Tropical Disease, Mohamed V Military Teaching Hospital, Rabat 10045, Morocco
Affiliation:
2Sequencing Unit, Center of Virology, Infectious and Tropical Disease, Mohamed V Military Teaching Hospital, Rabat 10045, Morocco
Affiliation:
1Team Research of Virology, Oncology, and Biotechnologies, Laboratory of Virology, Oncology, Biosciences, Environment and New Energies, Faculty of Sciences and Techniques Mohammedia, University Hassan II of Casablanca, Casablanca 28806, Morocco
Email: m.ennaji@yahoo.fr; mymustapha.ennaji@univh2c.ma
ORCID: https://orcid.org/0000-0001-5809-0270
Explor Med. 2024;5:641–655 DOI: https://doi.org/10.37349/emed.2024.00246
Received: May 30, 2024 Accepted: September 09, 2024 Published: October 18, 2024
Academic Editor: Ning Li, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Aim: The aim of this study is to investigate whether germline alterations of exon 5 of TP53 gene could be detected in the blood of known men with prostate cancer and to assess the potential association between the genomic alteration affecting this gene and clinicopathological characteristics of the patients.
Methods: Forty-eight blood samples from men diagnosed with prostate cancer were analyzed for TP53 germline mutations and confirmed by Sanger sequencing. The frequency and distribution of high-frequency mutations were analyzed according to the pathological criteria of the patients and a computational study was performed to assess the effect of new mutations.
Results: The Sanger sequencing revealed that 79% of the population studied carry mutations in TP53 gene. In summary, a total of 137 mutations have been identified in this gene, out of which 115 are new mutations. Frameshift mutations were the most frequent; the mutation c.392delA was recorded in fifteen cases (31%); the mutations c.383delC and c.432delG were observed at a frequency of 12.5% and 10% respectively. The most frequent missense mutation was the variant c.502C>A (p.His168Asn) identified in eleven patients (23%). One nonsense mutation was identified in one patient and resulted in a stop codon in position 126 (tyrosine). All codons affected by these alterations are part of the DNA binding domain of the protein TP53.
Conclusions: The germline mutation frequency observed in prostate cancer patients, and the new mutations recorded in TP53 gene, could be in favor of a potential association of genomic alterations in this gene and prostate cancer genesis, thereby constituting a tool, similar to other genes in the DNA repair pathway such as BRCA1 and BRCA2. This could contribute to the advancement of diagnosis and therapeutic strategies for prostate cancer.
The genetic alterations involved in prostate cancer (PC) are extremely complex, in fact, several studies have revealed the existence of different genetic subtypes of PC, particularly in localized forms considering aberrations on different oncogenic pathways [1, 2]. On the other hand, the evolution of the disease is marked by great heterogeneity, in fact, PC even diagnosed at the localized stage can lead to different clinical outcomes, considering in particular that relapses occur in 30% of men even with treatments like prostatectomy and/or radiotherapy [3]. The variability in disease progression suggests not only molecular changes impacting cancer cells but also the intricate interactions between cancer cells and their microenvironment [4]. Some studies have suggested a positive correlation between periprostatic adipose tissue (PPAT) thickness and the aggressiveness of PC [5, 6]. Another study investigated factors affecting the response to docetaxel in metastatic PC, it was found that adipocytes surrounding the tumor promote docetaxel resistance through a mechanism dependent on β-tubulin isoform 2B (TUBB2B) [7].
Considering these facts, conducting a detailed study of various clinical cases to identify genetic markers that characterize different disease stages becomes essential. These biomarkers will play a crucial role alongside established parameters [prostate-specific antigen (PSA), Gleason score] in further refining the stratification of PC. Such refinement is critical for selecting the most appropriate therapeutic strategies.
At the molecular level, numerous studies investigating signaling pathways altered in PC have revealed that genes involved in the DNA damage repair pathway are often affected by both somatic and germline mutations [8]. Among these are genes breast cancer gene 1 (BRCA1) and BRCA2, which act as tumor suppressors involved in the homologous recombination repair (HRR) process of double-strand breaks [9]. Indeed, BRCA germline mutations in PC patients have been associated with more aggressive disease and poorer clinical outcomes. Cancer-specific survival (CSS) and metastasis-free survival (MFS) at 5 years are also negatively affected, with a CSS of 8.6 years in BRCA mutation carriers compared to 15.7 years in non-carriers, and an MFS of 77% versus 93% [10]. Based on the finding that alterations in DNA repair pathways correlate with metastasis occurrence and poor prognosis, therapeutics such as PARP inhibitor [poly(adenosine diphosphate-ribose) polymerase inhibitors] targeting BRCA1 and BRCA2 genes have been developed to treat patients with metastatic castration-resistant PC (mCRPC) [10, 11]. However, since PARP inhibitor therapies do not work in all patients with DNA repair gene alterations, further investigation of other genes in this pathway is warranted to better understand the dysfunctions that may be associated with the occurrence of PC. One of the most important gene in DNA repair process is tumor protein 53 (TP53), often referred to as the guardian of the genome due to its role in defending against various external and internal stressors. It is a key gene regulating the transcription of several genes involved in important biological processes [12], which explains that gene dysfunction induced by various genetic alterations is found in more than 50% of human primary tumors [13]. The germline TP53 mutation is associated with Li-Fraumeni syndrome (LFS), an inherited autosomal dominant disorder, associated with a predisposition to several cancers like breast carcinomas, sarcomas, brain tumors, and adrenal cortical carcinomas [14].
Although mutations in the TP53 gene have traditionally been considered a late event in PC [15, 16], recent studies have reported significant frequencies of TP53 mutations in primary, and, particularly, in castration-naive metastatic PC [17, 18]. A study conducted on 175 primary PC patients, who later developed mCRPC, found that TP53 gene aberrations were the most frequent genetic events, detected in 25% of the studied population (primary PC) [19]. Another study on mutant TP53 clones from primary prostate tumors, suggested that these mutations could be the cause of metastatic spread with a lag period of 17 years [20]. Given these data, there is an emerging hypothesis that mutations in the TP53 gene may be an early event in certain forms of PC and may predispose to a lethal disease outcome.
Hence, the aim of this study was to evaluate the mutational profile of exon 5 of the TP53 gene, considering that mutation of this gene is mainly located in exons 5 and 8 [21]. The study group comprises 48 patients at various stages of PC to assess the mutational status of the TP53 gene in both advanced and localized forms.
Required ethical approval was obtained from the committee of biomedical research ethics in Morocco (No. 3/2018/April 30/2018). Blood samples were collected between June 2021 and February 2022 at the Urology Department, Military Hospital teaching Mohammed V, Rabat, Morocco, from a population of 48 subjects diagnosed with PC. Histological evidence has already been obtained to diagnose them with adenocarcinoma of the prostate. Samples were obtained according to standard protocols directly by physicians. Every sample was attached with clinical and pathological parameters: age, PSA concentration, Gleason score, and the stage of the disease. The clinical characteristics of PC patients are given in Table 1.
The clinical characteristics of prostate cancer patients
| Parameters | N (%) | |
|---|---|---|
| Age | < 60 | 6 (12.5) |
| ≥ 60 | 39 (81.25) | |
| Unknown | 3 (6.25) | |
| PSA (ng/mL) | ≤ 10 | 13 (27) |
| > 10 and < 20 | 18 (37.5) | |
| ≥ 20 | 14 (29) | |
| Unknown | 3 (6.25) | |
| Gleason score | < 7 | 13 (27) |
| 7 | 15 (31.25) | |
| > 7 | 17 (35) | |
| Unknown | 3 (6.25) | |
| Clinical stage* | T1 | 14 (29) |
| T2 | 24 (50) | |
| T3 | 4 (8) | |
| T4 | 3 (6.25) | |
| Unknown | 3 (6.25) | |
| Smoking | Yes | 22 (46) |
| No | 23 (48) | |
| Unknown | 3 (6.25) | |
| Alcohol | Yes | 14 (29) |
| No | 31 (65) | |
| Unknown | 3 (6.25) | |
PSA: prostate-specific antigen. * according to the American Joint Committee on Cancer Classification and Stage Group (AJCC, 7th edition).
DNA extraction took place in the oncology and virology laboratory in the faculty of sciences and techniques at Mohammedia, Morocco. Genomic DNA was extracted from a volume of approximately 200 μL using the Blood Kit Roche Applied System. The absorbance was measured using the NanoDrop spectrophotometer 2000 (Thermo Scientific) at 260/280 nm for purity check and concentration verification. Samples with a DNA concentration greater than or equal to 30–60 ng/μL were selected to perform the polymerase chain reaction. To evaluate the quality and integrity of the extracted DNA, a 268-base-pair fragment of the housekeeping β-globin gene was amplified using the GH20/PCO4 primer set as previously described [22].
The polymerase chain reaction (PCR) amplifications targeting exons 5 were performed using a specific set of primers described elsewhere [23]. The amplification reaction was conducted in “Perkin Elmer 2400 Thermal Cycler®, CA, USA” using the Master Mix Vazyme Green Taq Mix. The reliability and the quality of the extraction were assessed by the amplification of β-globin through the following steps: initial primary denaturation for 10 min at 94°C, 35 cycles of denaturation at 94°C for 45 s, hybridization at 54°C for 45 s, extension at 72°C for 1 min. All positive β-globin gene PCR products were subjected to another amplification targeting the exon 5 of TP53 gene by using the primers which sequences are: F-5’CAC TTG TGC CCT GAC TTT CAA C-3’ for the forward strand and R-5’-CAA CCA GCC CTG TCG TCT CTC-3’ for the reverse strand. The cycling program consisted of the following steps: initial denaturation at 94°C for 10 min; 35 cycles of 94°C denaturation for 1 min, hybridization at 56°C for 1 min, 72°C extension for 1 min; followed by final extension at 72°C for 10 min. PCR products corresponding to the 5th exon of TP53 gene which is 184 bases pair (bp) in length, were analyzed and resolved by running the samples through electrophoresis at 70V on a 1.5% concentrated agarose gel (DNA SUB CELTM, Bio-RAD, Italy) and visualization was done after addition of the Ethidium bromide.
The sequencing was performed at the National Center for Scientific and Technical Research (CNRST) in Rabat, Morocco. The purification step was performed by the use of the ExoSAP-IT Express PCR Product Cleanup system in order to remove primers and nucleotides not involved in the PCR reaction. After the purification step, the BigDye XTer-minator purification kit was employed for sequencing the purified products in the 3130 genetic analyzers machine (Applied Biosystems).
For the identification of mutations, the Refseq (NM_000546.6) of the TP53 gene was used as the reference sequence. The mutation search was performed using MEGA program, Nagahama server [24]. The novel mutations detected by this study were submitted to the ClinVar—NCBI database and were recorded as clinically relevant variants, type Condition ID: MedGen, Condition ID value: C4722327 (hereditary prostate cancer.1) by accession numbers of TP53 exon 5 (SCV004232051-SCV004232160). The resulting TP53 exon 5 DNA sequences were successfully submitted to GenBank under the following accession numbers (PP350183-PP350261).
All statistical analysis was generated using SPSS software (SPSS version 11.0, Chicago, IL). The clinicopathological characteristics were compared between the mutation carrier and non-mutation carrier groups, and the statistical analysis was performed by Chi-squared tests or Fisher exact test to analyze categorical data (when one of the theoretical numbers is less than 5). The difference was considered statistically significant when the P-value was ≤ 0.05.
In order to explore the effects of novel missense variants, two computational prediction tools were used: SIFT BLINK and Polyphen 2.0. SIFT (http://sift.bii.a-star.edu.sg/www/SIFT_seq_submit2.html) is a sequence homology-based tool that predicts whether an amino acid substitution in a protein will have a phenotypic effect [25]. The native TP53 protein, accession: NP_000537.3, 393 amino acids, and mutated TP53 protein’s sequences were submitted as input file to SIFT server. Output scores are in a range between 0 to 1, corresponding to a predicted effect from damaging (score 0) to neutral (score 1).
PolyPhen 2.0 (http://genetics.bwh.harvard.edu/pph2/uses) is another tool that uses a prediction method based on the calculation of the difference in PSIC (position-specific independent count) score between the wildtype and the mutant amino acid [26]. The sequences were submitted in FASTA format along with the positions of the native and mutant substitutions amino acids. A score > 0.85 indicates that the variant is probably damaging, if the score is > 0.15 possibility that the variant is damaging, and if below this threshold, the variant is considered benign.
The clinical characteristics of study subjects are summarized in Table 1. Individuals aged over 60 represent more than 81% of the population studied. Seventeen individuals (35%) have a Gleason score > 7, fifteen subjects (31.25%) have a score of 7, and thirteen patients (27%) have a Gleason score < 7. About the PSA value, 37.5% of the population have a PSA level ranging between 10 and 20 ng/mL. The analysis based on the clinical stage showed that fifty percent of patients are at stage T2, 29% of the population is at stage T1, and only 8% and 6.25% are at stage T3 and T4, respectively. The analysis of other collected parameters related to certain risk factors such as smoking and alcohol has shown that 46% of patients smoke, and 48% are non-smoker. Regarding alcohol consumption, 29% of patients drink alcohol, while 65% of the population doesn’t drink.
The mutations were arranged according to genotype, genome location, the effects on the amino acids, and frequency rate. Our research showed that some patients harbor known TP53 gene variants, but also identified new mutations of this gene. Among the population studied, 38 (79%) patients carry TP53 mutations. A total of 137 germline mutations of the TP53 gene were identified, including 115 new mutations. All the mutation carriers harbored at less than three mutations and some subjects exhibit a significant frequency of mutation such as case 43 with seventeen mutations. The effects of the identified alterations are of different types, but with a predominance of the frameshift type and missense mutations. Mutations were found in patients at different stages of the disease, but predominantly in stage T2 (Figure 1).
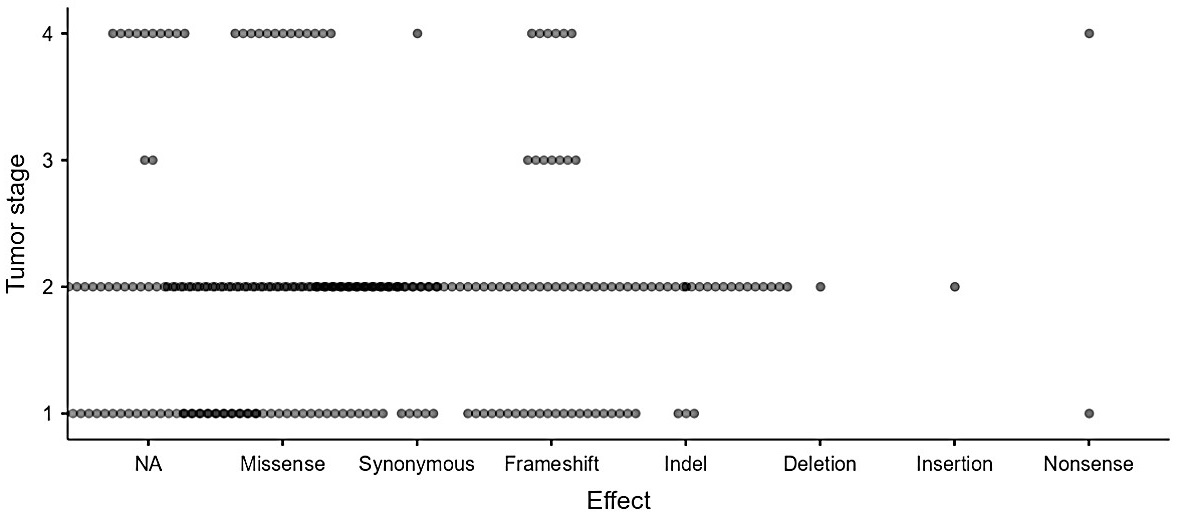
TP53 mutations profile by tumor stage in Moroccan prostate cancer patients. The presentation illustrates the distribution of the 137 mutations found in 38 patients according to the disease stage. The graph takes into account recurring mutations carried by more than one patient. The variation in circle colors represents the frequency of mutations at each cancer stage; the more common a type of mutation is, the darker the circle
Among the new germline mutations detected in the TP53 gene, we noted a significant frequency of frameshift mutation. Sixty-one new frameshift mutations were identified in 36 patients (95% of mutation carriers), the mutation c.392delA was recorded in fifteen cases (31%); the mutations c.383delC and c.432delG observed at a frequency of 12.5% and 10% respectively.
Eighteen missense new mutations of the TP53 gene were detected in 68% of the mutation carriers group, the most frequent was the variant c.502C>A (p.His168Asn) identified in eleven patients (23%). The mutation c.382C>T resulting in the substitution of proline at position 128 by serine was observed in six cases (12.5%). The two other mutations: c.398T>G (p.Met133Arg) and c.413C>G (p.Ala138Gly) were observed in four cases (8%), whilst the 12 remaining variants were observed only once. Regarding the synonymous mutations, seven variants were recorded but the mutant c.441T>G (p.Val147=) was the only recurrent variant, identified in three cases (6%). One nonsense mutation was identified in one patient and resulted in a stop codon at position 126 (tyrosine). All codons affected by these alterations are part of the DNA binding domain of the protein. The most common alterations revealed, genome locations, and phenotypes are given in Table 2.
TP53 exon 5 highly frequent new mutations detected in Moroccan prostate cancer patients with the corresponding effects according to Human Genome Variation Society (HGVS) nomenclature
| Effect | CDS change | Genome location | Protein change | Frequency, N = 48 |
|---|---|---|---|---|
| Deletion | c.534_536delCCA | Chr17:7675076 | NA | 1 (2%) |
| Frameshift | c.392delA | Chr17:7675220 | NA | 15 (31.25%) |
| Frameshift | c.383delC | Chr17:7675229 | NA | 6 (12.5%) |
| Frameshift | c.432delG | Chr17:7675180 | NA | 5 (10%) |
| Frameshift | c.396_397insT | Chr17:7675215 | NA | 4 (8%) |
| Frameshift | c.496_497insG | Chr17:7675115 | NA | 4 (8%) |
| Frameshift | c.388_389insC | Chr17:7675223 | NA | 3 (6.25%) |
| Frameshift | c.433delC | Chr17:7675179 | NA | 3 (6.25%) |
| Frameshift | c.379_380insT | Chr17:7675232 | NA | 2 (4%) |
| Frameshift | c.393delC | Chr17:7675219 | NA | 2 (4%) |
| Frameshift | c.413delinsTG | Chr17:7675199 | NA | 2 (4%) |
| Frameshift | c.428_429insT | Chr17:7675183 | NA | 2 (4%) |
| Frameshift | c.431_432delinsG | Chr17:7675180 | NA | 2 (4%) |
| Frameshift | c.532_533insC | Chr17:7675079 | NA | 2 (4%) |
| Frameshift | c.548_549insT | Chr17:7675063 | NA | 2 (4%) |
| Missense | c.502C>A | Chr17:7675110 | p.His168Asn | 11 (23%) |
| Missense | c.382C>T | Chr17:7675230 | p.Pro128Ser | 6 (12.5%) |
| Missense | c.398T>G | Chr17:7675214 | p.Met133Arg | 4 (8%) |
| Missense | c.413C>G | Chr17:7675199 | p.Ala138Gly | 4 (8%) |
| Missense | c.457C>G | Chr17:7675155 | p.Pro153Ala | 3 (6.25%) |
| Missense | c.550G>C | Chr17:7675062 | p.Asp184His | 2 (4%) |
| Missense | c.376_377delinsCT | Chr17:7675235 | p.Tyr126Arg | 1 (2%) |
| Missense | c.427G>C | Chr17:7675185 | p.Val143Leu | 1 (2%) |
| Missense | c.428_429delinsG | Chr17:7675183 | p.Val143Ala | 1 (2%) |
| Missense | c.428T>A | Chr17:7675184 | p.Val143Glu | 1 (2%) |
| Missense | c.431_432delinsGG | Chr17:7675180 | p.Gln144Pro | 1 (2%) |
| Missense | c.432_433delinsCA | Chr17:7675179 | p.Gln144His | 1 (2%) |
| Missense | c.443A>T | Chr17:7675169 | p.Asp148Val | 1 (2%) |
| Missense | c.454C>A | Chr17:7675158 | p.Pro152Thr | 1 (2%) |
| Missense | c.461_462delinsAT | Chr17:7675150 | p.Gly154Asp | 1 (2%) |
| Missense | c.476C>A | Chr17:7675136 | p.Ala159Asp | 1 (2%) |
| Missense | c.490A>C | Chr17:7675122 | p.Lys164Gln | 1 (2%) |
| Missense | c.496T>G | Chr17:7675116 | p.Ser166Ala | 1 (2%) |
| NA | c.559+13_559+12delinsAG | Chr17:7675040 | NA | 12 (25%) |
| NA | c.376-2delA | Chr17:7675238 | NA | 10 (21%) |
| NA | c.559+35G>A | Chr17:7675018 | NA | 8 (17%) |
| NA | c.376-6delC | Chr17:7675242 | NA | 7 (15%) |
| NA | c.376-12delC | Chr17:7675248 | NA | 6 (12.5%) |
| NA | c.376-3delC | Chr17:7675239 | NA | 5 (10%) |
| NA | c.376-6C>G | Chr17:7675242 | NA | 5 (10%) |
| NA | c.376-11T>C | Chr17:7675247 | NA | 3 (6.25%) |
| NA | c.376-36_376-35insC | Chr17:7675271 | NA | 2 (4%) |
| NA | c.376-4A>C | Chr17:7675240 | NA | 2 (4%) |
| NA | c.376-8delT | Chr17:7675244 | NA | 2 (4%) |
| Nonsense | c.378C>A | Chr17:7675234 | p.Tyr126Ter | 1 (2%) |
| Synonymous | c.441T>G | Chr17:7675171 | p.Val147= | 3 (6.25%) |
| Synonymous | c.375_376delinsCT | Chr17:7675236 | p.Thr125= | 1 (2%) |
| Synonymous | c.387C>T | Chr17:7675225 | p.Ala129= | 1 (2%) |
| Synonymous | c.390C>G | Chr17:7675222 | p.Leu130= | 1 (2%) |
| Synonymous | c.453C>A | Chr17:7675159 | p.Pro151= | 1 (2%) |
| Synonymous | c.492_493delinsTT | Chr17:7675119 | p.Lys164= | 1 (2%) |
| Synonymous | c.531C>A | Chr17:7675081 | p.Pro177= | 1 (2%) |
CDS: coding DNA sequences
Among the 137 detected mutations, twenty-two (16%) were identified in 34 cases (71%) and correspond to known variants (Table 3). The most frequent mutation type was the missense variants, observed in eighteen patients. Among ten missense changes, the variant rs1057519977 (p.Cys141Trp) was the most frequent and has been recorded in nine patients. The two missense variants rs1131691037 (p.Asn131Ile) and rs138729528 (p.Arg175Gly) were recorded at a frequency of 10% and the variants rs1555526335 (p.Tyr126Cys) and rs758781593 (p.Gln136His) at a frequency of 8%. Five other missense variants were also identified but only once: rs1555526226 (p.Val147Ile), rs28934875 (p.Ala138Pro), rs587782596 (p.Arg181Ser), rs730881999 (p.Ser127Cys), rs786203071 (p.Gln144Pro). One mutation resulting in the insertion of Proline (rs786202525) and another one resulting in a truncated protein in position 144 (rs757274881) were also identified once.
TP53 exon 5 frequency of known variants in Moroccan prostate cancer patients and their corresponding effects according to NCBI 1000 Genomes Browser and Human Genome Variation Society (HGVS) nomenclature
| Effect | Variant ID | CDS change | Genome location | Protein change | Frequency, N = 48 |
|---|---|---|---|---|---|
| Insertion | rs786202525 | c.532_533insCCC | Chr17:7675079 | p.Pro177_His178insPro | 1 (2%) |
| Missense | rs1057519977 | c.423C>G | Chr17:7675189 | p.Cys141Trp | 9 (19%) |
| Missense | rs1131691037 | c.392A>T | Chr17:7675220 | p.Asn131Ile | 5 (10%) |
| Missense | rs138729528 | c.523C>G | Chr17:7675089 | p.Arg175Gly | 5 (10%) |
| Missense | rs1555526335 | c.377A>G | Chr17:7675235 | p.Tyr126Cys | 4 (8%) |
| Missense | rs758781593 | c.408A>C | Chr17:7675204 | p.Gln136His | 4 (8%) |
| Missense | rs1555526226 | c.439G>A | Chr17:7675173 | p.Val147Ile | 1 (2%) |
| Missense | rs28934875 | c.412G>C | Chr17:7675200 | p.Ala138Pro | 1 (2%) |
| Missense | rs587782596 | c.541C>A | Chr17:7675071 | p.Arg181Ser | 1 (2%) |
| Missense | rs730881999 | c.380C>G | Chr17:7675232 | p.Ser127Cys | 1 (2%) |
| Missense | rs786203071 | c.431A>C | Chr17:7675181 | p.Gln144Pro | 1 (2%) |
| NA | rs1321881901 | c.559+31G>A | Chr17:7675022 | NA | 26 (54%) |
| NA | rs1032547645 | c.559+22G>A | Chr17:7675031 | NA | 3 (6.25%) |
| NA | rs1221388024 | c.376-4A>T | Chr17:7675240 | NA | 1 (2%) |
| NA | rs1466952182 | c.376-51C>T | Chr17:7675287 | NA | 1 (2%) |
| NA | rs775915220 | c.559+8G>A | Chr17:7675045 | NA | 1 (2%) |
| NA | rs786202799 | c.376-2A>G | Chr17:7675238 | NA | 1 (2%) |
| Nonsene | rs757274881 | c.430C>T | Chr17:7675182 | p.Gln144Ter | 1 (2%) |
| Synonymous | rs786203928 | c.433C>T | Chr17:7675179 | p.Leu145= | 2 (4%) |
| Synonymous | rs1131691034 | c.492G>A | Chr17:7675120 | p.Lys164= | 1 (2%) |
| Synonymous | rs761222871 | c.468C>T | Chr17:7675144 | p.Arg156= | 1 (2%) |
| Synonymous | rs876659481 | c.456G>A | Chr17:7675156 | p.Pro152= | 1 (2%) |
CDS: coding DNA sequences
The clinicopathological characteristics were compared between the mutation carrier and non-mutation carrier groups (Table 4). Fifteen patients of the mutation carriers have a PSA ranging between 10 and 20 ng/mL, while only three patients of non-carriers have this value (P = 0.614). Concerning the Gleason score, 13 patients among mutation carriers have a score greater than 7, while four among non-carriers have this score (P = 0.815). Regarding the clinical stage, 20 carriers of mutations are at the T2 stage, while four out of ten non-carriers are in this stage (P = 0.364). The smoking individuals among the mutation carriers were fifteen, while seven individuals belonged to the non-carriers group (P = 0.053). Regarding alcohol consumption, twelve of the mutation carriers are an alcohol consumer and six are from the non-carriers group (P = 0.010).
Assessment of the correlation between carrier and non-carriers of TP53 exon 5 mutations and clinicopathological features
| Parameters | No. cases, n (%)(N = 48) | Carriers, n (%)(N = 38) | Non-carriers, n (%)(N = 10) | P | |
|---|---|---|---|---|---|
| Age | < 60 | 6 (12.5) | 4 (11) | 2 (20) | 0.314 |
| ≥ 60 | 39 (81.25) | 32 (84) | 7 (70) | ||
| Unknown | 3 (6.25) | 2 (5) | 1 (10) | ||
| PSA (ng/mL) | ≤ 10 | 13 (27) | 10 (26) | 3 (30) | 0.614 |
| > 10 and < 20 | 18 (37.5) | 15 (39) | 3 (30) | ||
| ≥ 20 | 14 (29) | 11 (29) | 3 (30) | ||
| Unknown | 3 (6.25) | 2 (5) | 1 (10) | ||
| Gleason score | < 7 | 13 (27) | 11 (29) | 2 (20) | 0.815 |
| 7 | 15 (31.25) | 12 (32) | 3 (30) | ||
| > 7 | 17 (35) | 13 (34) | 4 (40) | ||
| Unknown | 3 (6.25) | 2 (5) | 1 (10) | ||
| Clinical stage | T1 | 14 (29) | 11 (29) | 3 (30) | 0.364 |
| T2 | 24 (50) | 20 (53) | 4 (40) | ||
| T3 | 4 (8) | 2 (5) | 2 (20) | ||
| T4 | 3 (6.25) | 3 (8) | 0 (0) | ||
| Unknown | 3 (6.25) | 2 (5) | 1 (10) | ||
| Alcohol | Yes | 14 (29) | 12 (32) | 6 (60) | 0.010 |
| no | 31 (65) | 28 (74) | 3 (30) | ||
| Unknown | 3 (6.25) | 2 (5) | 1 (10) | ||
| Smoking | Yes | 22 (46) | 15 (39.5) | 7 (70) | 0.053 |
| No | 23 (48) | 21 (55.25) | 2 (20) | ||
| Unknown | 3 (6) | 2 (5) | 1 (10) | ||
PSA: prostate-specific antigen
Protein sequence and mutational positions were submitted to SIFT Blink server, the server predicts if the amino acid substitution alters the protein function. The intolerant range ≤ 0.05 has been used as a limit of variants classification. More than this limit indicates as damaging to the protein function. 13 (72%) out of 18 missense variants were predicted to affect the protein function. Eight showed the highest score of 0.00. Five of the missense variants tested (28%) were predicted as tolerated. These results were combined with scores given by the Polyphen tool. The scores of this last were comprised from zero to one. Score (1.00) means that substitution is probably damaging, low scores were considered a benign effect. Out of 18 missense variants, 13 (72%) were predicted as probably damaging with values range: (0.965–1.00), two missense variants possibly damaged the protein with values of 0.466 and 0.768, the rest were benign. The results of missense variants effect prediction are given in Table 5.
Prediction of the effects of novel missense variants on the TP53 protein by SIFT and PolyPhen 2.0 tools
| CDS | Protein change | SIFT | Polyphen | ||
|---|---|---|---|---|---|
| Score | Effect | Score | Effect | ||
| c.502C>A | p.His168Asn | 0.02 | Affect protein function | 0.995 | Probably damaging |
| c.382C>T | p.Pro128Ser | 0.13 | Tolerated | 0.967 | Probably damaging |
| c.398T>G | p.Met133Arg | 0 | Affect protein function | 0.021 | Benign |
| c.413C>G | p.Ala138Gly | 0 | Affect protein function | 1 | Probably damaging |
| c.457C>G | p.Pro153Ala | 0.11 | Tolerated | 0.001 | Benign |
| c.550G>C | p.Asp184His | 0.03 | Affect protein function | 0.965 | Probably damaging |
| c.376_377delinsCT | p.Tyr126Arg | 0 | Affect protein function | 1 | Probably damaging |
| c.427G>C | p.Val143Leu | 0.09 | Tolerated | 0.466 | Possibly damaging |
| c.428_429delinsG | p.Val143Ala | 0 | Affect protein function | 1 | Probably damaging |
| c.428T>A | p.Val143Glu | 0 | Affect protein function | 1 | Probably damaging |
| c.431_432delinsGG | p.Gln144Pro | 0.04 | Affect protein function | 1 | Probably damaging |
| c.432_433delinsCA | p.Gln144His | 0.03 | Affect protein function | 0.999 | Probably damaging |
| c.443A>T | p.Asp148Val | 0.25 | Tolerated | 0.001 | Benign |
| c.454C>A | p.Pro152Thr | 0 | Affect protein function | 0.996 | Probably damaging |
| c.461_462delinsAT | p.Gly154Asp | 0.01 | Affect protein function | 1 | Probably damaging |
| c.476C>A | p.Ala159Asp | 0 | Affect protein function | 0.992 | Probably damaging |
| c.490A>C | p.Lys164Gln | 0 | Affect protein function | 0.998 | Probably damaging |
| c.496T>G | p.Ser166Ala | 0.44 | Tolerated | 0768 | Possibly damaging |
CDS: coding DNA sequences
Despite the great diversity of genes involved in oncogenesis, the transcription factor TP53 remains a key tumor suppressor and a master regulator of various signaling pathways involved in this process [27]. It is considered a powerful tumor suppressor because of its various roles including the ability to induce cell cycle arrest, DNA repair, senescence, and apoptosis, to name only a few. Furthermore, TP53 mutations were reported to occur in almost every type of cancer at rates varying between 10% (e.g., in hematopoietic malignancies) [28] and close to 100% (e.g., in high-grade serous carcinoma of the ovary) [29].
Germline mutation of TP53 is also associated with cancer predisposition emphasized notably by LFS characterized by a wide spectrum of tumor types occurring over a wide age range, starting at a young age [30].
Here, we identified 22 variants in the DNA-binding domain of the TP53 gene, in Moroccan PC patients, which were previously reported. The mutation c.423C>G (p.Cys141Trp) is one of the highly frequent variants recorded in 9 patients (19%). This mutant was reported in breast cancer and LFS [31]. The mutant c.392A>T detected in 5 patients, resulted in deleterious substitution of asparginine in position 131 by isoleucine, this variant was identified first in a family with a history of multiple malignancies [32]. Another missense variant identified at the same frequency is c.523C>G mutant and affects the arginine in position 175, considered one of the hot spot residues. This alteration was identified in a French family meeting LFS criteria [33], besides, another alteration at this same amino acid position (p.R175H) is a well-characterized TP53 hotspot mutation [34]. The nonsense variant c.430C>T identified in one case, generates a premature translational stop signal (p.Gln144*) in the TP53 gene. It is expected to result in an absent or disrupted protein product. This mutant has been observed in individuals with clinical features of LFS [35]. Within the known mutations identified the intronic variant c.559+31G>A was the most frequently recorded in 26 patients (54%).
Considering previous reports on TP53 gene, the majority of mutations reported are missense mutations [36]. Indeed, among the mutants identified in our study, missense variants were very frequent and have been recorded in 27 cases. Furthermore, 79% of the population studied was found to carry germline mutation all localized at the DNA-Bainding site, according to previous studies this site hosts 90% of the mutations between residues 110 and 290, and this could therefore lead to the inhibition of its transcriptional activity [37]. Although few data are available on the association of PC with germline TP53 mutations [38, 39], but a large study by Maxwell et al. [40] identified germline TP53 mutations in 38 PC patients (0.55% prevalence) with a relative risk of having germline TP53 significantly elevated at 9.1 (95% CI 6.2–14, P < 0.0001) compared with the non-cancer population database. In the same study, the incidence of PC in LFS was assessed, and PC was identified in 31 cases of 163 (19%).
Our study identified a high frequency of new germlines mutations affecting the TP53 gene which do not appear to be associated with LFS, a similar finding was reported by the Maxwell et al. study [40], which revealed that over half of the germline TP53 variants identified in PC patients are considered attenuated or hypomorphic variants and not typically associated with classic LFS. The actual effects of these variants on the TP53 protein are unknown. However, the two prediction tools SIFT and PolyPhen-2, employed to assess the impact of missense variants, revealed that more than half of these mutants are predicted to alter the function of the TP53 protein. Thus, these mutations lead to defects in the protein pathway, affecting its role as a tumor suppressor gene. The variants predicted as the most deleterious by both SIFT and Polyphen.2 tools namely: p.Ala138Gly, p.Tyr126Arg, p.Val143Ala, p.Val143Glu, p.Gln144Pro, p.Gln144His, p.Pro152Thr, p.Gly154Asp, p.Ala159Asp, p.Lys164Gln, p.His168Asn, p.Asp184His. The codon at position 143 could be considered a critical residue. A study by Dridi et al. [41] aimed to determine the dominant-negative effect of different TP53 mutations in the near-diploid LoVo colon carcinoma cell line in response to mitotic spindle inhibitors, demonstrated that the TP53-175H and p53-143A mutant clones re-enter S phase with no apparent arrest unlike the wild type showing a tetraploid G1 cell arrest. Regarding the functional significance of TP53 mutations, missense mutation can have dominant negative effects on the transactivation of other genes containing p53-specific responsive elements. According to Forrester et al. [42] generally minimal dominant negative effects can be attributed only to codons 143ala-, 175his-, 248trp-, 249ser-, 273his-mutations in PC cell line PC-3, lacking one base pair in codon 138.
Despite the absence of a correlation between the clinicopathological features and mutational status, we noticed a significant frequency of mutations in patients at localized stages of PC T1 (29%) and T2 (50%) (Figure 1). Although alterations of this gene have been mainly associated with advanced stages of PC and constitute a late event in carcinogenesis [43, 44], substantial aberrations in TP53 have previously been reported not only in advanced PC but also in primary PC and were associated with poor patient outcomes [19, 45–47]. Taken together, our results suggested that some patients may harbor germline genetic alterations in the TP53 gene, which are likely involved in the initiation or progression of the disease. Indeed, unlike somatic mutations, which do not have a hereditary character, germline mutations involve gametes by definition and are therefore passed down to the offspring [48]. Hence, the presence of these inherited alterations could increase the risk of developing hereditary cancer.
Germline mutations affecting other genes including the BRCA2 and BRCA1 genes were also identified by the Cancer Genome Atlas Research Network in 333 patients with primary PC, with respective rates of 13% and 0.3% [45]. Recently, another study involving a population of 620 PC patients, reported prevalence rates of 24.3% and 6.4% for BRCA1 and BRCA2 respectively [49]. Indeed, alterations in BRCA1/2 genes are included in gene panels used for patient stratification in the context of PARP inhibitors, which currently do not include TP53 [50].
In addition to genetic alterations affecting crucial genes like tumor suppressors TP53, BRCA1, and BRCA2 in the DNA damage repair pathway, as well as various other genes such as PTEN and PIK3ca in the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mTOR pathway [51], various molecular and biological aspects must be explored together, to better elucidate the onset and progression of the disease. Indeed, variations in cancer outcomes can be observed within the same phenotype, and the disease frequently progresses to severe forms, such as CRPC and mCRPC [48]. Recent research highlights the significant roles of circular RNAs (CircRNAs) in chemotherapy resistance in various cancers [52]. CircRNAs are known to be dysregulated in various malignancies, including PC [53, 54]. A recent study investigating the role of CircRNAs in CRPC identified four CircRNAs that are upregulated during the progression to castration resistance in patient-derived xenografts (PDXs) [55]. These CircRNAs can also be detected in the plasma of PC patients, suggesting their potential utility as biomarkers for CRPC [56]. Another study by Shen et al. [57] has revealed that CircRNAs are involved in determining the sensitivity of PC to docetaxel.
Another aspect that has recently been explored is the role of the tumor microenvironment (TME) in the disease. The TME, encompasses a diverse array of cell types, including cancer-associated fibroblasts (CAFs), immune cells, endothelial cells, and stromal cells. Additionally, non-cellular components, such as extracellular matrix proteins (ECM) related proteins, cytokines, chemokines, and extracellular vesicles (EVs) are integral [58]. These elements interact via a complex network and mounting evidence indicates that this crosstalk plays a crucial role in the onset and progression of CRPC [59].
Cross-referencing data relating to all characterized factors in PC, such as genetic alterations, interactions between different signaling pathways, other biomarkers like CircRNAs, and elucidating the role played by the TME, will help better characterize various pathological profiles of PC. Consequently, this will enable personalized management through targeted therapies.
Our results showed a substantial frequency of germline mutation in TP53 in Moroccan PC patients. Among these mutations, several known variants are associated with LFS, a similar finding was noted in another study, which reflects that adult cancers such as PC in LFS may be understudied and merit further attention. Furthermore, the identification of novel germline mutations of TP53 with high-frequency rate, some of which affect a hot spot codon, suggests that the role of the TP53 gene in PC development might be greater than expected before, and the possibility of finding PC clinical relevance with such alterations is present. Hence, we suggest further investigation of germline mutations of the TP53 by considering family history data, and ideally by paired tumor-normal testing in PC patients, to rule out somatic interference. This may allow reconsidering the gene panel testing in order to improve the personalized management of the disease considering other parameters for the stratification of the cancer.
BRCA1: breast cancer gene 1
CircRNAs: circular RNAs
LFS: Li-Fraumeni syndrome
mCRPC: metastatic castration-resistant prostate cancer
PARP: poly(adenosine diphosphate-ribose) polymerase
PC: prostate cancer
PCR: polymerase chain reaction
PSA: prostate specific antigen
TME: tumor microenvironment
TP53: tumor protein 53
Authors would like to thank the University Hassan II of Casablanca and the faculty of Sciences for all their efforts to support us during all the stages of this research. Thanks as well go to the team members of Laboratory: Virology, Oncology, Biosciences, Environment, and New BioEnergies of the FSTM. Many thanks to Mohammed V hospital in Rabat, Morocco for their help during sample collection.
KA: Conceptualization, Methodology, Formal analysis, Investigation, Project administration, Writing—original draft, Writing—review & editing. AL, RT, and KE: Resources. MME: Project administration, Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare no conflicts of interest.
The study protocol was approved by the Ethics Committee for Biomedical Research of the Faculty of Medicine and Pharmacy of Casablanca, Morocco (No. 3/2018/April 30/2018).
The informed consent to participate in the study was obtained from all participants.
Not applicable.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

