 Review
Review

Affiliation:
Clinical, Experimental, and Applied Physiology Unit, Department of Human Physiology, Gregory University Uturu, Abia state 441106, Nigeria
Email: osahmartinz@gmail.com
ORCID: https://orcid.org/0000-0002-2451-4466
Explor Med. 2025;6:1001289 DOI: https://doi.org/10.37349/emed.2025.1001289
Received: October 01, 2024 Accepted: February 09, 2025 Published: February 28, 2025
Academic Editor: Undurti N. Das, UND Life Sciences, USA
This review explored the physiological mechanisms underlying arginine vasopressin deficiency (AVP-D, formerly central diabetes insipidus) and AVP resistance (AVP-R, formerly nephrogenic diabetes insipidus), with a focus on water balance regulation. Vital components include the hypothalamic-pituitary-AVP axis, renal AVP responsiveness, and neural mechanisms of thirst regulation. Recent insights on thirst generation within circumventricular brain nuclei (subfornical organ, median preoptic nucleus, and organum vasculosum of the lamina terminalis) are discussed, along with the diagnostic utility of copeptin in polyuric states. This review highlighted the critical role of hypothalamic-pituitary integrity and renal AVP responsiveness in maintaining water-electrolyte homeostasis. Understanding these mechanisms provided the foundation for optimizing therapeutic strategies and advancing research on AVP-related disorders.
Arginine vasopressin deficiency [AVP-D, formerly central diabetes insipidus (DI)] and AVP resistance (AVP-R, formerly nephrogenic DI) are disorders characterized by impaired water balance regulation, leading to excessive thirst (polydipsia) and the production of large volumes of dilute urine (polyuria) [1]. These conditions arise from disruptions in the hypothalamic-pituitary-AVP axis or renal responsiveness to AVP, also known as antidiuretic hormone (ADH). AVP-D results from insufficient production or release of AVP from the hypothalamus or pituitary gland, while AVP-R stems from renal insensitivity to AVP, impairing water reabsorption despite adequate hormone levels [2–4].
The hypothalamic-pituitary-AVP axis plays a crucial role in maintaining fluid homeostasis by regulating AVP secretion in response to plasma osmolality and blood volume changes. Equally important is the neural control of thirst, which ensures appropriate water intake to complement AVP-mediated renal water conservation. The circumventricular brain nuclei, particularly the subfornical organ (SFO), median preoptic nucleus (MnPO), and organum vasculosum of the lamina terminalis (OVLT), serve as primary osmosensors that integrate signals from plasma osmolality and angiotensin II to drive thirst and AVP release. Recent studies utilizing optogenetics and neural circuit mapping have elucidated how these regions coordinate anticipatory and compensatory responses to dehydration. Notably, research by Pool et al. [5] and Augustine et al. [6] has provided insight into the cellular basis of these processes, while Kim et al. [7] described how AVP secretion is modulated even before detectable disturbances in osmolality. Also, advances in biomarker research have highlighted the role of copeptin, the C-terminal fragment of pre-proAVP, as a stable and reliable surrogate for AVP measurement. Copeptin has emerged as a valuable diagnostic tool for distinguishing polyuric states, particularly in differentiating AVP-D from primary polydipsia and AVP-R.
This review aimed to explore the physiological mechanisms underlying AVP-D and AVP-R, with a particular focus on the integration of thirst regulation, AVP dynamics, and renal function. It also emphasized the importance of recent insights into the neural control of water balance for understanding disease pathophysiology and guiding future therapeutic interventions.
The hypothalamic-pituitary-AVP axis plays a crucial role in maintaining water balance, blood pressure, and plasma osmolarity [7, 8]. The hypothalamus, specifically the supraoptic nucleus (SON) and paraventricular nucleus (PVN) detects changes in plasma osmolarity and blood volume through specialized osmoreceptors and baroreceptors [9]. In response to dehydration or a drop in blood pressure, AVP synthesis is upregulated in these nuclei and released into the bloodstream via the posterior pituitary gland.
Once secreted, AVP exerts its primary effects on the kidneys by binding to vasopressin 2 (V2) receptors (V2Rs) in the renal collecting ducts, stimulating aquaporin-2 (AQP-2) channels, which enhance water reabsorption. This mechanism reduces urine output and conserves water, counteracting the polyuria seen in AVP-D and AVP-R [1, 10–12]. Also, AVP acts on V1Rs in vascular smooth muscle, inducing vasoconstriction to stabilize blood pressure during fluid loss or hemorrhage [13].
The AVP axis also regulates plasma osmolarity, ensuring homeostasis by balancing water retention and excretion. When osmolarity rises (indicating dehydration), AVP secretion increases, promoting water conservation. Conversely, when osmolarity decreases, AVP release is suppressed, allowing greater water excretion to restore balance. This regulatory feedback mechanism ensures that water intake via thirst and renal conservation work in tandem to maintain fluid equilibrium [8, 12]. Figure 1 illustrates the components of the hypothalamic-pituitary-AVP axis, including AVP synthesis in the hypothalamus, its release from the posterior pituitary, and its effects on renal water conservation. Understanding this axis is fundamental to elucidating the pathophysiology of AVP-D and AVP-R, where disruptions in AVP production, release, or receptor function leads to impaired water balance.
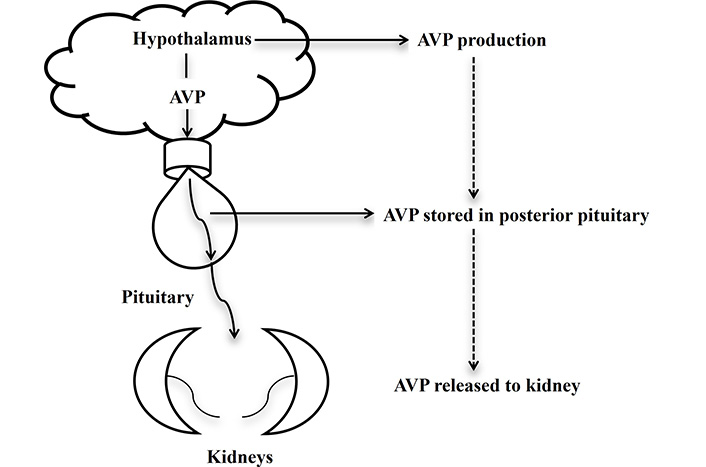
Hypothalamus-pituitary-AVP axis. AVP produced in the hypothalamus, stored in the posterior pituitary, and released to the kidney, induces H2O conservation. AVP: arginine vasopressin
The hypothalamus plays a central role in maintaining blood osmolality, blood pressure, and water retention by integrating sensory input from osmoreceptors and baroreceptors. Osmoreceptors, primarily located in the OVLT and SFO, detect changes in plasma osmolality, while baroreceptors in the carotid sinus and aortic arch sense alterations in blood pressure [14, 15]. When blood osmolality increases due to dehydration or an excess of solutes, or when blood pressure drops, osmoreceptors and baroreceptors stimulate AVP production in the SON and PVN of the hypothalamus. This leads to AVP release from the posterior pituitary gland into circulation [16]. AVP exerts its antidiuretic effects by binding to V2Rs in the renal collecting ducts, promoting AQP-2 channel insertion, which enhances water reabsorption, thereby reducing urine output and restoring fluid balance [17]. In addition, AVP acts on V1Rs in vascular smooth muscle, inducing vasoconstriction, which helps stabilize blood pressure in response to hypovolemia or hemorrhage [18]. Through these mechanisms, the hypothalamus ensures homeostatic balance by tightly regulating water retention, solute concentration, and circulatory stability in response to physiological demands. Dysfunction in these pathways underlies the pathophysiology of AVP-D and AVP-R, contributing to polyuria, polydipsia, and electrolyte imbalances [14–18].
Magnocellular neurosecretory cells, primarily located in the SON and PVN of the hypothalamus, are essential for the synthesis and secretion of AVP, also known as ADH [19]. These neurons produce AVP, which is transported along their axons to the posterior pituitary gland, where it is stored and subsequently released in response to physiological stimuli such as increased plasma osmolality, decreased blood pressure, or hypovolemia [19, 20]. AVP acts on the kidneys to enhance water reabsorption via AQP-2 channels, thereby conserving body water and maintaining fluid balance [19].
Recent studies have provided insights into the regulation of magnocellular neuron activity, particularly regarding thirst perception and anticipatory vasopressin release. Circumventricular organs; notably the SFO, MnPO, and OVLT, play critical roles in detecting osmotic and volemic changes and relaying this information to magnocellular neurons, thereby regulating AVP secretion and thirst responses. Pool et al. [5] demonstrated distinct modalities of thirst generation and their underlying neuronal mechanisms, while Augustine et al. [6] explored how the hypothalamus integrates thirst and sodium appetite regulation, emphasizing the role of the SFO, MnPO, and OVLT in coordinating homeostatic responses. Kim et al. [7] further elaborated on the concept of anticipatory vasopressin release, showing that magnocellular neurons not only respond to real-time osmotic disturbances but also anticipate future changes in osmolality, which is crucial for stabilizing plasma osmolality before predictable challenges, such as food or water intake.
Contrasting the functions of magnocellular and parvocellular neurosecretory cells within PVN highlights the specificity of their roles in hypothalamic regulation. While magnocellular neurons are specialized for AVP and oxytocin secretion, targeting the posterior pituitary to regulate water balance and parturition/lactation. Parvocellular neurons, in contrast, release corticotropin-releasing hormone (CRH) and other peptides, primarily modulating anterior pituitary functions, including the hypothalamic-pituitary-adrenal (HPA) axis [21].
These findings underscore the interplay between hypothalamic structures and peripheral signals, emphasizing the sophisticated mechanisms underlying AVP release and thirst regulation. Further exploration of these mechanisms may inform therapeutic strategies for disorders of water balance, such as AVP-D and AVP-R.
After AVP is synthesized in the magnocellular neurosecretory cells of the SON and PVN of the hypothalamus, it is packaged with a carrier protein, neurophysin II, to facilitate its axonal transport to the posterior pituitary gland [19, 22]. Neurophysin II binds to AVP, forming a stable complex that ensures the proper folding, stabilization, and efficient transport of the hormone along the hypothalamo-hypophyseal tract. This process safeguards AVP’s structural integrity and functional readiness for release upon physiological demand [23].
Upon reaching the posterior pituitary, AVP is stored within secretory vesicles until physiological stimuli such as increased plasma osmolality or decreased blood pressure, trigger its exocytosis into the bloodstream. Neurophysin II remains within these vesicles, highlighting its essential role in AVP transport, stabilization, and regulated release. This intricate mechanism ensures a rapid and efficient hormonal response to fluctuations in fluid balance, osmoregulation, and blood pressure homeostasis [23].
The neurophysin-AVP complex is necessary for maintaining precise water balance, osmolality regulation, and blood pressure control. Mutations in the AVP-NPII gene can disrupt this pathway, leading to impaired AVP secretion or function, contributing to AVP-D or AVP-R [24, 25]. These conditions manifest as disorders of water homeostasis characterized by excessive water loss leading to a polyuric state due to impaired AVP activity. Interestingly, neurophysin I, a related carrier protein, is involved in the transport of oxytocin, a hormone crucial for uterine contractions and milk ejection [26]. This functional parallel between neurophysins I and II underscores the specialized roles of hypothalamic carrier proteins in hormone stabilization and secretion.
Recent advancements in diagnostic techniques highlight the importance of copeptin, the C-terminal segment of the AVP precursor protein as a reliable biomarker in distinguishing the underlying causes of polyuric states. Unlike AVP, copeptin is more stable and easier to measure in the plasma, providing an indirect but accurate reflection of AVP secretion. A study demonstrated the utility of arginine or hypertonic saline-stimulated copeptin levels in differentiating AVP-D (formerly central DI) from AVP-R (formerly nephrogenic DI). Elevated or normal stimulated copeptin levels suggest preserved hypothalamic-pituitary function (indicating AVP-R), whereas low levels confirm AVP-D [27]. This distinction is crucial for tailoring therapeutic interventions to the underlying pathophysiology. The measurement of copeptin during controlled stimuli has emerged as a cornerstone in the diagnostic workup for polyuric disorders. It eliminates the variability associated with direct AVP measurements, offering a robust tool for clinical evaluation. Incorporating copeptin assessments into clinical practice enhances the accuracy of differential diagnoses, facilitating early and appropriate management of polyuric conditions, including AVP-D and AVP-R.
The posterior pituitary (neurohypophysis) is an extension of the hypothalamus, responsible for storing and releasing AVP, although it does not synthesize the hormone. AVP is produced in the hypothalamus by magnocellular neurosecretory cells in the SON and PVN [19]. These neurons transport AVP to the posterior pituitary, where it is stored in secretory vesicles at nerve terminals [11, 20].
When stimulated by increased blood osmolality or decreased blood pressure, nerve impulses from the hypothalamus trigger the release of AVP. This electrical signal causes the influx of calcium ions (Ca²⁺) into the nerve terminals, which increases intracellular Ca²⁺ levels. The elevated calcium promotes the fusion of AVP-containing vesicles with the cell membrane through a process mediated by SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins, essential for vesicular docking and exocytosis. This fusion releases AVP into the bloodstream [28–31].
Once in circulation, AVP travels to the kidneys, where it binds to V2Rs on the cells of the collecting ducts. This activates adenylyl cyclase, increasing cyclic adenosine monophosphate (cAMP) levels, which leads to the insertion of AQP-2 water channels into the membrane. This process enhances water reabsorption, helping to maintain water balance. Also, induces vasoconstriction, which contributes to blood pressure regulation [32, 33].
The kidney’s ability to respond to AVP is a crucial determinant of water homeostasis. AVP exerts its effects by binding to V2Rs located on the principal cells of the collecting ducts, initiating a cascade that activates the adenylyl cyclase-cAMP signaling pathway. This results in an elevation of intracellular cAMP, which promotes the insertion of AQP-2 water channels into the apical membrane of collecting duct cells. Consequently, water reabsorption is enhanced, allowing the body to conserve fluids, particularly under conditions of dehydration or hypovolemia [34, 35].
Disruptions in AVP signaling manifest in two distinct pathological states: AVP-D and AVP-R. AVP-D arises from inadequate production or release of AVP due to hypothalamic or pituitary dysfunction, leading to excessive water loss and an inability to concentrate urine. Management primarily involves desmopressin (DDAVP), a synthetic AVP analog that compensates for hormone deficiency and restores fluid balance [36, 37]. Conversely, AVP-R is characterized by renal insensitivity to AVP, often resulting from mutations in the V2R or downstream signaling defects. This impaired responsiveness prevents proper water reabsorption, culminating in persistent polyuria and dehydration. Unlike AVP-D, AVP-R does not respond to DDAVP, necessitating alternative therapeutic strategies. Thiazide diuretics, which induce mild hypovolemia, enhance proximal tubular sodium and water reabsorption, thereby reducing urine output independently of the AVP pathway [38].
Emerging research suggests that additional molecular pathways may modulate renal sensitivity to AVP, including variations in AQP-2 expression and intracellular trafficking mechanisms. These findings highlighted potential future therapeutic targets aimed at improving water retention in patients with AVP-related disorders. Maintaining kidney responsiveness to AVP is essential for fluid homeostasis, and further studies are needed to explore novel interventions that can enhance renal AVP signaling in both AVP-D and AVP-R. This process is critical in conserving body water, especially during dehydration or low blood volume. Without proper AVP signaling, the kidneys cannot reabsorb sufficient water, leading to excessive urine production (polyuria) and dehydration. This condition is characteristic of AVP-D and AVP-R, disorders marked by an inability to concentrate urine and retain water [36]. In AVP-D, which occurs when the hypothalamus or pituitary fails to produce or release adequate AVP. Treatment typically involves administering synthetic AVP like DDAVP to replace the missing hormone and restore water balance [37]. While in AVP-R, the kidneys are unresponsive to AVP due to mutations in the V2Rs or defects in the downstream signaling pathways. Treatment of AVP-R involves managing fluid intake and using medications like thiazide diuretics, which help reduce urine output by promoting sodium and water reabsorption in the nephron, bypassing the AVP pathway. Maintaining kidney responsiveness to AVP is crucial for ensuring the kidney effectively regulates water balance and prevents excessive fluid loss [38].
The kidneys play a fundamental role in maintaining homeostasis by regulating blood filtration, waste removal, electrolyte balance, acid-base equilibrium, and fluid homeostasis [39, 40]. In addition to excreting metabolic waste products such as urea and creatinine, they regulate blood pressure through the renin-angiotensin-aldosterone system (RAAS) and contribute to erythropoiesis by secreting erythropoietin in response to hypoxia [41–43]. These integrated functions ensure systemic stability, including the precise regulation of water balance, which is critical in preventing disorders such as AVP-D and AVP-R.
Water homeostasis is primarily governed by AVP, which modulates water reabsorption in the collecting ducts by stimulating V2R activation and AQP-2 insertion into the apical membrane of collecting duct cells. Under normal physiological conditions, the kidneys adaptively regulate urine concentration by responding to AVP-mediated signals, thereby preventing excessive water loss [36–38]. In AVP-D, where AVP secretion is impaired due to hypothalamic or pituitary dysfunction, and in AVP-R, where the kidneys fail to respond to AVP due to V2R mutations or downstream signaling defects, this adaptive mechanism is disrupted, leading to polyuria and dehydration [32, 33].
The kidneys contribute to osmoregulation by maintaining stable plasma sodium concentrations, a key determinant of water balance. Juxtaglomerular cells monitor blood pressure and volume, while macula densa cells detect sodium levels in the distal convoluted tubule (DCT), ensuring that deviations in blood osmolality trigger appropriate AVP secretion [44, 45]. Dysregulation of this process, such as impaired sodium sensing, can lead to inappropriate AVP secretion or resistance, further exacerbating fluid imbalances associated with AVP-D and AVP-R.
The RAAS system plays a complementary role in AVP-mediated water conservation. When blood volume decreases, renin secretion leads to aldosterone release, promoting sodium and water retention in the nephron to maintain circulatory homeostasis [41, 45]. RAAS dysfunction may contribute to excessive water loss or retention, mimicking AVP-D or AVP-R pathophysiology. Proper integration of RAAS and AVP signaling is essential to maintaining fluid balance and preventing maladaptive states of dehydration or volume overload.
Electrolyte imbalances, particularly hypercalcemia and hypokalemia, can impair renal responsiveness to AVP. Calcium overload interferes with AQP-2 trafficking, while potassium depletion disrupts V2R signaling, both of which contribute to AVP-R pathogenesis [44, 45]. By regulating sodium, potassium, and calcium homeostasis, the kidneys support optimal AVP function and reduce the risk of AVP-related disorders.
Therefore, the kidneys are central to the avoidance of AVP-D and AVP-R by finely regulating water balance, electrolyte homeostasis, and hormonal interactions. Their ability to integrate AVP signaling, osmoregulation, RAAS modulation, and electrolyte control is essential for maintaining fluid equilibrium and preventing the pathophysiological consequences of AVP dysfunction. Further research into renal adaptations and molecular pathways affecting AVP sensitivity may provide novel therapeutic strategies for managing AVP-related disorders.
The nephron, the functional unit of the kidney, facilitates essential renal processes. Each kidney contains approximately 1 to 1.5 million nephrons, which include the glomerulus (for filtration), proximal convoluted tubule (PCT) (for nutrient reabsorption), the loop of Henle (for urine concentration), DCT (for electrolyte regulation), and the collecting duct (which modulates water reabsorption in response to AVP) [46].
In AVP-D and AVP-R, nephron dysfunction, particularly in the collecting ducts, results in impaired urine concentration. Normally, AVP binds to V2Rs on collecting duct cells, activating adenylyl cyclase and increasing intracellular cAMP levels [38, 46]. This activates protein kinase A (PKA), which phosphorylates AQP-2, leading to its insertion into the apical membrane, facilitating water reabsorption. Water then moves from the tubular lumen into the nephron cells via AQP-2 and exits through AQP-3 and AQP-4 into the bloodstream [34, 35].
Water reabsorption is influenced by sodium and urea transport (UT). The Na⁺/K⁺-ATPase pump maintains an osmotic gradient essential for water movement through aquaporins. Any dysfunction in this pump disrupts osmotic balance, leading to excessive water loss and polyuria, as seen in AVP-D and AVP-R. Also, UT-A1 and UT-A3 in the inner medullary collecting duct enhance urine concentration by maintaining medullary osmolality [47, 48].
Urine formation, concentration, and dilution are essential processes for maintaining water balance in the body, with the countercurrent multiplier and exchanger systems playing pivotal roles in creating and maintaining the renal osmotic gradient. In AVP-D and AVP-R, the inability to properly regulate water reabsorption disrupts these mechanisms, leading to excessive urine output and dehydration. Understanding the physiological basis of these processes and the pathophysiology of AVP dysfunction is crucial for the effective management of these disorders and highlights the importance of maintaining the proper function of AVP, aquaporins, and the countercurrent mechanisms for water conservation [49–52].
Urine formation is a multi-step process involving glomerular filtration, tubular reabsorption, and tubular secretion. During glomerular filtration, blood is filtered through the glomerulus into the nephron, where water, electrolytes, and waste products are separated from the blood. In the subsequent process of tubular reabsorption, essential substances, including water and sodium, are reabsorbed into the bloodstream. Tubular secretion follows, where waste products are actively transported from the blood into the nephron for excretion.
Water reabsorption, a critical aspect of urine formation, is regulated to maintain water homeostasis. AVP, also known as ADH, plays a key role in this process by increasing water reabsorption in the collecting ducts. It does so by promoting the insertion of AQP-2 channels in the membranes of collecting duct cells, allowing water to be reabsorbed back into the bloodstream, thus concentrating the urine [49, 50].
The countercurrent multiplier system, located in the loop of Henle, is responsible for generating a hyperosmotic environment in the renal medulla. This is achieved by the active transport of sodium and chloride from the thick ascending limb of the loop into the surrounding interstitium, which creates a high osmolarity in the medulla. The thin descending limb of the loop, which is permeable to water but not solutes, allows water to passively exit the nephron due to the osmotic gradient, thereby concentrating the tubular fluid. The countercurrent exchanger, primarily in the vasa recta, preserves the osmotic gradient created by the countercurrent multiplier. As blood flows through the vasa recta, solutes, and water are exchanged between the blood and the interstitium, allowing for the continued concentration of urine without diluting the renal medullary gradient [49, 51]. The proper functioning of both systems is essential for efficient water conservation, particularly during dehydration or conditions of water scarcity.
In AVP-D (formerly central DI) and AVP-R (formerly nephrogenic DI), the kidneys lose the ability to concentrate urine, leading to excessive urine output (polyuria) and dehydration. AVP-D is characterized by insufficient production or secretion of AVP from the hypothalamus or posterior pituitary, impairing water reabsorption in the collecting ducts. Without adequate AVP signaling, water cannot be reabsorbed effectively, and urine remains dilute, resulting in dehydration [38]. In AVP-R, the kidneys fail to respond to AVP due to mutations in the V2R or the AQP-2 channels. Despite the presence of AVP, these defects prevent the normal insertion of AQP-2 channels into the collecting duct membranes, impairing water reabsorption. This leads to symptoms similar to AVP-D, including polyuria and dehydration [52].
The malfunction of AVP signaling and the impaired function of the countercurrent multiplier and exchanger systems in both AVP-D and AVP-R lead to a significant loss of the kidney’s ability to concentrate urine. In the absence of effective AVP action, both the countercurrent multiplier and exchanger are unable to maintain the renal osmotic gradient necessary for water reabsorption, exacerbating dehydration and resulting in the hallmark symptoms of these disorders [38, 49, 52].
In understanding the physiological basis underlying the development of AVP-D and AVP-R, it is crucial to differentiate between the AVP-D and AVP-R forms, as well as to recognize the specific dysfunctions in the AVP pathway. These points are summarized in Table 1, which provides an overview of mechanisms, including defects in AVP production, receptor function, AQP-2 channels, and the countercurrent mechanism.
Main points on physiological basis underlying development of AVP-D and AVP-R
| Conditions | Description | Reference(s) |
|---|---|---|
| AVP-D | Involves insufficient production or release of AVP from the hypothalamus or posterior pituitary gland, leading to reduced water reabsorption in the kidneys, resulting in dilute urine and dehydration. | [53] |
| AVP-R | The kidneys are unable to respond to AVP, despite normal hormone levels. This is often due to defects in AVP receptors or downstream signaling pathways in the renal tubules. | [38] |
| AVP pathway dysfunction | V2 receptor defects: in AVP-R, mutations in the V2 receptor (found in the collecting ducts of the kidney) prevent AVP from binding and triggering the molecular cascade required for water reabsorption. AVP-D: in AVP-D, insufficient AVP production or release results in a lack of hormonal signaling to the kidneys, preventing activation of downstream mechanisms needed for urine concentration. | [54, 55] |
| AQP-2 channel deficiency | AVP normally promotes the insertion of AQP-2 water channels into the apical membrane of collecting duct cells, allowing water reabsorption. Defects in AQP-2 gene expression or trafficking in AVP-R impair this process, leading to an inability to concentrate urine. | [56] |
| Disruption in countercurrent mechanism | The countercurrent multiplier and exchanger systems in the nephron create the osmotic gradient needed for water reabsorption. Disruptions in solute transporters (e.g., Na+/K+/2Cl– co-transporters) within the loop of Henle impair the creation of a hyperosmotic medullary environment, reducing the efficiency of water conservation in AVP-D and AVP-R. | [57] |
AQP-2: aquaporin-2; AVP: arginine vasopressin; AVP-D: AVP deficiency; AVP-R: AVP resistance; V2: vasopressin 2
The avoidance of AVP-D and AVP-R in the body involves several physiological mechanisms, including the release and action of AVP, the functionality of AQP-2 channels, and the maintenance of the medullary osmotic gradient. Table 2 summarizes these mechanisms, highlighting processes such as the regulation of urine concentration, AVP receptor activation, and solute transporter roles in supporting water reabsorption.
Main points on physiological basis underlying AVP-D and AVP-R avoidance
| Mechanism | Description | Reference(s) |
|---|---|---|
| AVP release and function | AVP is produced by the hypothalamus and released by the posterior pituitary in response to increased plasma osmolality or dehydration. It promotes water reabsorption in the kidneys, concentrating urine. | [30, 31] |
| Countercurrent mechanism | The countercurrent multiplier in the loop of Henle and the exchanger in the vasa recta create a hyperosmotic gradient in the renal medulla, essential for water reabsorption in the presence of AVP. | [49, 51] |
| Regulation of urine concentration and dilution | In the presence of AVP, collecting ducts become permeable to water, allowing reabsorption and urine concentration. Without AVP, the ducts remain impermeable, leading to dilute urine. | [52] |
| AVP receptor activation | AVP binds to V2 receptors in the kidney’s collecting ducts, activating the cAMP-PKA pathway, which promotes water reabsorption. Proper receptor function prevents excessive water loss, as in AVP-D. | [34, 35] |
| AQP-2 channels | AVP triggers the movement of AQP-2 water channels to the collecting duct cells’ apical membrane, allowing water reabsorption. Proper AQP-2 function protects against AVP-R caused by channel disruption. | [56] |
| Maintenance of the medullary osmotic gradient | The countercurrent multiplier in the loop of Henle concentrates solutes, creating a gradient that facilitates water reabsorption under AVP influence. The exchanger helps preserve this gradient. | [57] |
| Solute transporters | Na+/K+/2Cl– co-transporters in the thick ascending limb of the loop of Henle establish the osmotic gradient, supporting urine concentration and preventing excessive water loss. | [58] |
AQP-2: aquaporin-2; AVP: arginine vasopressin; AVP-D: AVP deficiency; AVP-R: AVP resistance; cAMP-PKA: cyclic adenosine monophosphate-protein kinase A; V2: vasopressin 2
Emerging evidence suggests that environmental and lifestyle factors significantly influence cellular and systemic physiology, potentially modulating the onset, progression, and outcomes of AVP-D and AVP-R. These factors include dietary habits, hydration practices, physical activity levels, exposure to environmental toxins, and psychosocial stressors. Excessive fluid intake, often linked to psychogenic polydipsia or lifestyle choices, can lead to adaptive down regulation of AVP secretion or renal sensitivity to AVP, exacerbating AVP-D, and AVP-R like-symptoms [5], exposure to nephrotoxic agents from contaminated water or industrial environments may impair renal responsiveness to AVP, contributing to AVP-R [6]. Chronias have been implicated in dysregulation of the HPA axis, indirectly affecting water homeostasis and hypothalamic-pituitary-renal axis functioning [7]. These environmental lifestyle factors vary across populations and individuals, contributing to the heterogeneity in AVP-D, AVP-R presentation, and therapeutic response. Targeted lifestyle modifications, such as optimizing hydration practices and minimizing exposure to nephrotoxins, could complement pharmacological therapies to improve patient outcomes.
Molecular pathological epidemiology (MPE) is an interdisciplinary framework that examines the interplay between molecular mechanisms, environmental factors, and disease pathology. This approach has been applied extensively in cancer and gastrointestinal research [59, 60] and its integration. MPE can help identify biomarkers reflecting individual variability in AVP-D and AVP-R mechanisms. For example, genetic polymorphisms in genes such as AVP, AQP-2, and V2R may influence susceptibility to AVP-D and AVP-R or therapeutic responses to DDAVP. Environmental exposures, such as nephrotoxicity factors, may drive epigenetic modifications like methylation changes in AVP-related pathways that could serve as predictive biomarkers for AVP-D and AVP-R risk and progression [5–7]. Integrating these molecular insights with epidemic data enables the development of tailored interventions, bridging the gap between basic research and clinical practice. For example, identifying epigenetic markers associated with AVP-R could inform individualized treatment plans, improving efficacy and reducing adverse outcomes.
Pharmacological interventions in AVP-D and AVP-R aim to manage symptoms and restore water balance, depending on whether the condition is AVP-D (deficiency of AVP) or AVP-R (resistance of the kidneys to AVP). The approaches primarily focus on addressing the underlying cause by either replacing AVP or improving the kidney’s response to it. In AVP-D, where AVP production or secretion is impaired, the main pharmacological approach is to replace the missing hormone [61, 62].
DDAVP: DDAVP is a synthetic analog of AVP or ADH that mimics the natural hormone’s effect on the kidneys. It binds to the V2Rs in the collecting ducts, initiating the cAMP-PKA pathway, which promotes the insertion of AQP-2 water channels into the apical membrane of the collecting duct cells. This facilitates water reabsorption and reduces urine output, helping to concentrate urine. DDAVP is considered highly effective for AVP-D (central DI) as it selectively targets the kidneys without the vasoconstrictive side effects associated with natural AVP [63]. In AVP-R (nephrogenic DI), the kidneys do not respond to ADH. Treatment focuses on reducing urine volume by targeting molecular pathways involved in water reabsorption or compensating for the lack of AVP responsiveness [64].
Thiazide diuretics: paradoxically, thiazide diuretics reduce urine output in AVP-R by causing a mild state of hypovolemia. This stimulates increased proximal tubular reabsorption of sodium and water before the urine reaches the distal nephron, reducing the total amount of water delivered to the distal collecting ducts. On a molecular level, thiazides inhibit the Na⁺/Cl⁻ symporter in the DCT, which alters electrolyte handling and indirectly improves water conservation [65].
Amiloride: amiloride is a potassium-sparing diuretic often used when AVP-R is caused by lithium toxicity. Lithium interferes with AVP signaling by blocking the cAMP pathway downstream of the V2R, which disrupts AQP-2 expression. Amiloride blocks the epithelial sodium channels (ENaC) in the collecting duct, reducing lithium uptake by the kidney cells and thereby mitigating its toxic effects. This helps preserve AVP signaling and AQP-2 activity [66].
Non-steroidal anti-inflammatory drugs (NSAIDs): NSAIDs, such as indomethacin, reduce urine volume by decreasing prostaglandin synthesis. Prostaglandins antagonize the action of AVP on the kidney, so inhibiting their synthesis enhances AVP’s effect on water reabsorption. NSAIDs work by inhibiting cyclooxygenase (COX) enzymes, reducing prostaglandin levels, and promoting more effective water reabsorption in the collecting ducts [67].
AQP-2 modulators: experimental therapies are exploring direct targeting of AQP-2 channels to enhance water reabsorption. While still in early development, these interventions aim to correct the mislocalization or dysfunction of AQP-2 channels seen in AVP-R [68].
Vaptans (V2 antagonists): although typically used for conditions like the syndrome of inappropriate AVP secretion (SIADH), vaptans (V2 antagonists) have been researched for their potential to regulate water balance. However, they are not suitable for AVP-D or AVP-R treatment since they inhibit water reabsorption, contrary to what is needed in AVP-D or AVP-R [69].
The management of AVP-D and AVP-R often involves pharmacological interventions targeting molecular pathways to restore water balance. These strategies include AVP replacement therapy with DDAVP, the use of thiazide diuretics, and adjunctive treatments like amiloride and NSAIDs. Overview of these interventions, detailing their mechanisms of action, such as mimicking AVP activity, modulating sodium handling in the nephron, and preserving AQP-2 functionality (Table 3).
Molecular and cellular basis of pharmacological interventions of AVP-D and AVP-R
| Pharmacological intervention | Mechanism of action | Reference |
|---|---|---|
| AVP replacement (desmopressin) | Mimics AVP function at V2 receptors, activating the cAMP-PKA pathway, leading to the mobilization of AQP-2 channels, allowing water reabsorption in the kidneys. | [61] |
| Thiazide diuretics | Indirectly increase water reabsorption by altering sodium handling in the nephron, which reduces the load on the distal tubules where AVP usually exerts its effects. | [63] |
| Amiloride | Blocks ENaC channels, preventing lithium-induced damage to the AVP signaling pathway, thereby preserving AQP-2 channel activity and promoting water reabsorption. | [64] |
| NSAIDs | Inhibit prostaglandin synthesis, which reduces the antagonistic effect of prostaglandins on AVP, enhancing the hormone’s water-conserving action in the kidneys. | [65] |
AQP-2: aquaporin-2; AVP: arginine vasopressin; AVP-D: AVP deficiency; AVP-R: AVP resistance; cAMP-PKA: cyclic adenosine monophosphate-protein kinase A; ENaC: epithelial sodium channels; NSAIDs: non-steroidal anti-inflammatory drugs; V2: vasopressin 2
The physiological basis of AVP-D and AVP-R centers on the failure of the body in preserving the integrity of the hypothalamic-pituitary axis, ensuring renal sensitivity to AVP, and maintaining electrolyte and fluid balance. Early diagnosis, pharmacological interventions, and lifestyle modifications play critical roles in preventing the onset of AVP-D and AVP-R or managing its symptoms. Understanding these physiological mechanisms formed basis that can guide researches and clinical approaches to both its prevention and treatment.
ADH: antidiuretic hormone
AQP-2: aquaporin-2
AVP-D: arginine vasopressin deficiency
AVP-R: arginine vasopressin resistance
Ca²⁺: calcium ions
cAMP: cyclic adenosine monophosphate
DCT: distal convoluted tubule
DDAVP: desmopressin
DI: diabetes insipidus
HPA: hypothalamic-pituitary-adrenal
MnPO: median preoptic nucleus
MPE: molecular pathological epidemiology
NSAIDs: non-steroidal anti-inflammatory drugs
OVLT: organum vasculosum of the lamina terminalis
PKA: protein kinase A
PVN: paraventricular nucleus
RAAS: renin-angiotensin-aldosterone system
SFO: subfornical organ
SON: supraoptic nucleus
UT: urea transport
V2: vasopressin 2
V2Rs: vasopressin 2 receptors
OMO: Conceptualization, Validation, Writing—original draft, Writing—review & editing.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

