Case Report
Case Report

Affiliation:
1Division of Metabolic Internal Medicine, University of Modena and Reggio Emilia, 41126 Modena, Italy
Affiliation:
1Division of Metabolic Internal Medicine, University of Modena and Reggio Emilia, 41126 Modena, Italy
Affiliation:
2Department of Internal Medicine, General, Emergency and Post-Acute, Division of Metabolic Internal Medicine, University Hospital of Modena, 41126 Modena, Italy
ORCID: https://orcid.org/0000-0002-4794-9809
Affiliation:
2Department of Internal Medicine, General, Emergency and Post-Acute, Division of Metabolic Internal Medicine, University Hospital of Modena, 41126 Modena, Italy
Email: maurantonio.mauro@aou.mo.it
ORCID: https://orcid.org/0000-0003-3500-1145
Explor Med. 2021;2:324–332 DOI: https://doi.org/10.37349/emed.2021.00050
Received: May 08, 2021 Accepted: June 24, 2021 Published: August 31, 2021
Academic Editor: Lindsay A. Farrer, Boston University School of Medicine, USA
Central pontine myelinolysis (CPM) is a rare manifestation of osmotic demyelination syndrome (ODS) which involves the pons and causes significant morbidity and mortality. CPM usually occurs in the setting of rapid correction of severe chronic hyponatremia. A rare case of CPM due to hyperglycemia in a 27-year-old man with type 1 diabetes is reported. During the patient’s hospitalization, his plasma glucose level showed a wide variability ranging from 38 mg/dL to 530 mg/dL; while plasma sodium level was constantly normal. At computed tomography (CT) scans, areas of hypodensity with a hyperdense ring were identified in the anterior part of the pons. At magnetic resonance imaging (MRI) scan, pontine abnormalities compatible with CPM were observed. According to laboratory tests, we concluded that CPM resulted from rapid and wide shifts in osmolar gradient owing to variability in plasma glucose levels. While universally recognized in several clinical settings, CPM is rarely observed in diabetic patients. Our report supports the notion that hyperosmolarity per se plays a key role in the pathogenesis of CPM, which may occur independently of sodium abnormalities.
Central pontine myelinolysis (CPM) is a rare manifestation of osmotic demyelination syndrome (ODS) which involves the pons. Especially in case of either malnutrition or alcoholic hepatitis, the most common cause of CPM is rapid correction of hyponatremia, which clinically is heralded by encephalopathy [1, 2]. In patients with diabetes mellitus, clinical conditions of hyperglycemia, hypovolemia, metabolic acidosis, and electrolyte imbalance require aggressive schedules of parental hydration and intravenous insulin therapy to correct hyperosmolarity state. However, such intensive management can also cause major shifts in plasma osmolality associated with rapid rises in plasma sodium concentrations which happen as plasma glucose values rapidly decline. CPM occurring in the context of these clinical manifestations has been reported in only few cases. We report on an unusual case of CPM due to hyperglycemia and normal serum sodium in a patient with type 1 diabetes (T1D); moreover, we extensively reviewed the pertinent literature on the pathogenesis of CPM.
A 27-year-old man with autoimmune anti-glutamic acid decarboxylase (anti-GAD) positive T1D was admitted to our medical guard on the grounds of systemic weakness, sensory loss at the lower extremities, throbbing headache, dizziness, photophobia, and chronic, unexplained diarrhea.
Our patient was diagnosed with autoimmune T1D at the age of 13 with microvascular complications: retinopathy submitted to laser photocoagulation and neuropathy treated with gabapentin and duloxetine. Moreover, owing to inadequate compliance to therapy he had suffered from some episodes of hypoglycemic coma a few years earlier. Furthermore, he had chronic unexplained diarrhea with blood, fecal and histologic tests negative for infectious causes and celiac disease. He also reported previous diagnosis of gastro-esophageal reflux disease and was hospitalized owing to acid reflux, heartburn, dyspnea, and chest pain treated with proton pump inhibitors. Our psychiatric consultant identified depression reactive to social and familiar deprivation given that the patient lived in Italy but his mother and his wife lived in Morocco.
At time of admission, he presented sensory loss at the lower right extremity associated with impairment of strength, mild-severe headache, nausea, dizziness, and photophobia, without vomiting. Strength, sensitivity, muscle tone, and reflexes were normal in the upper limbs. He presented leg weakness with deficient muscle tone: grade 4 hypostenia at right iliopsoas, grade 2 hypostenia of anterior tibial muscle, and grade 4 hypostenia of peroneous muscle. Plantar flexor reflexes were poorly elicitable.
His plasma glucose was 217 mg/dL, with glycosuria 500 mg/dL and HbA1c 115 mmol/mol. Serum sodium was 141 mEq/L, and hepatic and renal function tests were normal. Urinary C-peptide was below to normal range, suggestive of a beta-cell pancreatic insufficiency; albuminuria was present (Table 1).
Blood and urine tests in decompensated diabetes and pancreatic insufficiency
| Parameters | Patient value | Reference range |
|---|---|---|
| HbA1c | 115 mmol/mol | 20–38 mmol/mol |
| Glycosuria | 500 mg/dL | < 180 mg/dL |
| Urinary C-peptide | < 0.1 ng/mL | 1.1–4.4 ng/mL |
| Urinary C-peptide 24 h | undetectable | 24–207 microg/24 h |
| Albuminuria | > 300 mg/g | < 30 mg/24 h |
| Diuresis | 1700 mL/24 h | not applicable |
Electromyography was compatible with moderate peripheral sensitive and motor axonal polyneuropathy. Analysis of cerebral spinal fluid showed high total protein and immunoglobulin-G (IgG) oligoclonal band at protein electrophoresis; the search for Borrelia and other infectious agents was negative. Autoimmune diseases, Whipple disease, gangliosidosis, and cancer were all ruled out. To identify the cause of chronic diarrhea, the patient underwent screening for coeliac disease, autoimmune disease, infectious gastroenteritides (in particular Salmonella species, Shigella, and Campylobacter), and inflammatory bowel diseases. Given that all these tests were negative, diarrhea was attributed to vegetative neuropathy.
Magnetic resonance imaging (MRI) angiography (Angio-MRI) scanning disclosed regular signal in the vascular system at the level of Willis polygon while MRI scan of brain showed abnormal signal centrally in the pons: hyperintense pontine lesions on long repetition time (TR) sequence, hypointense on T1-weighted scans, with increased diffusion coefficient, with an ovalar morphology and a cranio-caudal diameter of 14 mm with transverse fibers of the pons involved, without abnormalities in the peripheral part of the pons or mass effect. Moreover, imaging showed extensive atrophy both in the basilar area and the tegmentum of pons, and dilated fourth ventricle (Figure 1).
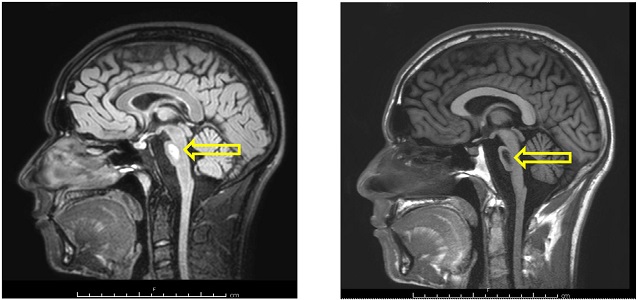
MRI scan of brain: signal abnormalities in the central part of the pons. Hyperintense pontine lesions on long TR sequence (on left), hypointense on T1 scans (on right)
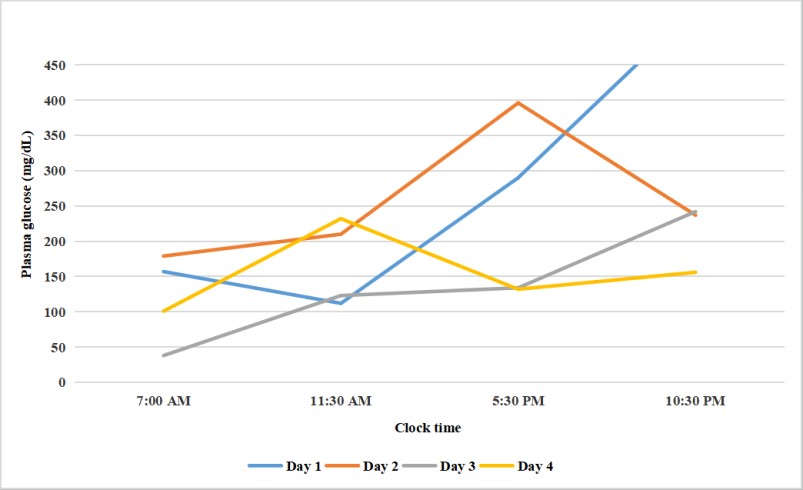
Our patient was diagnosed with CPM. During the hospitalization, his plasma glucose showed a wide variability ranging from 38 mg/dL to 530 mg/dL while sodium plasma levels were always normal (Figure 2).
With appropriate insulin therapy, glycemic stabilization was achieved which allowed full recovery of general and neurological conditions at discharge.
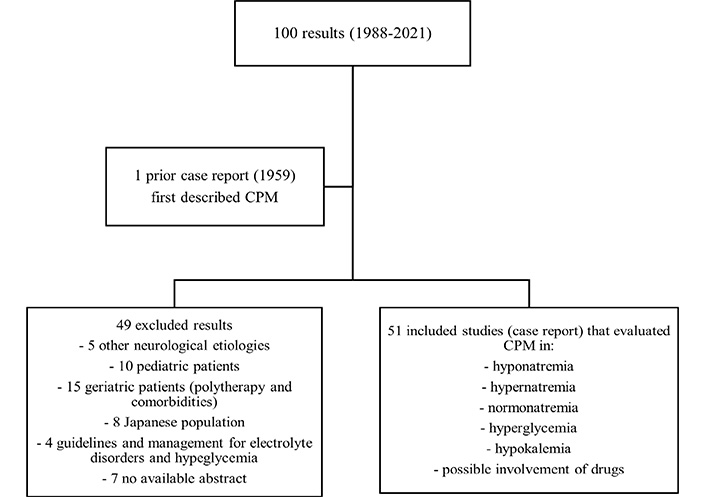
We systematically searched PubMed database using the following keywords “Central Pontine Myelinolysis”, “Osmotic demyelination syndrome”, “Diabetes Mellitus” and “Hyperglycemia”. Studies that evaluated the effects of hyponatremia, hypernatremia or normonatremia, and hyperglycemia on CPM were included. Criteria for exclusion: studies that considered pediatric and/or geriatric and/or Japanese population and studies that described ODS due to other neurological etiologies (Figure 3).
ODS is an uncommon neurological disorder caused by damage to the myelin sheath of nerve fibers. CPM is the typical presentation, reflecting greater susceptibility of pontine white matter tracts, consisting of focal, bilaterally symmetric region of myelin destruction; however, extra-pontine involvement (extra-pontine myelinolysis or EPM) is common [1, 2].
CPM was first described in 1959 by Adams and Victor who reported on four patients with quadriplegia and pseudo-bulbar palsy, three of these were chronic alcoholics and all were malnourished [3].
As is shown in Table 2, CPM can occur in a variety of clinical settings [1–24].
Systematic review—CPM in a variety of clinical settings
| CPM etiology | Reported in |
|---|---|
| Alcohol and malnutrition | Adams RD (1959) [3]Case report on four malnourished patients |
| “Any relatively hypertonic insult” | McKee AC (1988) [4]Retrospective study on 3,528 patients submitted to general autopsy |
| Rapid correction of hyponatremia | Brown WD (2000) [1] Review article |
| Severe hyperemesis gravidarum-induced hyponatremia | Lee IW (2010) [5]Case report on pregnant woman |
| Hypophosphatemia | Qadir F (2005) [6]Case report on a young lady with prolonged febrile illness and severe hypophosphatemia |
| Hypokaliemia | Shintani M (2016) [7]Case report on a 43-year-old diabetic man with nephrotic syndrome |
| Hypokalemic in nephrogenic diabetes insipidus | Davenport C (2010) [8]Case report on a 63-year-old man with nephrogenic diabetes insipidus |
| Liver transplantation | Singh N (1994) [9]Clinical review of a subgroup of patients after liver transplantation |
| Lithium toxicity | Fabisiak DB (2002) [10]Case report on patient in lithium therapy for bipolar disorder |
| Lymphoma | Doni E (2016) [11]Case report on patient with diffuse large-B cell lymphoma |
| Acquired metabolic encephalopathies | Sharma P (2009) [12]A pictorially review of the more classic MRI features of several acute toxic and acquired metabolic encephalopathies |
| Intravenous sodium bicarbonate therapy | Chang KY (2014) [13]Case report on a 40-year-old woman with a bicarbonate deficit |
| Hyperglycemic with rebound hypernatremia | O’Malley G (2008) [14]Case report on a 49-year-old woman with hyperglycemic hyperosmolar state |
| Hyperglycemic with normal natremia | Donnelly H (2016) [2] Burns JD (2013) [15]Guerrero WR (2013) [16]Hirosawa (2018) [17]Corps Fernández D (2017) [18]Fasano A (2018) [19]Case reports on patients with hyperglycemia |
| Binge eating attack induced hyperglycemia in the absent of a relevant hyponatremia | Pliquett RU (2018) [20]Case report on a type 2 diabetes patient with hyperglycemia after a binge-eating attack |
| Chronic kidney disease | Talluri S (2017) [21]Case report on a patient with chronic kidney disease and hyperosmolar hyperglycemia |
| Fluctuating hyperglycemia in patient treated with interferon and insulin therapy | Gourine M (2016) [22]Case report on a diabetic young patient chronic hepatitis as predisposing factor for rapid changes in osmolality |
| Acute hypoglycemia | Madey JJ (2013) [23]Case report in diabetic patient treated with levofloxacin Purucker E (2000) [24]Case report in patient with myocardial infarction following severe hypoglycemia |
CPM, most often reported in association with rapid correction of severe chronic hyponatremia, has also been described associated with other electrolyte derangements such as hypophosphatemia, hypokalemia as well as in the setting of liver transplantation and lithium toxicity [6, 7, 9, 10].
In chronic hyponatremia, electrolytes are lost from intracellular space flowing into cerebrospinal fluid over a few days during which brain cells become isotonic to the extracellular fluid to maintain cell volume through a process called “cerebral adaptation”. As a result, given that they can neither synthesize nor transport osmotic substrates at such rapid rates (which leads to osmotic demyelination), oligodendrocytes will shrink up as a consequence of cell volume loss during rapid correction of tonicity [25].
Clinical manifestations are variable: various degrees of encephalopathy are commonly observed however either totally asymptomatic or mildly symptomatic cases can also occur. Typically patients with ODS present with parkinsonism, quadriparesis, “locked-in” syndrome, coma, bulbar palsy, or, less frequently, with dysphagia, dysarthria, facial paresis, ataxia, nystagmus, tremor, lethargy, confusion, behavioral disturbances, and/or disorientation [26, 27].
In the past, brainstem auditory evoked potentials were used, but modern imaging has superseded its use, and brain MRI is the current accepted standard diagnostic technique because of a greater sensitivity and a superior capacity for the demonstration of the lesions of EPM, than CT. At MRI, hyperintense lesions on T2 weighted, and hypointense lesions on T1 weighted images are suggestive for CPM [25].
Other than supportive treatment, there is no effective treatment for CPM. Supportive treatment should be provided to all patients for at least 6–8 weeks before making any conclusions regarding irreversibility of disease. In cases where CPM is caused by rapid correction of severe chronic hyponatremia lowering serum sodium may be beneficial if initiated early. In literature there is some evidence supporting the beneficial role of plasma exchange techniques that allow removal from blood circulation of myelin toxic substances. Conversely, the use of steroids and intravenous immunoglobulins is not supported by evidence [25].
McKee et al. [4] have suggested that myelinolysis can result from any “relatively hypertonic insult”. These authors have speculated that CPM can result from any circumstances wherein serum and extracellular space become hypertonic faster than brain cells can compensate.
Reports of ODS associated with hyperglycemic hyperosmolar state with rebound hypernatremia have been published [14]. Over the last few years, CPM has also been reported in diabetic patients exhibiting deranged metabolic control even in the absence of electrolyte disturbances [2, 15].
In 2015, Saini et al. [28] reported on a case of CPM secondary to hyperglycemia per se rather than secondary to its treatment. The patient presented with ataxia and pseudobulbar manifestations and followed a sub-acute course over a two-week time.
Hyperglycemia, by leading to an increased plasma osmolality, has been blamed to cause pontine demyelination both in type-2 diabetes and in a T1D patient [15, 28, 29]. This complication has also been described in a patient with latent autoimmunity diabetes displaying significant glucose fluctuations [30].
Although the possible pathogenic mechanisms responsible for development of CPM in patients with diabetes are still a matter of debate, a few hypotheses have been proposed in earlier studies. In patients with poorly controlled diabetes, hyperosmolarity resulting from increased blood glucose with greater osmolar shifts might cause brain edema. An explanation might be that in chronically uncontrolled diabetes, osmotic stress induces protein damage and aggregation within brain oligodendrocytes.
More specifically, it has been postulated that serum hyperosmolarity induces the release of nitric oxide and inflammatory mediators and this may damage tight junctions. Furthermore, toxic substances released by damaged endothelial cells may injure oligodendrocytes [31].
In the setting of hyperosmolar hyperglycemia, several theories have been proposed to explain the pathogenesis of osmotic demyelination: rapid changes in osmolality associated with the fluctuations in serum glucose, subacute changes in brain cells secondary to hyperglycemia, and hypertonic insult associated with hyperglycemia [31].
Moreover, risk factors for CPM in diabetics include concurrent diabetic nephropathy requiring dialysis, infection with hepatitis C virus, and arterial hypertension [32].
The only treatment for non-sodium-dependent osmotic demyelination is supportive care [27].
In this case report, diagnosis of CPM was based on the evidence of injury to central pons, which was clinically compatible with myelinolysis due to metabolic derangement. Generally, CPM is caused by osmotic demyelination due to rapid correction of hyperosmolarity in hypernatremia. Only in very rare cases does this condition occur in patients with diabetes and normal serum sodium concentration: in these, it would seem plausibly caused by the hyperosmotic effects of glucose which carries oligodendrocyte fluids into the extracellular compartment. In our case, this effect was also presumably facilitated by concurrent dehydration caused by chronic diarrhea.
In literature, CPM has been attributed to sudden, severe, and sustained hyperglycemia, especially when another risk factor is present such as malnutrition, alcohol abuse, chronic diseases, and liver cirrhosis, which are all listed as risk factors for CPM. Reports of hypoglycemic-induced CPM/ODS are rare; glucose deprivation can lead to cytotoxic edema secondary to the failure of ion pumps in the cell membrane that manifests itself with MRI changes typical of CPM [24]. Furthermore, over-treatment resulting in hyperglycemia will activate the nicotinamide adenine dinucleotide phosphate (NADPH) pathway in neurons, causing cytotoxic oxidative stress. Our literature research shows the possible involvement of drugs, although there is no often a causal correlation with CPM/ODS; however, the hypothesis of drug influence should be kept in mind in unexplained neurological manifestation suggestive for CPM [10, 22]. For example, it is known that fluoroquinolones can cause or exacerbate hypoglycemia as consequence of direct stimulation of insulin secretion from the β-cells of the pancreas and typically occurs 4–6 days after the start of antibiotic therapy: this can play a crucial role in diabetic patients [24].
There are only few other similar published cases of CPM complicating the management of the correction of the variability of glucose levels in patient with diabetes. Conversely, there are more cases of CPM due to other predisposing factors (either chronic comorbidities or drugs). Despite the variability of etiologies, many patients were diabetics and this might highlight the close link of CPM with diabetes. CPM should always be considered in the differential diagnosis of neurological manifestations in patients with diabetes.
In conclusion, CPM, a potentially fatal condition, follows a favorable course provided that it is promptly recognized and managed. Our case suggests that, in diabetic patients presenting with signs and symptoms of CPM, recent episodes of decompensated glucose control as well as a large variability of glucose serum levels must both be taken into consideration to reduce the risk of brain damage. In conclusion, it is necessary to monitor neurological and cognitive status in patients with diabetes and/or other clinical conditions in which both an inappropriately rapid correction of osmolarity and any hypertonic insult in normal osmolarity status could potentially cause CPM.
CPM: central pontine myelinolysis
MRI: magnetic resonance imaging
ODS: osmotic demyelination syndrome
T1D: type-1 diabetes
SDA wrote the first draft of the manuscript; SDA and AACC wrote section of the manuscript; SDA, PA, and MM contributed to manuscript revision; all authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
The consent to participate in the description of the case report is informed.
Consent to publication of the privacy issues has been obtained.
Not applicable.
Not applicable.
© The Author(s) 2021.
Copyright: © The Author(s) 2021. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 5316
Download: 59
Times Cited: 0