 Review
Review

Affiliation:
1Department of Veterinary Pathobiology, University of Missouri, Columbia, MO 65211, USA
†The two authors contributed equally to the work.
ORCID: https://orcid.org/0000-0003-2567-029X
Affiliation:
2Department of Surgery, University of Missouri, Columbia, MO 65211, USA
†The two authors contributed equally to the work.
Email: yangmin@health.missouri.edu
ORCID: https://orcid.org/0000-0002-4895-5864
Explor Med. 2021;2:494–510 DOI: https://doi.org/10.37349/emed.2021.00066
Received: August 22, 2021 Accepted: October 15, 2021 Published: December 31, 2021
Academic Editor: Amedeo Lonardo, Azienda Ospedaliero-Universitaria di Modena, Italy
The article belongs to the special issue Exploring Chronic Liver Disease
Non-alcoholic fatty liver disease (NAFLD) as the most common chronic liver disease poses a significant impact on public healthcare and economic risk worldwide. As a multifactorial disease, NAFLD is usually associated with many comorbidities such as obesity, insulin resistance, hypertension, hyperlipidemia, diabetes, and cardiovascular disease. Without effectively preventive intervention, the advanced stage of NAFLD, non-alcoholic steatohepatitis (NASH), can progress to cirrhosis and hepatocellular carcinoma (HCC). However, there is no approved therapeutic treatment. Excessive fat accumulation in the liver is the hallmark of NAFLD, which can lead to mitochondrial dysfunction and endoplasmic reticulum (ER) stress. Dysfunction of two organelles also induces the upregulation of reactive oxygen species (ROS), activation of the unfolded protein response (UPR), and disruption of calcium transport, which promote NAFLD progression. Herein, this review summarized the current understanding of the roles of mitochondrial dysfunction and ER stress in the pathogenesis of NAFLD. Specifically, this review focused on the key molecules associated with the ER-mitochondria communication and different treatment options by targeting ER stress and mitochondrial dysfunction to treat NAFLD or NASH. Clinical trials to evaluate the therapeutic efficacy of representative agents, such as natural products, metabolites, and modulators of stress, have been reviewed and analyzed. Overall, recent findings suggest that targeting ER stress and mitochondrial dysfunction holds a promise for NAFLD treatment.
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease, posing significant problems to public health and increasing the economic burden worldwide [1]. The pathogenesis of NAFLD ranges from non-alcoholic fatty liver (NAFL) to non-alcoholic steatohepatitis (NASH) with the progression of liver inflammation, associated with or without liver fibrosis [2, 3]. NAFLD/NASH is commonly associated with other comorbidities, including obesity, insulin resistance, type 2 diabetes (T2D), hyperlipidemia, hypertension, and cardiovascular disease [4, 5]. Without effective treatment, NAFLD/NASH can lead to liver cirrhosis and hepatocellular carcinoma (HCC). However, the underlying mechanisms in the progression of NAFLD are not fully understood, consisting of multiple hits such as oxidative stress (OS)-induced mitochondrial dysfunction, endoplasmic reticulum (ER) stress, endotoxin [lipopolysaccharide (LPS)]-induced release of inflammatory cytokines, and iron overload [6–10].
Factors that trigger NAFLD can be divided into two origins, including external factors (e.g., adipose tissue and intestinal products) and intrahepatic factors (e.g., cell death and inflammation). Emerging evidence suggests that mitochondrial dysfunction and/or ER stress play essential roles in the pathogenesis of NAFLD [11, 12] and NASH [13]. Excessive lipid accumulation causes liver inflammation and metabolic dysfunction. Alteration of lipid species is also related to mitochondrial function. Change of lipid species is associated with mitochondrial function. For example, cardiolipin and ubiquinone were accumulated in NAFL with a protective effect on mitochondrial function, whereas acylcarnitine was increased in NASH with a decreased mitochondrial function [9]. Mitochondrial dysfunction further contributes to the disturbance of reactive oxygen species (ROS) production, inflammation, and cell death [11].
Neighboring mitochondria, ER is an important organelle for regulating protein synthesis and storage. The transient generation or accumulation of toxic lipids in hepatocytes can cause ER stress [14]. In response to ER stress, activation of unfolded protein response (UPR) is usually triggered, an adaptive pathway to maintain protein folding homeostasis [10]. Under a non-resolvable condition, persistent ER stress causes pathological consequences, including fat accumulation, insulin resistance, inflammation, and apoptosis [12, 15]. ER stress has been reported to contribute to the etiology of steatosis, NASH, and HCC [15]. Therefore, understanding the role of ER stress in the pathogenesis of NAFLD provides new therapeutic strategies.
Besides their independent functions, mitochondria and ER mutually impact each other. Disturbance of their interaction is implicated in many human diseases, including liver illnesses [16]. For instance, liver-specific ablation of the mitochondrial protein mitofusin 2 (Mfn2) in mice can trigger liver inflammation, triglyceride accumulation, fibrosis, and cancer [17]. Similarly, Mfn2 expression was downregulated in liver biopsies from NASH patients. Hepatic Mfn2 deficiency can decrease ER-mitochondrial phosphatidylserine (PS) transfer and phospholipid synthesis, leading to ER stress and liver diseases [17].
Herein, the current understanding of mitochondrial dysfunction and ER stress in NAFLD pathogenesis was summarized, specifically focusing on some important molecules involved in ER-mitochondria communication. Then, some potential therapeutic options by targeting ER stress and/or mitochondrial dysfunction to treat NAFLD were discussed. Finally, the current clinical trials that evaluate the potential effects of relative therapeutic agents were reviewed.
Hepatocytes account for 60–80% of the liver mass and play essential roles in liver function [18], including nutrient metabolism, storage of vitamins, detoxification, bile acid production and secretion, and protein synthesis. Intracellular mitochondria occupy about 20% of the cytoplasmic area in hepatocytes [19] and display diverse functions, such as maintaining metabolic (carbohydrate, lipid, and protein metabolism) and ROS homeostasis [20]. Beta-oxidation is processed to break down fatty acids in mitochondria, the important energy generators, and it can impact lipid accumulation in hepatocytes, as well as the subsequent production of ROS and ER stress [21]. Abnormal lipid metabolism causes mitochondrial dysfunction and ER stress in the pathogenesis of NAFLD. For instance, free cholesterol accumulation in the liver induces mitochondrial dysfunction, ROS production, and triggers UPR in the ER, resulting in ER stress and cell apoptosis. ER stress and mitochondrial dysfunction ultimately promote hepatocyte death and ongoing liver injury [22].
Mitochondria and adjacent ER are connected by mitochondria-associated membranes (MAMs) that regulate cell death and the exchange of lipids and lipid-derived molecules [23, 24]. Interruption of the integrity of MAMs happens during mitochondrial dysfunction and ER stress. Furthermore, persistent ER stress causes hepatocyte apoptosis and liver injury by impacting calcium (Ca2+) homeostasis [25] and accelerating the accumulation of toxic levels of ROS [26], leading to NAFLD progression. A schematic figure shows an example of how mitochondrial dysfunction and ER stress in hepatocytes function in the pathogenesis of NAFLD (Figure 1).
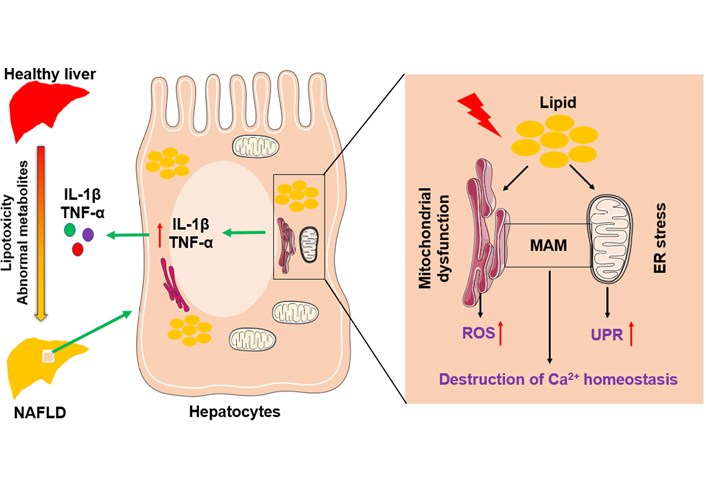
The role of mitochondria and endoplasmic reticulum in NAFLD pathogenesis. Abnormal lipid and glucose metabolism caused by diet and other environmental factors can lead to lipid accumulation in hepatocytes, resulting in liver injury and steatosis. Hepatic lipid accumulation causes mitochondrial dysfunction and ER stress, accompanied by the increase of ROS and the activation of the unfolded protein response. In addition, disruption of the integrity of MAM destroys Ca2+ homeostasis. Above factors adverse liver inflammatory by inducing overexpression of pro-inflammatory cytokines, such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α)
Liver non-parenchymal cells (NPCs) consist of Kupffer cells (KCs), liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs), and other lymphocytes. All of them are implicated in the pathogenesis of NAFLD [27]. As large abundant NPCs, LSECs as liver gatekeeping cells protect liver injury and filter the transport of nutrients, including lipids and lipoproteins [28]. Under the pathological condition, liver injury causes LSEC defenestration, inflammation, and impaired lipid transport. Meanwhile, inflammation also can increase ROS production and cytokines (e.g., TNF-α) in LSECs accompanying with alteration of mitochondrial permeability, resulting in the activation of KCs and HSCs [29, 30]. Meanwhile, lipid metabolism is critical for the survival and function of T cells, such as regulatory T cells (Tregs). For example, fatty acid-binding protein 5 (FABP5), a lipid chaperone, is required for the uptake and intracellular trafficking of lipid. Inhibition of FABP5 expression in Tregs caused impaired lipid metabolism and mitochondrial integrity, and induced loss of cristae structure [31]. ER-mitochondria communication in macrophages modulates fatty acid metabolism that is associated with inflammation and ER stress [32]. ER stress can further induce the activation of inflammasomes and production of proinflammatory cytokine IL-1β in KCs by triggering UPR, leading to hepatic inflammation and steatosis [33]. Furthermore, ER stress plays an important role in HSC activation and hepatic fibrosis [34, 35]. Overall, ER and mitochondria play pivotal roles in hepatocytes and liver NPCs and are involved in the pathogenesis of NAFLD.
Given the critical roles of mitochondria and ER in NAFLD pathogenesis [36], they are the therapeutic targets [37]. ER and mitochondria communicate with each other via MAMs to exchange Ca2+ and lipids [38]. Some important proteins associated with ER-mitochondria communication, such as glucose-regulated protein 75 (GRP75) and Mfn2, impact hepatic lipid and cholesterol metabolism in NAFLD [39].
Recent studies show that intracellular Ca2+ signaling plays a key role in the regulation of lipid and carbohydrate metabolism in hepatocytes [40], such as modulating the phosphorylation and dephosphorylation of enzymes in glucose metabolism [41]. Disrupting Ca2+ signaling can increase lipid accumulation via inhibiting lipolysis and activating lipogenesis, resulting in HCC progression [42]. These changes are associated with ER stress, generation of ROS, and mitochondrial dysfunction. Besides, Ca2+ signaling is implicated in the MAM [43]. Proteins such as sarcoendoplasmic reticulum calcium ATPase (SERCA) located in the MAM control the influx of cytosolic free Ca2+ into mitochondria to prevent overload of Ca2+ that can induce mitochondrial damage, ER stress, and overexpression of ROS [44]. Another study also showed that matrine treatment can significantly suppress ER stress and restore mitochondrial function via directly inhibiting SERCA to regulate Ca2+ level [45]. Therefore, agents targeting Ca2+ transporters, channels, or binding proteins are potential options for NAFLD treatment.
Mfn proteins include two homologous transmembrane proteins, Mfn1 and Mfn2. Both Mfn1 and Mfn2 play important roles in mitochondrial fusion, but they also have their distinct functions [46]. Studies have shown that Mfn2 is implicated in ER-mitochondria crosstalk. For example, acute Mfn2 depletion can inhibit mitochondrial uptake of Ca2+ released from the ER in mouse embryo fibroblasts and enlarge the distance between ER and mitochondria [47]; however, this phenomenon was not shown in Mfn1-depleted cells. Hepatic Mfn2 protein expression was decreased in mice fed with a high fat-high sugar diet compared to that in mice fed with a normal chow diet [39]. Meanwhile, this study further showed that silencing Mfn2 expression mediated by small interfering RNAs (siRNAs) in human liver cancer cell line Huh7 cells can decrease ER-mitochondria interactions and mitochondrial oxidative capacity, resulting in triglyceride (TG) storage [39]. Furthermore, Mfn2 expression was reduced in the livers of human patients and mice with NASH, and Mfn2-deficient mice showed increased liver inflammation, fat accumulation, fibrosis, and HCC compared to wild-type controls [17]. A molecular mechanism study revealed that Mfn2 was implicated in mitochondrial PS and phosphatidylethanolamine (PE) synthesis [17].
C-X-C motif chemokine receptor 3 (CXCR3) has been found to be upregulated in the liver tissues of human NAFLD patients and mice with diet-induced NASH models [48]. Overexpression of CXCR3 in steatohepatitis was positively associated with liver inflammation and infiltration of macrophages and T cells, as well as ER stress. The expression of Mfn1 was reduced in CXCR3 knockout (CXCR3−/−) mice, which was associated with improved mitochondrial integrity compared to wild-type mice while they were fed either methionine and choline-deficient diet for 4 weeks or high fat-high carbohydrate-high cholesterol diet for 12 weeks [48].
Mitochondrial fission, an effector stimulating ROS production in the metabolic process, plays an important role in NAFLD progression [49]. Dynamin-related proteins (DRPs) and cofactors regulate mitochondrial fission [50]. DRP1 (also known as DLP1) is a guanosine triphosphatase (GTPase) that controls mitochondrial and peroxisomal fission [51]. Upregulation of DRP1 has been shown to be positively associated with liver metabolic dysfunction in animal models, accompanying an increased expression of peroxisome proliferator-activated receptor-gamma (PPARγ) and sterol regulatory element-binding protein 1 (SRBEP1) [52]. Given the decreased mitochondrial fission, hepatic steatosis and oxidative stress were suppressed in DLP1-K38A mutant mice while feeding a high-fat diet containing 60% of daily caloric intake compared to that in wild-type mice [49].
GRP75 is a tethering protein of the MAM interface, which regulates Ca2+ transfer between mitochondria and ER [53]. GRP75 has a similar function with Mfn2, silencing GRP75 can alter MAM integrity and promote TG accumulation in Huh7 cells [39]. GRP78 is mainly located in ER but also shown in mitochondria and other locations, which plays multiple roles in maintaining cellular homeostasis [54], such as control of UPR and macroautophagy. One study showed that hepatic overexpression of GRP78 in ob/ob mice with infection of adenoviruses can inhibit liver triglyceride and cholesterol content and improve insulin resistance [55]. Molecular studies showed that overexpression of GRP78 decreased the expression of sterol regulatory element-binding protein-1c (SREBP-1c) and reduced ER stress markers, such as activating transcription factor 6α (ATF6α) and X box-binding protein-1 (XBP-1) [55].
Voltage-dependent anion channel 1 (VDAC1), an outer mitochondrial membrane protein, plays a pivotal role in cellular homeostasis of energy metabolism and crosstalk of mitochondria and the rest of the cell [56, 57], including Ca2+ homeostasis. Multi-omics analysis showed that VDAC1 was associated with cardiolipin composition shift in mitochondria membrane lipids and mitochondrial dysfunction in NAFLD [58]. Overdose of acetaminophen can cause hepatocyte ferroptosis, associated with mitochondrial dysfunction and suppression of tricarboxylic acid cycle (TCA) and fatty acid β-oxidation, which may be driven by VDAC1 oligomerization [59]. Therefore, VDAC1 is a therapeutic target for NAFLD by suppressing mitochondrial dysfunction.
Mitochondrial uncoupling affects mitochondrial proton influx and ATP generation [60], which is implicated in many chronic diseases such as obesity, cardiovascular diseases, and cancer liver metastasis [60, 61]. Mitochondrial uncoupling proteins play an essential role in NAFLD and NASH [61], and Dr. Goedeke and Dr. Shulman [62] report that mitochondrial uncouplers are therapeutic targets for metabolic-associated fatty liver disease (MAFLD) and NASH. For example, disruption of mitochondria proton flux was shown in the livers of rats fed with a high-fat or high-fat and high-cholesterol diet compared to the livers of control rats fed with a chow diet. This abnormal potential of the mitochondrial membrane was associated with increased expression of uncoupling protein-2 (UCP-2) in the liver [63].
In addition, genetic factors impact the progression of NAFLD and NAFLD-related HCC [64, 65], such as the rs738409 C > G single nucleotide polymorphism (SNP) variant of patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene and the rs58542926 C > T genetic variant of transmembrane-6 superfamily member 2 (TM6SF2) gene. Increasing evidence also shows that NAFLD patients have an increased proportion of liver mitochondrial DNA (mtDNA) mutations compared to those in healthy controls [66], such as gene mutations in oxidative phosphorylation (OXPHOS) chain. For example, feeding a western diet caused liver injury and alteration of liver mtDNA, evidenced by downregulation of mitochondrial OXPHOS, reduction in part of the fusion machinery Mfn1, and upregulation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) inflammatory singling pathway [67]. Mutations of mtDNA and SNPs could be potential diagnostic markers and treatment targets for NAFLD, which is reviewed by another publication recently [68], which will not be discussed here.
Current pharmacological options are limited in the treatment of NAFLD; therefore, new treatment options are urgently needed. Changes in lifestyle and caloric consumption help to prevent NAFLD development. However, it is hard to treat advanced-stage NASH. Here, we summarize some ongoing research studies in NAFLD treatment, specifically for interventions that can inhibit ER stress and mitochondrial dysfunction.
Physical exercise can prevent NAFLD development through modulating mitochondria function by increasing cytochrome c content, enzyme activities, and fatty acid oxidation [69]. Excessive fructose taken from foods and beverages can augment lipogenesis and insulin resistance [70] and lead to mitochondrial dysfunction [71], resulting in the development of NAFLD. Therefore, a change of diet and/or physical activity can prevent NAFLD by maintaining mitochondrial function.
Gut microbiota-derived components and metabolites are involved in the pathogenesis of liver diseases [72]. Alteration of gut microbiota through bariatric surgery [73], such as sleeve gastrectomy and Roux-en-Y gastric bypass (RYGB), has been shown to ameliorate hepatic inflammation, NAFLD, and NASH [74, 75]. However, the underlying mechanisms are not completely clear. Ezquerro et al. [74] reported that bariatric surgery increases the hormone ratio of acylated/desacyl ghrelin, which is associated with diminished hepatic steatosis, apoptotic cells, and inflammation. In addition, surgical treatment can reduce mitochondrial dysfunction, oxidative phosphorylation complexes I and II, and ER stress markers [e.g., ATF4 and C/EBP homologous protein (CHOP), as well as chaperone GRP78]. Moreover, acylated ghrelin ameliorates palmitate-induced hepatocyte steatosis and inflammation, associated with a reduction of ER stress [74]. Another study showed that RYGB can preserve liver mitochondrial respiratory chain complex I activity and reduce OS and TNF-α expression [76].
Some natural products and their derivates with anti-inflammatory and antioxidant activity have been applied to treat NAFLD [77, 78]. For example, a study showed that treatment of aqueous açai extract (AAE) in vitro has a high capacity to inhibit ROS production in HepG2 cells. AAE administration in vivo protected liver damage and inflammation, evidenced by a decrease of alanine aminotransferase (ALT), the number of inflammatory cells, and serum level of TNF-α. In addition, AAE treatment protected mitochondrial function and reduced OS by reducing lipid peroxidation and modulating the expression of antioxidant enzymes (e.g., glutathione reductase and catalase) [79]. Studies in rat primary hepatocytes and NAFLD model identified that isosteviol (ISV), a derivative of stevioside extracted from the plant Stevia Rebaudiana Bertoni, can prevent free fatty acid (FFA)- and high-fat diet (HFD)-induced hepatic injury through modulating protein kinase C-β/Src-homology-2-domain-containing transforming protein 1-mediated OS and ER stress, respectively, and it decreased ER-mitochondrial interaction [80]. Another study showed that intragastrical administration of Polygonatum kingianum (PK), a Chinese herb, significantly inhibited HFD-induced increase of ALT, aspartate aminotransferase (AST), total cholesterol (TC), and low-density lipoprotein (LDL) cholesterol in serum, and liver TC and triglyceride in rats. PK also promoted mitochondrial function by inhibiting HFD-induced increase of malondialdehyde and mitochondrial UCP-2 and HFD-induced decrease of cytochrome c and up-regulating carnitine palmitoyl transferase-1 (CPT-1), which alleviated NAFLD [81].
Melatonin, a hormone produced primarily by the pineal gland, has been found to protect liver function in obese subjects. In a diet-induced mouse NAFLD model, melatonin supplementation with drinking water ameliorated hepatic steatosis and metabolic dysfunction by suppressing the expression of microRNA (miR)-34a-5p and SREBP-1-dependent on silent information regulator 1 (SIRT1) signaling [82]. In addition, it can reduce ER stress evidenced by decreased GRP78 expression and modulate the change of mitochondrial shape. Another study showed melatonin supplementation can halt DRP1-mediated mitochondrial fission but recover mitophagy via blockade of nuclear receptor subfamily 4 group A member 1 DNA-dependent protein kinase catalytic subunit-p53 (NR4A1-DNA-PKcs-p53) pathway, displaying a protective effect to hepatocytes and mitochondrial function [83]. In ob/ob mice, melatonin treatment significantly reduced glycemia and steatosis and ameliorated the morphology and organization of these organelles. Treatment with melatonin also improved mitochondrial and ER function, including partially restoring ATP synthase β function and decreased GRP78 and CHOP expression in hepatocytes [84].
Azoramide, a small-molecule modulator of UPR with antidiabetic activity [85], has been shown to reverse the fructose-induced increase of body and liver weights, insulin resistance, ER-stress markers (GRP78 and XBP-1), lipid profile, AST, ALT, and histopathological changes in mice [86]. Anti-T2D medicines such as empagliflozin also can reduce the expression of genes involved in hepatic lipogenesis and ER stress (CHOP and ATF4) [87].
Metabolic abnormalities such as dyslipidemia and lipotoxicity contribute to liver injury and inflammation, promoting NAFLD progression to NASH, cirrhosis, and HCC [88]. Metabolic modulation is a potent effective pharmacotherapy for NAFLD. Analysis of hepatic proteomics and plasma lipidomics indicated that 6-month treatment with n-3 polyunsaturated fatty acids (PUFA), consisting of 64% alpha-linolenic, 21% eicosapentaenoic, and 16% docosahexaenoic acid (DHA), significantly ameliorated markers of lipid metabolism, ER stress, and mitochondrial dysfunction in NASH patients [89]. Maresin 1 (MaR1), a DHA-derived metabolite, has anti-inflammatory and anti-OS properties [90]. MaR1 treatment improved insulin sensitivity and attenuated adipose tissue inflammation in ob/ob mice and diet-induced obese mice [91]. Furthermore, MaR1 treatment inhibited hepatic lipid synthesis and steatosis via activating 5’ adenosine monophosphate-activated protein kinase (AMPK)/SERCA2b-mediated suppression of ER stress in HFD-fed mice [92].
Saturated palmitic acid (PA) can induce hepatocyte lipotoxicity to induce NASH. Treatment with monounsaturated oleic acid (OA) markedly improved PA-induced cellular apoptosis, OS, ER stress, mitochondrial dysfunction, as well as inflammation in hepatocytes. In rats, half replacement of HFD with OA producing olive oil decreased NASH symptoms, while full replacement of HFD with olive oil profoundly reversed NASH, reducing hepatocyte ballooning, liver inflammation, and liver fibrosis [93].
Over the past decade, miRs have been recognized to be involved in the pathogenesis of NAFLD and NASH [94]. For instance, in a fast-food diet-induced NASH model, miR-21 ablation inhibited the progression of hepatic steatosis, lipoapoptosis, and fibrosis [95], via modulating the expression of PPARα. Increased expression of the miR-21 signaling pathway was found in liver biopsies of NAFLD patients. In addition, supplementation of farnesoid X receptor (FXR) agonist obeticholic acid was helpful to markedly improve metabolic symptoms of NASH, showing a potential therapy for NAFLD [95]. Similarly, miR-34a levels were significantly upregulated in liver tissues of HFD-fed mice, associated with the downregulation of SIRT1. Suppressing miR-34a expression with inhibitor reduced lipid accumulation and improved the degree of steatosis, ameliorating the development of NAFLD [96]. Overall, potential treatment agents that can function on energy metabolism have effects on ameliorating mitochondrial dysfunction and ER stress (Table 1).
Treatment options for NAFLD
| Molecules | Target | Function | References |
|---|---|---|---|
| AAE | OS | AAE protected mitochondrial function and reduced OS by reducing lipid peroxidation and modulating the expression of antioxidant enzymes (e.g., glutathione reductase and catalase). | [79] |
| ISV | Oxidative and ER stress | ISV, a derivative of stevioside extracted from the plant Stevia Rebaudiana Bertoni, prevents FFA- and HFD-induced hepatic injury through modulating protein kinase C-β/Src-homology-2-domain-containing transforming protein 1-mediated oxidative and ER stress. ISV also decreased ER-mitochondrial interaction. | [80] |
| PK | Mitochondrial UCP-2 | PK promoted mitochondrial function by inhibiting HFD-induced increase of mitochondrial UCP-2 and HFD-induced decrease of cytochrome c and by up-regulating CPT-1, which alleviates NAFLD. | [81] |
| Melatonin | Mitochondrial fission | Melatonin supplementation halted mitochondrial fission but recovered mitophagy via blockade of NR4A1-DNA-PKcs-p53 pathway, conferring a protective effect to hepatocytes and mitochondrial function. | [83] |
| n-3 PUFA | ER stress and mitochondrial dysfunction | Analysis of hepatic proteomics and plasma lipidomics indicated that 6-month treatment with n-3 PUFA significantly ameliorated markers of lipid metabolism, ER stress, and mitochondrial functions in patients with NASH. | [89, 97] |
| MaR1 | SERCA2b | MaR1, a DHA-derived metabolite, inhibited hepatic lipid synthesis and steatosis via activating AMPK/SERCA2b-mediated suppression of ER stress in HFD-fed mice. | [81, 92] |
| Monounsaturated OA | ER stress, mitochondrial dysfunction, OS | Treatment with monounsaturated OA markedly improved PA-induced cellular apoptosis, OS, ER stress, mitochondrial dysfunction, as well as inflammation in hepatocytes. | [93] |
| Astragaloside IV (AS-IV) | ER stress | Treatment with AS-IV, a bioactive compound isolated from Astragali Radix, attenuated FFA-induced hepatic ER stress in a dose-dependent manner, evidenced by the reduction of the critical markers, GRP78, CHOP, and phospho-protein kinase R-like endoplasmic reticulum kinase (p-PERK). These results suggest that AS-IV is a promising therapeutic agent for hepatic steatosis and NAFLD. | [89, 97] |
| R-Tf-D-LP4 | VDAC1 | R-Tf-D-LP4 treatment eliminated hepatocyte ballooning degeneration, inflammation, and liver fibrosis associated with steatosis, NASH, and hepatocarcinoma, and it restored liver pathology-associated enzyme and glucose levels. | [98] |
| Silymarin | ER stress | Silymarin ameliorated NAFLD via inhibiting ER stress proteins GRP78 and XBP-1. | [99] |
Clinical trials have been performed to investigate different interventions in patients with NAFLD, including natural products, lifestyle change, dietary supplementation, bariatric surgery, and therapies modulating signaling pathways involved in lipid and glucose metabolism. Some representative trials are summarized in Table 2 (data selected from https://clinicaltrials.gov/).
Clinical trials for the intervention of NAFLD
| NCT number | Title | Intervention | Phase | Outcome measures |
|---|---|---|---|---|
| NCT01677325 | The clinical trial of NAFLD treated by traditional Chinese medicine | Chinese herb Yiqi Sanju Formula (YQSJF) | I | Computed tomography (CT) ratio of liver/spleen |
| BMI (body mass index) and insulin sensitivity homeostatic model assessment (HOMA) index | ||||
| Liver function | ||||
| Lipid profile and non-esterified fatty acid (NEFA) | ||||
| Adiponectin, leptin, IL-6, TNF, and hs-C-reactive protein (CRP) | ||||
| NCT00230113 | Effect of n-3 PUFA supplementation in NAFLD patients | n-3 fatty acid diet supplementation | II | Reduction of intrahepatic fat content as determined by magnetic resonance spectroscopy |
| Change in lipid profile and serum aminotransferase levels | ||||
| Change in levels of proinflammatory cytokines | ||||
| Change in insulin resistance determined by HOMA | ||||
| NCT04019561 | A study to evaluate the safety and pharmacodynamic efficacy of MEDI0382 in obese subjects with NAFLD/NASH | MEDI0382, a peptide targeted on glucagon-like peptide-1 (GLP-1) | II | Assess the safety and tolerability of MEDI0382 |
| Change in hepatic fat, liver volume, and fat volume | ||||
| Change in visceral adipose tissue and subcutaneous adipose tissue | ||||
| Change in circulating markers of hepatic inflammation | ||||
| And others | ||||
| NCT02098317 | DHA and Vitamin D in children with biopsy-proven NAFLD | DHA plus Vitamin D | III | Improvement in NAFLD activity score (NAS) and safety |
| Improvement of laboratory parameters of metabolic syndromes, such as lipids and gluco-insulinemic profile | ||||
| NCT03970031 | A study of MSDC-0602K to assess glycemic control and cardiovascular outcomes in patients with pre-T2D or T2D and NAFLD/NASH | MSDC-0602K, a modulator of the mitochondrial pyruvate carrier | III | Change in glycosylated hemoglobin A1c (HbA1c) from baseline to week 26 |
| Change in the weighted average of standardized AST, cytokeratin 18 (CK-18), and HbA1c values | ||||
| NCT02973295 | Silymarin in NAFLD | Silymarin, a flavonolignan extracted from the milk thistle [Silybum marianum (L.) Gaernt.] | IV | Change (Reduction) of parameters of liver steatosis defined by controlled attenuation parameter (CAP) and liver fibrosis defined by liver stiffness measurements (LSM) during the 6 months |
| Change in liver enzymes in a period of 6 months | ||||
| Change in insulin resistance in a period of 6 months | ||||
| Change in lipidogram in a period of 6 months | ||||
| NCT02564679 | Sleeve gastrectomy in adolescents with complicated morbid obesity and NAFLD | Laparoscopic sleeve gastrectomy (LSG) | NA | Improvement of metabolic parameters |
| Improvement of liver parameters | ||||
| Improvement of liver histology | ||||
| NCT04049396 | Berberine and NAFLD | Berberine, a chemical found in several plants such as European barberry and Oregon grape | NA | Change from baseline of ALT, AST, alkaline phosphatase (ALP), TC, fasting blood sugar (FBS), TG, LDL, and high-density lipoprotein (HDL) |
| NCT04073368 | Study of the safety and tolerability of AXA1125 and AXA1957 in subjects with NAFLD | Dietary supplement AXA1125 or AXA1957 | NA | Incidence of study product emergent adverse events (AEs) and serious adverse events (SAEs) |
| Percent change in liver fat | ||||
| Change in HOMA-insulin resistance (HOMA-IR) | ||||
| Change in plasma glucose, insulin, ALT, and AST | ||||
| NCT01680640 | Investigation of synbiotic treatment in NAFLD | Synbiotic, a mixture of probiotics and prebiotics | NA | Change in biomarkers for NAFLD and change in liver fat |
| Change in adipose tissue inflammation, satiety, and risk factors for T2D | ||||
| NCT03737071 | The effects of low carbohydrate diet on liver fat content and mitochondrial fluxes | Low carbohydrate diet | NA | Change in hepatic triglyceride content |
| Change in hepatic mitochondrial flux | ||||
| Change in beta-hydroxybutyrate production rate | ||||
| Change in glucose production rate | ||||
| NCT03193125 | Effects of carnitine supplementation on liver mitochondria fatty acid processing | Dietary supplement of L-carnitine | NA | Change from baseline in liver mitochondrial fatty acid processing during high-fructose drink challenge |
| NCT03151798 | Nutrient overload, insulin resistance, and hepatic mitochondrial dysfunction | Lifestyle treatment | NA | Liver mitochondrial gene expression |
| Liver mitochondrial fat oxidation | ||||
| Histology to determine the amount of fibrosis in the liver | ||||
| NCT02457702 | Mitochondrial function in patients with severe liver disease | Orally administered labeled glycerol ([U-13C3]glycerol) | NA | Detection of 13C in plasma glucose by nuclear magnetic resonance of a blood sample |
| NCT03587727 | Hepatic fat content and mitochondrial flux in obese youth before and after bariatric surgery | Weight loss surgery or bariatric surgery | NA | Changes in hepatic mitochondrial function |
| Changes in insulin resistance/pancreas/gastrointestinal hormones | ||||
| Changes in NAFLD status via tissue | ||||
| NCT03480594 | Effect of fatty liver on TCA cycle flux and the pentose phosphate pathway (HP FFF) | Hyperpolarized [13C]pyruvate injection | I | Bicarbonate/lactate ratio |
| Labeled glycerol fraction | ||||
| The fraction of [U-13C]glycerol that has passed through mitochondrial pathways prior to gluconeogenesis and the fraction of glucose, derived from [U-13C]glycerol, that has passed through the pentose phosphate pathway | ||||
| NCT02344186 | Effects of liraglutide on endoplasmic (ER) stress in obese patients with T2D | Liraglutide, a GLP-1 receptor agonist | NA | Changes in unfolded protein response markers |
NA: not applicable
NAFLD, the most common chronic liver disease, is a deleterious factor for other metabolic disorders, such as diabetes, obesity, and cardiovascular diseases. It is a complex and multifactorial disease with severity from NAFL to NASH, which can be initiated by genetic and non-genetic factors. Both mitochondria and ER are critical organelles for cellular metabolic function and energy production. Emerging evidence indicates that mitochondrial dysfunction and ER stress are involved in the initiation and progression of NAFLD. Herein, we summarize some recent findings for ER-mitochondria crosstalk in NAFLD, the associated molecular mechanisms, and treatments targeting mitochondrial dysfunction and ER stress. In addition, clinical studies have been processed to investigate the application of some potential interventions for NAFLD treatment. Better investigating of the precise cellular mechanisms involved in the ER and mitochondrial functions provides new therapeutic targets for NAFLD.
AAE: aqueous açai extract
ALT: alanine aminotransferase
AST: aspartate aminotransferase
ATF6α: activating transcription factor 6α
Ca2+: calcium
CHOP: C/EBP homologous protein
CXCR3: C-X-C motif chemokine receptor 3
DHA: docosahexaenoic acid
DLP/DRP: dynamin-related protein
ER: endoplasmic reticulum
FFA: free fatty acid
GRP75: glucose-regulated protein 75
HCC: hepatocellular carcinoma
HFD: high-fat diet
HOMA: homeostatic model assessment
HSCs: hepatic stellate cells
IL-1β: interleukin-1β
ISV: isosteviol
KCs: Kupffer cells
LSECs: liver sinusoidal endothelial cells
MAMs: mitochondria-associated membranes
MaR1: maresin 1
Mfn2: mitofusin 2
miR: microRNA
mtDNA: mitochondrial DNA
NAFL: non-alcoholic fatty liver
NAFLD: non-alcoholic fatty liver disease
NASH: non-alcoholic steatohepatitis
NPCs: non-parenchymal cells
OA: oleic acid
OS: oxidative stress
PA: palmitic acid
PK: Polygonatum kingianum
PUFA: polyunsaturated fatty acids
ROS: reactive oxygen species
SERCA: sarcoendoplasmic reticulum calcium ATPase
T2D: type 2 diabetes
TC: total cholesterol
TG: triglyceride
TNF-α: tumor necrosis factor-α
UCP-2: uncoupling protein-2
UPR: unfolded protein response
VDAC1: voltage-dependent anion channel 1
XBP-1: X box-binding protein-1
MY and CZ contributed conception, drafted the review, and contributed equally.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2021.
Copyright: © The Author(s) 2021. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Maria Gabriela Delgado ... Jean François Dufour
Maria Giulia Cornacchia ... Gaetano Serviddio
Alessandro Mantovani ... Andrea Dalbeni
Sheikh Mohammad Fazle Akbar ... Yoichi Hiasa
Amedeo Lonardo
Chunye Zhang ... Ming Yang
Katerina Delladetsima ... Stratigoula Sakellariou
