Review
Review

Affiliation:
Department of Epidemiology, Faculty of Naval Medicine, Second Military Medical University, Shanghai 200433, China
†These authors share the first authorship.
Affiliation:
Department of Epidemiology, Faculty of Naval Medicine, Second Military Medical University, Shanghai 200433, China
†These authors share the first authorship.
Affiliation:
Department of Epidemiology, Faculty of Naval Medicine, Second Military Medical University, Shanghai 200433, China
†These authors share the first authorship.
Affiliation:
Department of Epidemiology, Faculty of Naval Medicine, Second Military Medical University, Shanghai 200433, China
Affiliation:
Department of Epidemiology, Faculty of Naval Medicine, Second Military Medical University, Shanghai 200433, China
Affiliation:
Department of Epidemiology, Faculty of Naval Medicine, Second Military Medical University, Shanghai 200433, China
Affiliation:
Department of Epidemiology, Faculty of Naval Medicine, Second Military Medical University, Shanghai 200433, China
Email: gcao@smmu.edu.cn
Explor Med. 2021;2:516–526 DOI: https://doi.org/10.37349/emed.2021.00068
Received: August 19, 2021 Accepted: November 20, 2021 Published: December 31, 2021
Academic Editor: Derek M. Dykxhoorn, University of Miami Miller School of Medicine, USA
The processes of cancer and embryonic development have a partially overlapping effect. Several transcription factor families, which are highly conserved in the evolutionary history of biology, play a key role in the development of cancer and are often responsible for the pivotal developmental processes such as cell survival, expansion, senescence, and differentiation. As an evolutionary conserved and ubiquitously expression protein, CCCTC-binding factor (CTCF) has diverse regulatory functions, including gene regulation, imprinting, insulation, X chromosome inactivation, and the establishment of three-dimensional (3D) chromatin structure during human embryogenesis. In various cancers, CTCF is considered as a tumor suppressor gene and plays homeostatic roles in maintaining genome function and integrity. However, the mechanisms of CTCF in tumor development have not been fully elucidated. Here, this review will focus on the key roles of CTCF in cancer evolution and development (Cancer Evo-Dev) and embryogenesis.
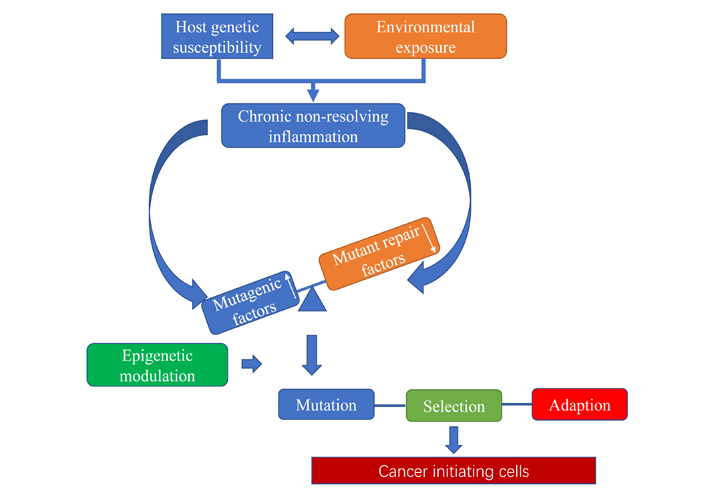
CCCTC-binding factor (CTCF) is a multivalent 11-zinc finger (ZF) protein that binds tens of thousands of sites in the human genome [1]. Initially identified as transcriptional regulator of chicken MYC proto-oncogene (c-myc gene) in 1990 [2, 3], CTCF was later found to mediate gene imprinting at the H19 imprinted maternally expressed transcript/insulin-like growth factor 2 (H19/Igf2) locus [4]. Follow-up studies indicated that CTCF interferes with the interaction between enhancers and promoters because of its binding sites frequent presence in various chromosomal structural boundaries, such as the topologically associated domains (TADs) [5, 6]. Increasing evidence indicated that CTCF protein is highly conserved from Drosophila to humans [7]. CTCF is one of the key regulatory proteins in vertebrates, and its knockout leads high in utero embryonic lethality [8]. CTCF can participate in a variety of biological processes and have different functions, including insulator function [9], regulation of gene transcription [10], gene imprinting (Figure 1) [11], X chromosome inactivation [12, 13], affecting messenger RNA (mRNA) alternative splicing as well as nucleosome rearrangement and DNA replication [14, 15]. Recently, high-throughput chromosome conformation capture (Hi-C) indicated that CTCF has dynamic roles as regulator of imprinted loci and governing higher-order chromatin architecture other than enhancer blocking activity [5, 16]. The latest evidences revealed that CTCF is directly involved in the transcriptional modulation of various key factors of cellular cycle control, apoptosis, senescence, and differentiation [17, 18]. Indeed, ectopic expression of CTCF in several human tumoral cell lines inhabits cell division and clonogenicity [18]. Somatic mutations at CTCF-binding sites, which can abrogate the CTCF-mediated spatial folding of chromosomes [19], have been profoundly found in various human cancers [20]. Interestingly, CTCF also has a key role in the establishment of three-dimensional (3D) chromatin structure during human embryogenesis [21]. The basic framework of the hypothesis of cancer evolution and development (Cancer Evo-Dev) is shown in Figure 2. The complex interactions between host genetic susceptibility and environmental exposure [such as hepatitis B virus (HBV) infections] induced immune system disorder. The dysfunctional immune system activates and maintains chronic inflammation with the permanent existence of HBV in host. Under the inflammatory microenvironment, the imbalance of mutagenic factors and mutant repair factors, cooperating with HBV integration and epigenetic modulations, promotes HBV-related somatic mutations and HBV mutations. A majority of hepatocytes with genomic mutations and HBV variants are eliminated in the survival competition in inflammatory microenvironments. Only a small part of cells survive by alternating survival signaling pathways and exhibit “stemness” characteristics. The survivals gradually evolved to be cancer stem cells, which drives the development and evolution of cancer [22]. According to the Cancer Evo-Dev hypothesis, CTCF is considered as an essential regulator during cancer cells “backward evolution” and “retro-differentiation”.
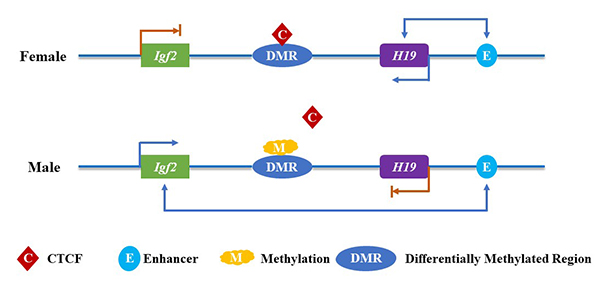
Gene imprinting. The methylation of the paternal allele DMR affects the binding of CTCF, and the enhancer downstream of H19 can interact with the Igf2 over a long distance to promote its expression. On the contrary, the maternal allele DMR is not methylated, which means CTCF can bind to it and then block the interaction between the downstream enhancers of H19 and the promoters of Igf2. As a result, the expression of Igf2 is turned off, but H19 can still be expressed under the action of downstream enhancers
There is a growing consensus that a hierarchical chromatin structure is established within the nucleus in the interphase of the cell cycle [23, 24]. The spatial folding of chromosomes and their organization, which is largely mediated by CTCF, have profound effects on gene expression [25, 26]. Genetically inducible CTCF deletion in several specific cell types, including oocytes, lymphocytes, and cardiomyocytes, leads to organ-specific failure [27, 28]. Knocking out the CTCF of the oocyte seriously disrupts the entry of the fertilized oocyte into the blastocyst stage [28]. Furthermore, homozygous deletion of CTCF at the whole embryo level results in embryo death [8], which is characterized by aberrant enhancer-promoter interactions and transcriptional dysregulation [29]. However, albeit numerous studies have explored the function of CTCF binding, the in vivo roles of CTCF during embryonic development are largely unknown.
Recent studies revealed that TAD structures are totally lost in human 2-cell embryos, are at a low level in 8-cell embryos, and then gradually established during embryonic development [21]. As an essential hierarchical structural feature of chromatin organization, A/B compartmentalization is absent in human 2-cell embryos and reformed during embryogenesis [21, 30, 31]. Unlike in mature mouse sperm, TAD structures are absent in human sperm. Previous studies have shown that depletion of CTCF can lead to the disruption of TADs, suggesting that the lack of CTCF may contribute to the loss of TAD structures in human sperm [21, 32]. Immature TAD boundaries gained at the 2-cell embryos tend to locate around housekeeping genes, which may promote the expression levels of housekeeping genes. In addition, both CTCF expression and TAD establishment in human embryos require human zygotic genome activation (ZGA). Further studies suggested that CTCF expression is required, but is not the only factor needed, for TAD establishment during human ZGA [21].
CTCF can induce both cell cycle progression or arrest to accomplish its function [17], which is dependent by cell-specific CTCF-binding DNA sequences (CTSs), protein partners and chromatin long-range interactions [33]. CTCF can repress cell growth and colony formation suggesting a suppressor role of CTCF [34, 35].
It is reported that CTCF depletion activates DNA damage response and increases the risk of chromosomal instability [36]. DNA damage signaling, Mre11 (MRE11 homolog, double strand break repair nuclease)-Rad50 (RAD50 double strand break repair protein)-Nbs1 (nijmegen breakage syndrome 1) complex and CTCF DNA-binding domain promote CTCF enrichment at DNA damage sites [36]. As the core DNA-binding protein of homologous recombination (HR), RAD51 is overexpressed in a variety of tumors [37]. CTCF participates in HR repair of DNA double strand breaks by interacting with RAD51 and promoting the formation of RAD51 repair foci [38]. CTCFL (CTCF like), otherwise known as Brother of the Regulator of Imprinted Sites (BORIS), is a male system-specific protein with the same 11-zinc finger structure as CTCF [38]. In non-small cell lung cancer (NSCLC), BORIS suppresses DNA damage and promotes cisplatin resistance by enhancing the mismatch repair system of cancer cells [39].
Gene methylation detection showed that DNA methylation at the CpG sites in the CTCF coding gene caused the CTCF binding sites to be blocked [40]. The function of CTCF in regulating gene imprinting suggested that the binding of CTCF to the target gene is methylation sensitive [4]. The aberrant DNA methylation of key regulate regions can hinder CTCF binding, which may lead to epigenetic silencing of tumor suppressor loci or lead to activation of oncogenes [41]. Large tumor suppressor (LATS) kinase can be activated by a variety of stress responses, such as glucose deficiency, and then phosphorylates downstream pathways to promote cell survival, while abnormal LATS activation promotes tumorigenesis. LATS kinase directly phosphorylates CTCF at the ZF junction, inhibiting its DNA binding activity [42]. Recent studies indicated that DNA methylation-regulated alternative cleavage and polyadenylation (APA) requires CTCF and the cohesin complex [43]. Epigenetic inactivation of Ras association domain family 1 isoform A (RASSF1A) and E-cadherin (CDH1) in breast cancer is associated with alternations in CTCF recognition sites [44]. Aberrant DNA methylation can inhibit CTCF-mediated silencing of BCL6 transcription repressor (BCL6) gene, thus increasing the expression of proto-oncogene BCL6 in lymphoma [45]. In addition, alternations in CTCF gene in endometrial cancer can promote tumorigenesis by promoting cell survival and changing cell polarity [46].
CTCF hemizygous deletions are frequently found in various human tumors [47]. Loss of a single CTCF allele (Ctcf+/−) significantly increases the risk of cancer and enhances malignant progression [40]. CTCF hemizygosity dysregulates cancer-related pathways, such as Ras, Ras-mitogen-activated protein kinase (Ras-MAPK) and extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathways [48]. Ctcf+/− mice remain a disorder methylation status, and even lead to a modest overall increase levels of genome-wide DNA methylation in lung [40]. Chromosome 16q22.1 is a common deletion region in various epithelial cancers, and CTCF is located on this region. In prostate cancer cells, knockdown of CTCF results in hypermethylation of the CTCF/cohesin-binding sites (CBSs). Prostate and breast cancers with insufficient CTCF copy number show increased DNA hypermethylation events in vivo [49]. In addition, high frequency mutations of CBSs also lead to disorders of specific sites hypermethylation and promote the occurrence and development of cancer [43].
CTCF has been shown to modulate the expression of various cancer-associated genes. c-myc plays vital roles in tumorigenesis, embryogenesis, and somatic cells reprogramming [50–52]. CTCF exerts divergent roles on c-myc regulation by binding various sequences at c-myc promoter [1]. In myeloid cells, overexpression of CTCF decreases c-myc levels and growth rate, which promotes myeloid differentiation and induces cell cycle arrest in numerous tumoral cells [34, 53]. In most malignant tumors, telomerase is activated to prevent telomere shortening, which confers high proliferative capacity to tumoral cells [54, 55]. As one of the telomerase components, human telomerase reverse transcriptase (hTERT) is only expressed in telomerase-positive cells, and depletion of CTCF induced hTERT transcription in TERT-negative cells [56, 57]. However, CTCF is not able to exert a transcriptional repressive function because of DNA-methylation or BORIS-binding in TERT-positive cells [58]. Moreover, in cancer cells, methylation of CTSs at hTERT exon inhibits CTCF-binding, which prevents hTERT repression [59]. Retinoblastoma (RB) is considered a tumor suppressor gene and CTCF maintains RB gene promoters at an active epigenetic status [60, 61]. The inactivated of RB gene family leads to the loss of cell cycle control and cancer. Other evidences showed that CTCF is also involved in an epigenetic balance at the cyclin-dependent kinase inhibitor 2A locus (CDKN2A) and tumor protein p53 (TP53) gene promoters [58, 62]. In sum, by binding various CTSs, CTCF can modulate c-myc, hTERT, RB, and other tumor-related genes, which emphasize the cell-type dependency of its tumor suppressor role.
Chromosome architecture has different levels: euchromatin (A) and heterochromatin (B) compartments are corresponding to megabase-scale, TADs are corresponding to sub-megabase scale and smaller loop structures correspond to tens of kilobases level [63]. As a sister chromatid cohesin molecule, cohesin was also found to modulate higher-order chromosome architecture by cooperating with CTCF. CTCF interacts with the cohesin through two different domains at the N-terminal. Amino acid 222–231 binds to the cohesin’s conserved essential surface, which is a composite interface formed by stromal antigen (SA) and RAD21 cohesin complex component (RAD21) subunits. This motif is known as YxF motif in CTCF. Amino acid 23–27 binds to the cohesin protein PDS5A subunit. This motif is known as YxR motif in CTCF [64, 65]. Interestingly, CTCF assists cohesin to bind specific sites on chromosomes while its binding is independent of the presence of cohesin on chromatin [66]. Recent evidence indicated that cohesin catalyzes the folding of the genome into loops that are anchored by CTCF and CTCF enables chromatin loop formation by protecting cohesin against loop release [65]. CTCF/cohesin-mediated chromatin loops play an essential role in the maintenance of genome integrity [67]. Previous studies showed that mutations at CBSs and somatic mutations in the cohesin subunits were frequently detected in various cancers, while aberrant overexpression of the cohesin complex was also frequently found in several human malignancies [20, 68, 69]. Mutations in cohesin subunits and CBSs may promote genomic instability by perturbing proper long-range chromatin interactions. In cancer cells with chromosomal instability, CTCF/cohesin-mediated chromatin organization and DNA replication play an essential role in gene stable amplification [70]. As a member of cohesin family gene, stromal antigen 2 (STAG2) is one of the most commonly mutated genes in cancer. In Ewing’s sarcoma, STAG2 disfunction is related to aggressive behavior. STAG2 loss of function orchestrates oncogenic transcription factors by changing CTCF-anchored loop extrusion [71]. RAD21 is an essential subunit of the cohesin complex. Interestingly, depletion of RAD21 in epithelial cancer cells induces epithelial to mesenchymal transition (EMT) while overexpression of RAD21 in mesenchymal cancer cells induces mesenchymal to epithelial transition (MET)-specific expression patterns, suggesting that dynamic cohesin-mediated chromatin structures are responsible for the initiation and regulation of essential EMT-related cell fate changes in cancer [72].
CTCF is constitutively and widely expressed in normal tissues while BORIS protein exists normally only in the testis [73]. BORIS belongs to the cancer/testis gene family, which is only expressed in malignant tumors except male germ cells. The abnormal expression levels of BORIS RNA and protein, which are affected by DNA-methylations, related with the size of tumors and the degree of malignancy, otherwise the knockdown of BORIS induced apoptosis in tumorous cells [74]. CTCF and BORIS do not compete even they have the same recognition sites in normal somatic cells. However, expression of BORIS in BORIS-negative cells not only interferes with the normal functions such as growth inhibition of CTCF, but also leads to cell dysfunction, which leads to tumorigenesis due to the competitive binding of BORIS/CTCF gene family [75]. Recent studies indicated that ectopic expression of BORIS activates cancer testes antigens (CTA) and components of cancer relevant signaling pathways [76]. The test of sterile BORIS−/−CTCF+/− (compound mutant, CM) male mouses confirmed that combined depletion of BORIS/CTCF will lead to defection of meiotic recombination, increasing of apoptosis, and malformed spermatozoa [77]. Interestingly, BORIS/CTCF heterodimeric sites are enriched in both cancer and germ when the promotors and enhancers of cells are activated [78]. The low expression of spermatogenesis genes and aberrant expression of sterile genes in CM mouses indicated that joint action of BORIS/CTCF is essential for spermatogenesis program by restraining pre-meiotic genes and activating post-meiotic genes [77].
Cancer development is characterized by an evolutionary process of “mutation-selection-adaptation”, some highly conserved genes are highly expressed in the embryo, not expressed or low expressed in normal adult tissues, but highly expressed in cancer tissues, leading to reverse cell differentiation, malignant proliferation and enhanced migration capacity [22]. Recent evidences revealed that in normal prostate tissue, CTCF expression was negative to low, while in prostate cancers, CTCF expression was seen in 7,726 of 12,555 (61.5%) tumors and was considered low in 44.6% and high in 17% of cancers [79]. CTCF expression is a feature of poor prognostic in prostate cancer, but CTCF is a dissatisfactory candidate biomarker because of its low predictive power [79]. Similarly, CTCF is frequently up-regulated in partial primary hepatocellular carcinoma (HCC) compared with non-neoplastic liver. Overexpression of CTCF is associated with shorter disease-free survival in patients and the absence of CTCF can lead to decreased motility and invasiveness of HCC cells [80]. In addition, chromosomal ring anchors bound by CTCF and cohesin are prone to continuous DNA breakage, and regions of translocation break points in various cancers are enriched in these anchors [67]. Continuous DNA break and repair provide opportunities for genetic mutations, and in the context of numerous mutations, cancer is constantly selecting and adapting. Furthermore, multiple cancer types accumulate CBS mutations and CBSs are major mutational hotspots in the noncoding cancer genome, which emphasize the significant role of CTCF in cancer evolution and development. In summary, CTCF hemizygotic mutations and CBSs mutations disrupt genomic stability and, in combination with epigenetic modifications such as methylation, promote the mutation and adaptive selection of cancer cells, thereby promoting the evolution and development of cancer.
CTCF, a highly conserved and multifunctional protein, contributes to formation of multi-dimensions genome and control of central signals to transcriptional networks [5]. CTCF plays an essential role in embryonic development and cancer development, but its molecular mechanism has not been fully elucidated. CTCF exerts a tumor suppressor role by regulating several key factors related to growth and development and regulating the higher-order structure of chromosomes, but its high expression in various cancer cells may promote the malignant proliferation and migration, and often predicts a poor prognosis [33, 79]. Although roles of CTCF in carcinogenesis have been intensively explored, more researches are needed to better understand the significant functions and mechanisms of CTCF in embryonic development and cancer development.
With the continuous development of modern bioinformatics and biotechnology, the combined use of a variety of high-throughput analysis techniques such as chromatin immunoprecipitation-chip (ChIP-chip), ChIP-qPCR, and so on can constantly find out the corresponding binding sites and regulatory models of CTCF with DNA, protein and RNA, which can further clarify the action mechanism of CTCF. The developed highly specific epigenome editing technology based on clustered regularly interspersed short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9) as a broad prospect because it can modify the genome of specific regulatory elements [81]. For example, targeted editing of the DNA methylation status of CTCF binding sites can alter the expression of CTCF, thereby altering the expression of target genes by affecting the structure of advanced chromatin [82]. Therefore, the highly specific epigenome editing technology based on CRISPR/Cas9 may become an attractive epigenome-based cancer therapy in the next few years to regulate the occurrence and development of tumors.
BORIS: Brother of the Regulator of Imprinted Sites
Cancer Evo-Dev: cancer evolution and development
CBSs: cohesin-binding sites
c-myc: chicken MYC proto-oncogene
CTCF: CCCTC-binding factor
CTSs: CCCTC-binding factor-binding DNA sequences
HBV: hepatitis B virus
hTERT: human telomerase reverse transcriptase
Igf2: insulin-like growth factor 2
LATS: large tumor suppressor
RB: retinoblastoma
STAG2: stromal antigen 2
TADs: topologically associated domains
ZL, XZ and SC participated in the manuscript design, and writing. JF, ZW and YC contributed to the figures. GC extensively revised the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by grant 2015CB554006 from the National Key Basic Research Program of China (GC); grants 81673250, 91529305, and 81520108021 from the National Natural Science Foundation of China (GC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2021.
Copyright: © The Author(s) 2021. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.