
Abstract
The redox status in pathogenesis is critically regulated by careful balance between the generation of reactive oxygen species (ROS) and their elimination. Increased ROS level above the cellular tolerability threshold results in apoptotic or necrotic cell death. ROS belongs to a group of highly reactive compounds that have evolved to play key roles in cellular signaling pathways. It’s widely assumed that a reasonable amount of ROS is essential for a variety of biological processes. Elevated levels of ROS are known to cause various pathologic conditions like neurological disorders, cardiovascular conditions, inflammation, autoimmunity, and cancer. ROS is well known to initiate and assist in progression of tumor by promoting proliferation and survival of cancer cells and thus facilitates pro-tumorigenic signaling in tumor microenvironment. As cancer cells become more resilient to the effects of ROS manipulating drugs, increased antioxidant capacity attenuates their susceptibility to cancer treatment. Excessive environmental stress, on the other hand, can cause cancer cells to die. This review summarizes various molecular mechanisms including the role of checkpoint inhibitors that can be harnessed to develop effective therapeutic strategies for targeting ROS related signaling in cancer.
Keywords
Redox homeostasis, reactive oxygen species signaling, cancer, metastasis, angiogenesis, checkpoint inhibitors, therapeutic relevanceIntroduction
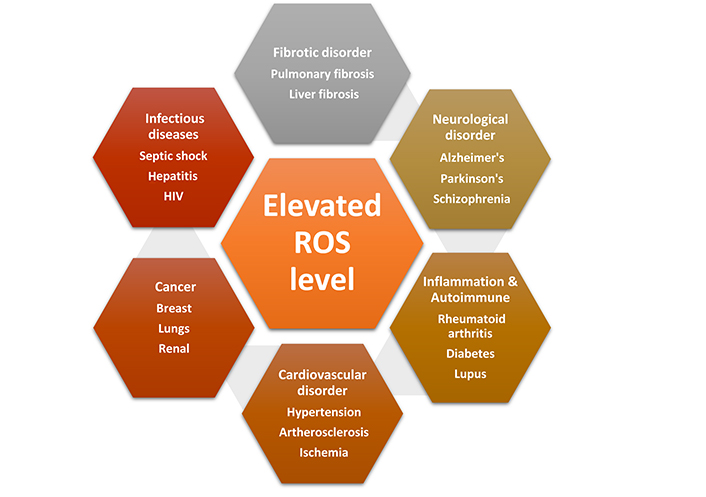
The reactive oxygen species (ROS) are short-lived oxygen derivatives that are unstable and highly reactive and formed as a byproduct of cellular metabolic processes. Superoxide anion (O2), hydrogen peroxide (H2O2), and hydroxyl radical (HO•) are examples of radical and non-radical forms that are generated by partial reduction of oxygen. These molecules play a pivotal role in cell signaling and are essential for normal cellular functions both in normal cells as well as in cancer cells [1]. In a biological setting, ROS are produced as a natural consequence of oxygen metabolism and play a key role in various signaling pathways and homeostasis. In order to promote normal cellular physiological function and survival, redox homeostasis regulation is maintained in the cell [2]. Any sort of elevated intracellular ROS concentration contributes to various pathological conditions (Figure 1). However, the cancer cells harbor high intracellular ROS level compared to normal cells due to high aerobic glycolysis that causes oxidative stress [3]. High level of ROS mostly accumulated due to imbalance in the ROS production and its elimination process. High ROS level alters various signaling pathways that affect cellular metabolism. However, in the cancer cells, the heightened level of ROS is countered by upregulated anti-oxidative defense mechanisms [4, 5]. Whereas, further rise in the ROS level is accompanied with down regulation of cellular antioxidant defense systems in tumor malignancy through various molecular targets including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and nuclear respiratory factor 2 (Nrf2) [6, 7]. High ROS level damages cellular DNA [8], proteins [9], and lipids [10], which promotes genomic instability and oncogenesis. High oxidative stress succumbs the cell to apoptosis, but robust antioxidant capacity of the cancer cells is increased in order to prevent the formation of high ROS and preserve the redox balance. High level of ROS scavenging enzymes calibrates the ROS production and allows activation of pro-tumorigenic signaling pathways without inducing cell death in tumor [4]. Comparatively, tumor cells instead of normal cells have altered redox milieu, and this property of cancer cells make them more sensitive to ROS alteration or redox manipulation [11]. The elimination of ROS by scavenging enzymes or increased ROS production may be effective options for cancer therapies [12]. In this review, we have discussed and focused on pro- and anti-tumorigenic signaling capabilities of ROS and ways to manipulate these factors for cancer treatment.
Homeostasis of ROS and redox control
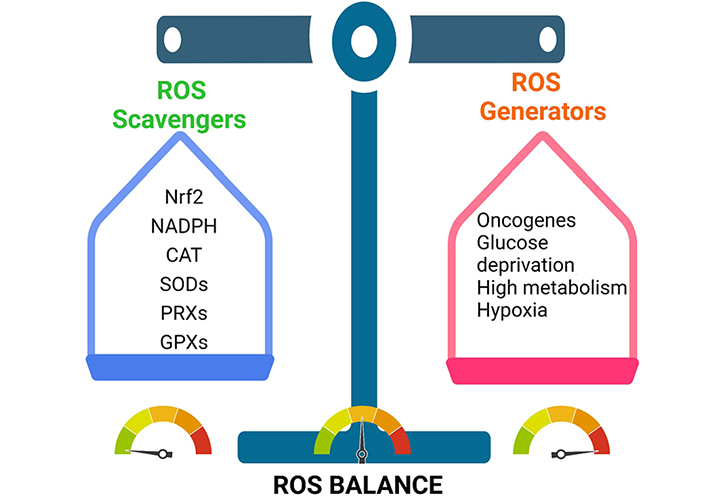
Continuous generation and detoxification of ROS is tightly controlled by redox homeostasis in the normal cells. The inequality between the manufacturing of oxidants and their removal by protective mechanisms or antioxidants is called the oxidative stress. This event eventually leads to accumulation of ROS in the cell that results in disturbance in redox steadiness, which could trigger harm to vital cell components such as proteins, lipids, and DNA, and potentially elevate the risk of somatic mutations and neoplastic transformation [8, 13]. These effects of ROS are also believed to play a part in the ageing process [14]. Numerous kinds of free radical ROS and non-free radical ROS present in the cells. The major source of ROS production appears to be located inside the mitochondrial electron transport chain where electrons can evade their path and react with oxygen [15]. The stimulation of growth factor receptors which activate reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase can also produce ROS and convert NADPH to superoxide. A strong antioxidant system that scavenges ROS keeps the cellular redox equilibrium in check. Majority of superoxides are transported to the mitochondrial matrix, where superoxide dismutase enzymes dismutate it to H2O2 manganese superoxide dismutase (MnSOD) or superoxide dismutase 2 (SOD2). Some superoxides are transported to the cytosol, where they are dismutated to H2O2 by the cytosolic enzyme SOD1 [16]. Glutathione (GSH) is the most important nonenzymatic component of intracellular antioxidant defenses as it regulates NADPH intracellular levels to maintain redox equilibrium [nicotinamide adenine dinucleotide phosphate (NADP)/NADPH and reduced glutathione (GSSG)/oxidized glutathione (GSH) ratios] [17]. The discrepancy between NADPH synthesis and consumption determines the intracellular NADPH level. The pentose phosphate pathway (PPP) and mitochondrial metabolism are the main sources of intracellular NADPH. The major consumer of intracellular NADPH is H2O2 detoxification by glutathione peroxidases (GPX) and peroxiredoxin (PRX) and fatty-acid synthesis [18]. Thus, production and consumption of NADPH inside the cells should be coordinated to maintain NADPH homeostasis. This is especially crucial when solid tumor forms and the cancer cells are under energy stress during metastasis.
Role of ROS in cancer development
Oxidative stress has been associated to cancer development and progression by increasing the incidence of DNA mutations, resulting in DNA damage, genomic instability, and cell proliferation [19–21]. Cancer cells produce more spatially concentrated ROS than normal cells, which hyperactivates the cell signaling pathways required for cellular transformation and carcinogenesis (Figure 2). When opposed to normal cells, one of the most distinguishing characteristics of cancer cells is their constant pro-oxidative state, which can lead to intrinsic oxidative stress [22, 23]. Overproduction of ROS and low levels or deactivation of antioxidant machinery causes increased oxidative stress in cancer cells. The capacity of tumor cells to regulate the expression of endogenous antioxidant enzymes that keeps the steady level of ROS below the threshold would otherwise cause tumor cell death. As a result, cancer cells have devised strategies to protect themselves from intrinsic oxidative stress and have developed a sophisticated adaptation strategy that involves effective reorganization of antioxidant functions and allows the up-regulation of pro-survival molecules [24]. Aside from increasing the incidence of mutations, ROS may have a role in cancer onset by dampening the pro-tumorigenic signaling pathways. The disulfide bonds in certain proteins are oxidized by ROS which can affect their activity, especially in the tyrosine phosphatases superfamily [25]. The tumor suppressor gene phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inactivation by oxidation is perhaps the finest illustration related with cancer growth [26, 27]. The phosphatidylinositol 3-kinase/Ak strain transforming (PI3K/Akt) signaling pathway, which is likely the most often activated signaling pathway in cancer cells, is hyperactivated when PTEN is inhibited by ROS. Another example of ROS induced phosphatase inhibition is mitogen-activated protein kinase (MAPK) which increases extracellular signal-regulated kinase (ERK) activity. Activation of these pathways would increase intracellular ROS levels by inhibiting the transcription factor forkhead box transcription factor O (FOXO), which boost the production of antioxidants such as SOD2, catalase, and sestrin3 [28].
Cancer cells maintain their tumorigenic ROS levels by balancing ROS production and scavenging. This enables them to stay inside the tumorigenic range. CAT: catalase
ROS: a dynamic cancer metastasis driver
Metastasis entails the unfurling of cancer cells from primary tumor site to the adjacent tissues and to the faraway organs, and is the main cause of illness and death [29, 30]. Studies discover that metastasis is not an autonomous program but is highly complex and multistep event, which occurs due to high mutational burden in cancerous cells and crosstalk between malignant cells and their non-malignant counterparts [31]. It follows due to the up-regulation of various transcriptional factors such as NF-κB, ETS proto-oncogene 1 (ETS-1, transcription factor), TWIST, activator protein 1 (AP-1), Snail and zinc finger E-box binding (ZEB); the metalloproteases via Matrix metallopeptidase 9 (MMP-9) and MMP-2; various chemokines and/or cytokines like transforming growth factor beta (TGF-β) [32, 33]. ROS performs a critical role in cancer cells migration and invasion. ROS are largely formed during mitochondrial electron transport in aerobic respiration and have several deleterious effects [34]. Epithelial to mesenchymal transition (EMT) is the major reason for metastasis, where epithelial cells lose their cell-cell adhesion property, polarity, subsequently leads to cell mobility [35]. Numerous findings have established that the ROS is the major player of EMT. TGF-β can also cause tumor milieu to produce ROS, which causes oxidative stress [36]. TGF-β1 synchronizes with urokinase-type plasminogen activator (uPA) and MMP-9 proteins to accelerate the cell movement and invasion to the adjacent or distant region of the body through ROS-dependent processes [37]. Another study uncovered that ROS increases the tumor cells migration by causing hypoxia, mediated by MMPs and cathepsin expression [38, 39]. TGF-β1 induced EMT in MDA-MB-231C and MCG-10A cell lines requires NADPH oxidase 4 (NOX4) dependent ROS generation [40]. p53 is involved in cell migration and uses the cytokine TGF-β1 [41]. Pelicano et al. [42] found that mitochondrial failure causes high levels of ROS, which affects the expression of C-X-C motif chemokine ligand 14 (CXCL14) via AP-1 signaling, resulting in cell mobility via increasing intracellular Ca2+ levels. ROS can stimulate Nrf2, which activates Klf9 (Kruppel like factor 9), which then activates ERK1/2, resulting in increased intracellular ROS generation in cancer cells. Thus, by utilising antioxidants that target Klf9, premalignant cancer cells proliferation can be suppressed [43, 44]. Cancer metastasis is also aided by mitochondrial Ca2+ and the transporter mitochondrial calcium uniporter regulator 1 (MCUR1) which is transcriptionally active in hepatocellular carcinoma (HCC), promoting EMT via ROS/Notch1/Nrf2 pathways. As a result, the MCUR1 protein may be a good target for HCC therapy [45]. NOX2 creates ROS which drives metastasis by downregulating the functional features of natural killer (NK) cells. According to Aydin et al. [46], blocking of NOX2 could re-establish interferon gamma (IFN-γ) dependent NK cell-mediated killing of myeloma cells. Vimentin, another protein that speeds up EMT and metastasis, also plays a role in triggering of cancer and its progression. Hypoxia-inducible factor 1-alpha (HIF-1α) induced oxidative stress that regulates vimentin gene transcription aided cancer cell invasion and migration by facilitating invadopodia formation [47]. Vimentin suppression by RNA interference reduces cancer cell metastasis and tumor volume [48]. ROS can also trigger changes in epigenetics in the promoter area of E-cadherin and other tumor suppressor genes leading to tumor development and metastasis. Increased Snail expression causes hyper-methylation in the promoter gene. Snail uses histone deacetylase 1 (HDAC1) and DNA methyltransferase 1 (DNMT1) to cause DNA methylation [49].
ROS as an angiogenesis regulatory mediator
During the early stages of cancer progression, new blood vessels were restructured from the pre-existing blood vessels through a process called angiogenesis which helps tumor cell proliferation and survival by supplying the nutrients and oxygen [50, 51]. Cancer proliferation triggers through ROS, facilitates the process of angiogenesis which results in increased metabolic rate, resulting in the generation of high ROS levels [51, 52]. High levels of ROS in the tumor microenvironment due to oxidative stress stimulates the cancer cells, which begins to secrete the angiogenic modulators [53]. ROS production spearheaded via endogenous and exogenous pathways, which stimulates various growth factors, including vascular endothelial growth factor (VEGF) and HIF-1α transcription factors resulting in stimulating tumor cell metastasis and cancer progression [54, 55]. The signaling surge through ROS facilitation has been well documented to maintain the secretion of VEGF, initiation of PI3K/Akt/mammalian target of rapamycin (mTOR) pathways and hypoxia dependent or independent mechanisms (by stabilizing the HIF-1α which enhances the production of VEGF). The hypoxia independent pathway also prime angiogenesis through oxidation of lipid ligands which induces activation of NF-κB via toll-like receptors (TLRs) (Figure 3) [56]. The Janus kinase 2 and c-Jun N-terminal kinase pathways regulate HIF-1 regulation via adenosine monophosphate-activated protein kinase (AMPK) in response to H2O2. AMPK can be thought of as a key contributing factor of HIF-1 functions in response to H2O2, and it may also play a role in the complex HIF-1 regulatory mechanisms (Figure 4) [57]. In addition to that, the rat sarcoma virus (Ras) signaling pathway has also been connected in up-regulation of VEGF secretion [58]. Mutant p53 is found to be linked in regulation of angiogenic response during the tumor progression via ROS mediated activation of VEGF-A and HIF-1 in human colorectal carcinoma cell line HCT116 [59]. ROS mediated angiogenesis has been studied by many investigators to identify the signaling cascade mechanisms, modulating the cancer progression. A research was conducted using MDAMB-231 breast adenocarcinoma cells and deferoxamine (DFO) reported to induce HIF-1α via ERK1/2 phosphorylation pathways which also aid in tumor cell migration and metastasis [60]. Angiogenesis activation occurs via ROS mediated by hypoxia dependent and independent pathways. Hypoxia dependent pathways increase the VEGF expression via PI3K regulatory subunit/Akt serine/threonine kinases/mTOR, PTEN, and MAPK. Signaling cascades via HIF-1α and ribosomal protein S6 kinase β1 (p70S6K1) releases various growth factors, cytokines, and upregulated MMPs, leading to the occurrence of angiogenesis.
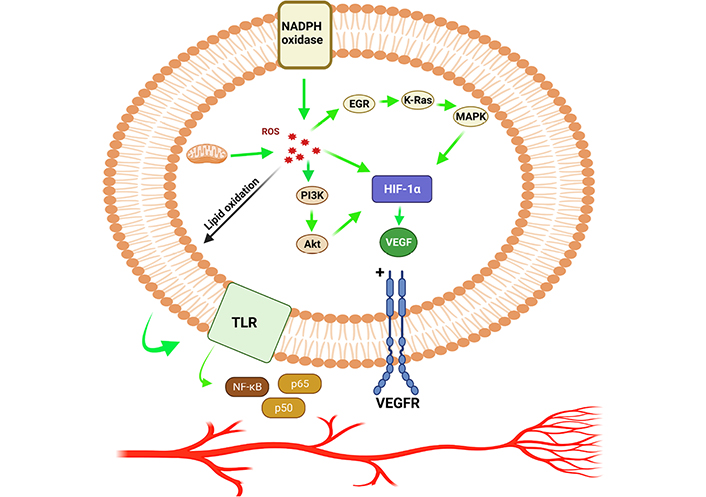
HIF-1α promotes angiogenesis by allowing ROS to speed up the process. EGR: early growth response; K-Ras: Kirsten rat sarcoma virus; VEGFR: vascular endothelial growth factor receptor
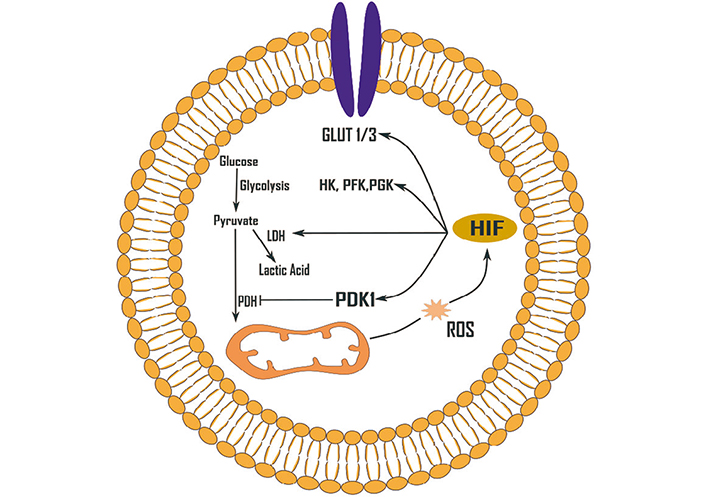
Effects of ROS (H2O2) on glycolysis up-regulation mediated by HIF-1. HIF-1 can be activated by a variety of ROS. GLUT 1/3: glucose transporter 1/3; HK: hexokinase; LDH: lactate dehydrogenase; PDH: pyruvate dehydrogenase; PDK1: pyruvate dehydrogenase kinase 1; PFK: phosphofructokinase; PGK: phosphoglycerate kinase
Han and his colleagues [61] uncovered that the increased concentrations of epidermal growth factor (EGF) could triggers the H2O2 production which stimulates p70S6K1 through PI3K/Akt pathways that lead to the triggering of VEGF and HIF-1α signaling. Similarly, Liu et al. [62] stated that EGF also triggers elevated levels of H2O2 in ovarian tumor cells which in turn initiate Akt/p70S6K1 pathways, thus resulting in enhanced VEGF expression. The researchers also found evidences that overexpression of catalase and rapamycin together reduced angiogenesis. H2O2 has also been shown to activate the PI3K/Akt/mTOR signaling cascade and Ras by inactivating phosphatase and PTEN via reversible oxidation in phosphatases of the cysteine thiol group [63]. It has also been discovered that when the tumor microenvironment is inflammatory, the macrophages produce more ROS, and these macrophages have M2 phenotypic markers [64]. ROS in angiogenesis could be a potential target with antioxidants which also involves the associated signaling pathways and have clinical significance in treatment of cancer patients by downregulating neo-vascularization. Many other tactics are also important to restore redox differences to improve the diseases outcome. Due to the failure of interventional trials with small antioxidants, more study is needed to investigate disease-specific ROS having therapeutic importance for the future development of prophylactic measure.
ROS and HIF-1α signaling have long been known of playing a role in cancer pathogenesis. The role of ROS mediated signaling via HIF-1α in cellular proliferation and angiogenesis is crucial. A growing amount of evidence implies that a number of intermediates are involved in HIF-1α regulation, mediated by ROS.
Checkpoint inhibitors, ROS generating medicines, and regulatory linkages in ROS for cancer therapy
Programmed cell death protein 1 (PD-1) is a surface receptor, expressed on highly differentiated effector T cells which act as a negative regulator, and become exhausted and anergic due to chronic antigenic exposure [65]. In cancers, programmed death ligand 1 (PD-L1) is expressed by the tumor cells which impair antitumor effector functions of PD-1+ T cells [66]. Checkpoint blockade is a novel therapeutic option to reinvigorate effector functions of exhausted T lymphocytes by obstructing PD-1–PD-L1 interactions [jointly designated as PD-(L)1] [67]. Anti-PD-1 therapy remain ineffective in a large number of cancer patients likely due to initial lukewarm response or disappearance of durable responses [68–70]. In early-stage exhaustion, PD-1+ T cells demonstrated less reliance on oxidative phosphorylation (OXPHOS) and glycolysis. Long-term chronically exhausted PD-1 high T lymphocytes, however, showed a dependence on glycolysis due to dysfunctional mitochondria [71]. Tumor development and metastasis is a gain of immune escape mechanisms which occurs through induction and recruitment of immunosuppressive cells. This includes regulatory T (Treg) cells, myeloid-derived suppressor cells, tumor-associated macrophages, and increased expression of various immunosuppressive molecules, such as PD-1 and PD-L1. CD8+ T cells only differentiate into heterogeneous cytolytic T lymphocytes (CTLs), including tumoricidal effectors and long-term memory cells. Mitochondrial activation is reported to be critically important for T cell proliferation and memory response in vitro [72] and in vivo [73]. T cell receptor engagement induces Ca2+ release, which in turn augments mitochondrial functional roles including the tricarboxylic acid (TCA) cycle and ROS generation [74]. ROS generated in mitochondrial complexes and the superoxide converted from ROS activates CD4+ and CD8+ T cells via nuclear factor of activated T cells (NFAT) activation and interleukin-2 (IL-2) production [75]. PD-1 deficiency and PD-1 blockade in tumor model study indicated that mitochondrial functions, including ROS generations, are upregulated in vivo following obliteration of PD-1 signaling [66]. Besides that, PD-1 blockade also activates AMPK and mTOR, which in turn upregulates the expression of peroxisome proliferator-activated receptor (PPAR)-gamma coactivator 1α (PGC-1α), resulting in increased mitochondrial activity together with ablation of PD-1, which suppresses tumor growth [66]. ROS are primary effectors of cytotoxicity induced by treatment with anti-cancer drugs. Immunosurveillance role of ROS relies on classic receptors such as TLRs, but also onsensing the metabolic environment. In malignancies, the subtle balance of internal redox environment between free radicals produced or quenched by cellular antioxidants and enzyme systems is grossly dysregulated. In this aberrant situation, it favors oxidative stress that significantly alters the tumor microenvironment and suppresses the effector T cell functions and induction of T cell death. In this condition, ROS exerts significant influence on the expression of PD-1 and its ligand PD-L1, but the mechanism of crosstalk between ROS and PD-(L)1 is not always clear. Increased metabolic activity in mitochondria could lead to high level of ROS, specifically in drug resistant cases including cisplatin resistant tumor cells as well as in patients who do not respond to cisplatin [76, 77]. Elevated ROS level in tumor cells has also been implicated in EMT which played critical role in PD-L1 expression in neoplastic cells [78–80]. In addition to cisplatin, 5-fluro uracil (5-FU), carboplatin and paclitaxel contributes to acquired PD-L1 expression in solid tumors including small cell lung cancer [81, 82]. Therapeutic interventions like photodynamic and sonodynamic methods allow the activated sensitizers to interact with biological substrates or molecular oxygen and generate ROS which induces cell death by apoptosis and necrosis (Figure 5) [83–85]. Combination regimen of anti-PD-L1 and photodynamic or sonodynamic therapy could be an attractive option for treating highly refractory and advanced pancreatic cancers with controlled ROS generation and maximum therapeutic benefits. Recently, we have shown that small biomolecular drug conjugate between 5-FU and bilirubin produced very high ROS generation in highly metastatic Dalton’s lymphoma (DL) cells as well as in doxorubicin resistant variant of DL cells [86]. Besides PD-1, T cell immunoglobulin and mucin-domain-containing protein-3 (Tim-3) regulate macrophage derived ROS and ROS mediated liver inflammation in mouse model of nonalcoholic steatohepatitis [87]. Enhanced Tim-3 expression in subpopulation of hepatic macrophages of mice with methionine and choline deficient diet indicated evidences, supporting the important role of Tim-3 in alleviating liver injury by regulating ROS production and proinflammatory cytokine secretion from macrophages [87]. This result suggests that Tim-3 plays a protective role against liver injury by regulating macrophage activation. Conditional deletion of Tim-3 in dendritic cells led to increased accumulation of ROS resulting in Nod-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasome activation. Following inhibition of inflammasome activation, or abrogation of downstream effector cytokines like IL-1β and IL-18, the protective anti-tumor immunity was minimized [88].
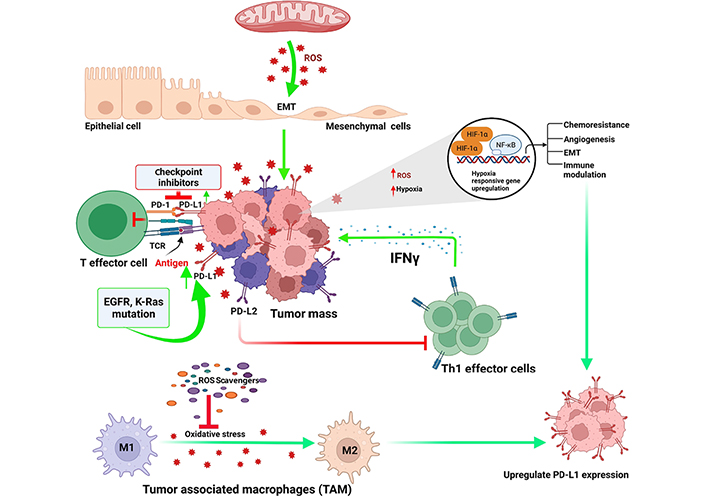
Regulatory links between oxidative stress and check point inhibitors in tumor microenvironment. TCR: T-cell receptor; Th1: T helper type 1; EGFR: epidermal growth factor receptor
Combination therapy in ROS regulation
The contrasting feature of normal cells to that of cancer cells is the rate of ROS production and redox regulation. Combinational therapy solves two glitches: (i) decrease the toxicity of chemotherapeutics to the normal cells, and (ii) augment the efficacy of anti-tumor regimen. The metabolic modulation is a good choice for combining with ROS-generators or ROS-scavenging inhibitors in combination therapy. Targeting the enzymes involved in ROS regulation between cancer cell and normal cell is an effective strategy for designing the combination therapy. For example, by regulating the redox reactions, specifically through the production of reducing counterparts like nicotinamide adenine dinucleotide hydrogen (NADH) and NADPH, ROS mediated killing of the cancer cells occur. The metabolic pathways involved in redox modulation are mostly related to glutamine metabolism that plays a key role in redox regulation and antioxidant response. Glutamine is the precursor for glutamate and is needed for GSH production and therefore antioxidant responses. The metabolism of glutamine has been essential for the survival of cancer leading to the belief that few cancer cells are glutamine obsessed [89]. Inhibition of glutaminase 1 (GLS1), the enzyme that converts glutamine to glutamate for entry into the TCA cycle inhibits oncogenic transformation [90, 91]. The anticancer impact of the medicine L-asparaginase was previously assumed to be due to its role in restricting asparagine levels; however, current research has revealed that the drug’s anticancer effect is due to its effect on glutamine levels [90]. Finally, in vitro and in vivo studies have revealed that the alternative glutamine route, regulated by the aspartate transaminase glutamic-oxaloacetic transaminase 1 (GOT1), is necessary for KRAS-driven pancreatic ductal adenocarcinoma (PDAC) growth [92]. GOT1 is a crucial enzyme in the aspartate-malate shuttle, generating pyruvate and boosting the NADPH/NADP+ ratios which keep GSH levels low and redox homeostasis under check. Inhibiting the GOT1 results in reduction of the ratios of reduced GSH to oxidised GSH, that increases the ROS levels and suppression of PDAC growth. Because glutamine metabolism is involved in NADPH production and GSH formation, glutamine pathway inhibitors could be useful anti-cancer partners. Other approach is to counter ROS through drug repurposing since the synthesis of new anticancer medications is often associated with higher costs, a longer development cycle, and a high probability of failure [93]. One of the promising strategies is to address existing cancer treatment obstacles using ROS-based repurposing medicines.
Future perspectives
Production and scavenging of ROS and its regulation in cellular environment is a tricky issue to understand the manipulation and extraction for beneficial aspects. Delicate methodology needs to be adopted to define meticulous balance between ROS formation and elimination, which may have great benefit to cancer therapy. Normal cells functioning requires a delicate equilibrium in ROS redox process in order to maintain physiological homeostasis. Ineffective neutralization of overmuch ROS could result in serious pathophysiology including cancer. Aberrant elevation in ROS leads to neoplastic growth and progression of metastatic tumor, spearheaded by signaling events including PI3/Akt/mTOR, MAPK, PTEN, MMPs and VEGF/VEGFR etc. Substantial hike in ROS levels is likely to intervene in cancer progression via augmentation in apoptosis, resulting in cell death, induced by traditional therapeutic interventions including chemotherapy. Chemotherapy induces apoptosis via ROS by targeting diverse cellular targets including a wide range of cell signaling pathways. ROS also play roles in drug resistance via activation of NF-κB, which triggers pro-inflammatory cytokine secretion. Thus, bipartite roles of ROS can be targeted as a therapeutic option for tumor growth suppression by impeding inflammation, curtailing angiogenesis, and inhibition of metastasis. Meticulous balance between ROS generation and their elimination potentially has great benefit for cancer therapy. Beneficial or detrimental effect of ROS also depends on functional attributes of immune responses. Neoplasia develops an elevated ROS level, caused by enhanced metabolism, which in turn results in immunosuppressive state of the tumor. Compared to normal cells, higher intracellular ROS in tumor cells shifts the redox balance rendering vulnerability in tumor cells to external sources of ROS. Intracellular ROS is also linked to immunogenic cell death and M2 to M1 polarization in macrophages is believed to be critical for immunomodulation in tumor inhibition. Thus, effective ROS modulation may enable distinction of region of interest in intracellular and extracellular microenvironment which could enable better control over the process for achieving better outcomes. The role of nanomaterials as ROS modulating device intrinsically or as a carrier is another approach with considerable promise with respect to better pharmacokinetics, pharmacodynamics and biodistribution. Nanomedicine may provide an effective platform to calibrate and manage these critical issues of drug delivery mechanisms. Future research on ROS may be focused on but not necessarily restricted to the following frontiers for therapeutic success (i) critical and decisive role of ROS in disease state; (ii) calibrated balance in ROS production and elimination; (iii) ROS level and immunological state; and (iv) precision medicine for extracellular and intracellular ROS level. Thus, an effective tumoricidal approach needs a comprehensive analysis of redox analysis with relation to malignancies for the larger benefit of the patients.
Conclusions
Malignant transformation of cancer exhibits high intrinsic generation of ROS level which controls the tumorigenic phenotype and propels the tumor progression. The redox mechanism in cancer cells by upregulating the antiapoptotic and antioxidant moieties enhances the survival and develops resistance to the anticancer agents. Currently, very little information is known regarding sensing of intracellular oxidative stress which elicits ROS-induced signaling to regulate the survival and antioxidant enzymes repertoire gene expression. The reliance of tumor cells on their antioxidant enzymes makes them exposed to agents that target the antioxidant systems. There is a convincing possibility for treatments intended to markedly enhance the intracellular ROS to execute the cancer cells by diminishing their antioxidant capabilities. This may be attained by harnessing compounds that deter the antioxidant system by inhibiting these signaling pathways which play roles in upregulating the antioxidants in cancer. Consequential surge in ROS may provoke cancer cell death through random damaging capabilities of ROS by inducing the apoptosis via specific death signaling pathways. The benefit of such approach is that the normal body cells are not considerably affected since they harbor basal levels of ROS and are less reliant on antioxidants. Although it is conceivable that a basal level toxicity in neoplastic cells is not achieved and that the supplementary surge in ROS causes additional mutations or drives tumor metastasis. Therefore, inhibitors combination against these antioxidant systems with such prooxidant properties is required to elevate the intracellular ROS concentrations within tumor cells and it may be crucial to devastate the antioxidant systems beyond the threshold of toxicity level. It becomes more apparent, that comprehensive knowledge of ROS mediated signaling in transformed cells is vital to develop novel strategies for ROS modulation based therapeutic intervention in order to kill the cancer cells selectively and overwhelm the challenge of drug resistance. Utilization of antibodies or checkpoint inhibitors opens up new frontiers for treatment of nearly 50% of unresponsive or less responsive cancer patients. Occurrence of bigger tumor mutational burden further necessitates a broad-spectrum protocol for therapy because of great degree of refractivity to PD-1 therapy. Thus, in order to overcome the lack of response, a combination of various types of treatment including inhibitors against Tim-3, lymphocyte-activation gene 3 (LAG-3) etc. in association with chemotherapy and or radiotherapy may produce desired response. A necessary striking synergistic effect may not be achieved. However, the most important key for successful treatment would be the presence of optimally activated T cells in tumor microenvironment with controlled ROS activity which may represent fruitful ground for long-term molecular anticancer strategies.
Abbreviations
| Akt: | Ak strain transforming |
| AMPK: | adenosine monophosphate-activated protein kinase |
| EMT: | epithelial to mesenchymal transition |
| ERK: | extracellular signal-regulated kinase |
| GOT1: | glutamic-oxaloacetic transaminase 1 |
| GSH: | glutathione |
| H2O2: | hydrogen peroxide |
| HIF-1α: | hypoxia-inducible factor 1-alpha |
| IL-2: | interleukin-2 |
| MAPK: | mitogen-activated protein kinase |
| MMP-9: | matrix metallopeptidase 9 |
| mTOR: | mammalian target of rapamycin |
| NADPH: | nicotinamide adenine dinucleotide phosphate |
| NF-κB: | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NOX4: | nicotinamide adenine dinucleotide phosphate oxidase 4 |
| Nrf2: | nuclear respiratory factor 2 |
| PD-1: | programmed cell death protein 1 |
| PD-L1: | programmed death ligand 1 |
| PI3K: | phosphatidylinositol 3-kinase |
| PTEN: | phosphatase and tensin homolog deleted on chromosome 10 |
| ROS: | reactive oxygen species |
| SOD2: | superoxide dismutase 2 |
| TGF-β: | transforming growth factor beta |
| Tim-3: | T cell immunoglobulin and mucin-domain-containing protein-3 |
| TLRs: | toll-like receptors |
| VEGF: | vascular endothelial growth factor |
Declarations
Acknowledgments
Authors acknowledge Sumeet Singh (Guru Nanak Dev University, Amritsar) for assistance in drawing figures. Figures were created using platform BioRender.com (https://biorender.com/).
Author contributions
RS searched literature, analyzed the data and wrote the first draft; PPM conceived, designed and edited the article. Both authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was funded by minor grant of Institution of Eminence (IoE) by the Ministry of Human Resource and Development, Government of India, (No. R/Dev/D/IOE/Incentive/2021-22/32275) to Partha Pratim Manna by Banaras Hindu University. RS thank UGC, India, for senior research fellowship [19/06/2016(i)EU-V]. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2022.