 Original Article
Original Article

Affiliation:
1Department of Obstetrics and Gynecology, University of Mississippi Medical Center, Jackson, MS 39216, USA
Affiliation:
2Department of Pharmacology, University of Mississippi Medical Center, Jackson, MS 39216, USA
Affiliation:
2Department of Pharmacology, University of Mississippi Medical Center, Jackson, MS 39216, USA
Affiliation:
2Department of Pharmacology, University of Mississippi Medical Center, Jackson, MS 39216, USA
3Department of Emergency Medicine, University of Mississippi Medical Center, Jackson, MS 39216, USA
Affiliation:
2Department of Pharmacology, University of Mississippi Medical Center, Jackson, MS 39216, USA
Affiliation:
2Department of Pharmacology, University of Mississippi Medical Center, Jackson, MS 39216, USA
Affiliation:
2Department of Pharmacology, University of Mississippi Medical Center, Jackson, MS 39216, USA
Affiliation:
2Department of Pharmacology, University of Mississippi Medical Center, Jackson, MS 39216, USA
Affiliation:
2Department of Pharmacology, University of Mississippi Medical Center, Jackson, MS 39216, USA
Affiliation:
4Experimental and Clinical Research Center, Max-Delbrück-Centrum für Molekulare Medizin, 13092 Berlin, Germany
Affiliation:
4Experimental and Clinical Research Center, Max-Delbrück-Centrum für Molekulare Medizin, 13092 Berlin, Germany
Affiliation:
2Department of Pharmacology, University of Mississippi Medical Center, Jackson, MS 39216, USA
Email: bblamarca@umc.edu
Explor Med. 2022;3:99–111 DOI: https://doi.org/10.37349/emed.2022.00077
Received: August 25, 2021 Accepted: December 09, 2021 Published: February 25, 2022
Academic Editor: Akiko Mammoto, Medical College of Wisconsin, USA
The article belongs to the special issue Angiotensins—A Century of Progress
Aim: Preeclampsia (PE) is a hypertensive disorder of pregnancy associated with activated CD4+ T cells and autoantibodies to angiotensin II type 1 receptor (AT1-AA). We have previously shown that CD4+ T cells isolated from women with PE cause hypertension, increased tumor necrosis factor alpha (TNF-α), endothelin-1, and soluble fms-like tyrosine kinase-1 (sFlt-1) when injected into pregnant nude-athymic rats compared to CD4+ T cells from normal pregnant (NP) women. However, the role of PE CD4+ T cells to cause AT1-AA as a mechanism of hypertension is not known. Our goal was to determine if PE CD4+ T cells stimulate AT1-AA in pregnant nude-athymic rats.
Methods: CD4+ T cells were isolated from human NP and PE placentasand injected into nude-athymic rats on gestational day (GD) 12. In order to examine the role of the PE CD4+ T cells to stimulate B cell secretion of AT1-AA, a subset of the rats receiving PE CD4+ T cells were treated with rituximab on GD 14 or anti-CD40 ligand (anti-CD40L) on GD 12. On GD 19, mean arterial pressure (MAP) and tissues were obtained.
Results: MAP [114 ± 1 mmHg (n = 9)] and AT1-AA [19.8 ± 0.9 beats per minute (bpm, n = 4)] were increased in NP nude + PE CD4+ T cells compared to NP nude + NP CD4+ T cells [98 ± 2 mmHg (n = 7, P < 0.05) and 1.3 ± 0.9 bpm (n = 5, P < 0.05)]. Rituximab (103 ± 2 mmHg, n = 3, P < 0.05) and anti-CD40L (102 ± 1 mmHg, n = 3, P < 0.05) lowered MAP compared to NP nude + PE CD4+ T cells. Circulating a proliferation-inducing ligand (APRIL) and placental angiotensin-converting enzyme 2 (ACE-2) activity was increased in response to PE CD4+ T cells.
Conclusions: These results show that placental CD4+ T cells play an important role in the pathophysiology of PE, by activating B cells secreting AT1-AA to cause hypertension during pregnancy.
Preeclampsia (PE) is a multi-system disorder that complicates approximately 2–8% of all pregnancies and 12% of pregnancies in Mississippi [1]. The diagnostic criteria for PE have varied slightly over the years, but the hallmark of the disease is still new onset hypertension after week 20 of pregnancy, accompanied with organ dysfunction, often being proteinuria or edema [1]. The clinical complications of PE include fetal growth restriction, preterm birth, placental abruption, hemolysis, elevated liver enzymes and low platelets (HELLP) syndrome, eclampsia, cardiovascular disease, and end-organ damage [2]. Despite making significant advances in understanding the pathophysiology of this disease, the only definitive treatment remains delivery of the fetus and placenta [2].
Although healthy pregnancy occurs with a baseline state of inflammation to ensure tolerance toward the fetus, women with PE have been shown to have chronic inflammation with increased levels of CD4+ T cells and pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin-17 (IL-17), while anti-inflammatory factors such as regulatory T cells and IL-10 are suppressed [3–6]. Our lab has previously shown that adoptive transfer of CD4+ T cells from the reduced uterine perfusion pressure (RUPP) rat model of PE into normal pregnant (NP) rats leads to the development of a PE-like phenotype in the NP rats, including elevated mean arterial pressure (MAP), increased cytolytic natural killer (NK) cells, increased agonistic autoantibodies to the angiotensin II type I receptor (AT1-AA), and placental and mitochondrial dysfunction [7–22]. Studies outside our lab by Zenclussen et al. [23] have shown that adoptive transfer of splenic cells cultured into pro-inflammatory T helper type 1 (Th1) like cells into pregnant mice leads to a PE-like response with increased blood pressure, abnormal renal function, and increased inflammatory markers. Furthermore, we have shown that adoptive transfer of CD4+ T cells isolated from placentas of patients with PE leads to a PE-like response in pregnant nude-athymic rats [13]. In these initial studies, adoptive transfer of human placental CD4+ T cells into pregnant nude-athymic rats increased MAP, circulating pro-inflammatory cytokines (TNF-α and IL-17), soluble fms-like tyrosine kinase-1 (sFlt-1), and renal endothelin-1 expression when compared to nude-athymic rats that received CD4+ T cells from NP women [13]. However, the role of CD4+ T cells to communicate with B cells in this model of PE pathophysiology was unknown.
Although we know the importance of inflammatory mediators during PE, there are very few studies examining the role of B cells in causing the pathophysiology of PE. As important members of the adaptive immune system, B cells aid in maternal-fetal protection while helping to orchestrate shifts in the immune profile. During murine pregnancy, Muzzio et al. [24] observed significant changes in pre/pro-B cells, immature B cells, and B cell development throughout pregnancy including those cells found in the peritoneal cavity, splenic, circulation, and lymph nodes during pregnancy. In humans, other groups have noted similar changes in transitional B cell populations [25, 26]. Importantly, Jensen et al. [27] published a study examining B1 or B2 cells to produce the AT1-AA from PE patients nine years ago, and since then there hasn’t been much follow up on the role of B cell in PE.
B lymphocytes are the producers of all five classes of antibodies against foreign antigens. Two major B cell populations have been described: B1 cells and B2 cells [28, 29]. While B1 and B2 cells both produce antibodies, B1s are first produced in the fetus and most undergo self-renewal in the periphery. B1s produce natural antibodies, primarily of the Immunoglobulin Mu (IgM) type which are characterized as polyreactive and having low affinity [28–30]. The conventional B cells are the B2 cells. They are produced after birth and replaced in the bone marrow, migrate to the spleen where they differentiate and they recirculate through secondary lymphoid organs. B2 cell activation, proliferation, and differentiation is mediated upon antigen exposure [28]. For full activation and maturation of B cells, interaction with activated T lymphocytes via the CD4 receptor, major histocompatibility complex (MHC) II, and the costimulatory molecule, CD40-CD40L, must occur. B2 cell receptors then undergo antigen dependent selection which includes somatic mutation, gene conversion, and class switching [29]. This process allows the B cell receptor (BCR) to exhibit a high degree of specificity responsible for the effective memory response [28–30]. Thus, antigen induced antibodies [immunoglobulin G (IgG)] are specifically designed and are primarily produced by B2 cells. Eventually, the B2 cells will differentiate into antibody-secreting plasma cells while a small amount will remain in the spleen and lymph nodes as memory cells.
This type of B cell activation occurs using a T cell dependent pathway. For T cell dependent B cell activation, CD4+ (“helper”) T cells interact with endogenous B cells to promote maturation of the B cells, which in turn leads to antibody production [8]. Stimulation of CD40 by CD40L leads to B cell proliferation, differentiation, and antibody production which can be inhibited by pharmaceutical monoclonal antibodies. Subsequent activation of a different receptor, CD20, stimulates mature B cells to differentiate into plasma cells which enter the circulation and produce antibodies. A proliferation-inducing ligand (APRIL) is a cytokine integral in the survival of antibody producing plasma cells [31–33]. While APRIL is primarily produced by myeloid lineage cells [31–33], regulation of myeloid cells by activated CD4+ T cells could contribute to a pro-B cell environment in PE. We have previously shown that inhibiting the CD40-CD40L interaction in normal pregnant rat recipients of RUPP CD4+ T cells, attenuated the resulting hypertension by lowering levels of AT1-AA [8]. This study demonstrated the importance of CD4+ T cells to stimulate B cells to cause hypertension in response to placental ischemia with blockade of CD40-CD40L B cell costimulatory pathway. An additional mechanism that we have shown to lower the AT1-AA and hypertension in the RUPP model of PE is B cells depletion via administration of antibodies targeting the CD20 receptor, such as rituximab [19].
In this study, we sought to determine if these same pathways could be utilized in lowering hypertension in response to PE CD4+ T cells. Moreover, we sought to determine if human PE CD4+ T cells communicate with endogenous B cells to cause AT1-AA secretion as a mechanism of the pathogenesis of PE. Preeclamptic women have increased AT1-AA which we believe could lead to increased activation of the renin-angiotensin-aldosterone system (RAAS) and upregulation of angiotensin-converting enzyme 2 (ACE-2). Normal pregnant women upregulate components of the RAAS at varying time points throughout their pregnancy so that they can compensate for the increase in plasma volume. Previous studies of circulating RAASS components demonstrated that plasma Ang I, Ang II, and renin activity were higher in NP subjects compared to PE while Ang-(1–7) was significantly decreased in PE compared NP [34]. Other components of the RAASS, except serum ACE, were reduced in PE compared to NP, however, placental ACE-2 was not examined in this study. Moreover, circulating Ang II remained elevated in PE compared to NP. With the increase in AT1-AA and Ang II, a compensatory response to increase ACE-2 at the placental level could occur. Other studies examining the RAAS expression in the placenta focus more on hypoxia or external factors and show that sustained angiotensin II type 1 receptor (AT1R) activation can occur with the AT1-AA and this may be a mechanism of long-term vasoconstriction in PE women [34]. Therefore, one additional question answered in this study was if PE women, who also have elevated AT1-AA, have increased placental ACE-2 activity and is this associated with activation of B cells, AT1-AA and hypertension in PE CD4+ T cell recipient pregnant rats.
Sixteen [9 with PE > 32 weeks gestation (GA), 7 with NP > 34 weeks without PE] pregnant women undergoing delivery at the Wiser Hospital for Women and Infants at the University of Mississippi Medical Center (UMMC) in Jackson, MS, were enrolled in the study. PE was defined according to the 2020 American College of Obstetrics and Gynecology (ACOG) guidelines, which included systolic blood pressure ≥ 140 mmHg and/or diastolic blood pressure ≥ 90 mmHg after 20 weeks of gestation in combination with proteinuria (defined as ≥ 300 mg protein in a 24-hour collection) in a previously normotensive patient [29]. Patients exclusion criteria were excluded from the study if the patients had multi-fetal gestation, a history of chronic hypertension, pre-existing immune disorders (e.g., systemic lupus erythematosus), or current substance use of alcohol/tobacco/nicotine/illicit drugs. All PE patients were managed according to the standard of care while in the hospital and treated with antihypertensive agents (labetalol, hydralazine, and/or nifedipine) as well as magnesium sulfate infusion to prevent eclamptic convulsions. Prior to enrollment delivery, all patients signed a UMMC Institutional Review Board approved consent. All placentas and blood collected were processed within 30 min of delivery.
Timed-pregnant nude-athymic rats (weighing 200–250 g) from Envigo (Indianapolis, IN, USA) were used for this study. Nude-athymic rats were chosen as an animal model because they lack a thymus, do not produce endogenous T cells, and will not reject human T cells. Moreover, these rats contain the other components of the immune system and are ideal to examine the effect of CD4+ T cells from PE patients to cause activation of downstream factors such as B cells secreting AT1-AA. Animals were housed in a temperature-controlled (23°C) barrier suite with a 12:12-h light–dark cycle. Because these animals were considered immunocompromised, no other rat breeds were housed in the suite, and all personnel working with these animals donned appropriate personal protective equipment (PPE) before entering the barrier suite. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at UMMC and executed in accordance with NIH Guidelines for Care and Use of Laboratory Animals.
Immediately after sample collection, placental tissue samples from NP and PE patients were collected from the maternal side of the placenta, rinsed in PBS + 5% penicillin-streptomycin (Pen-Strep) to remove surface blood, then placed in 4% collagenase I, 0.25 U Deoxyribonuclease I, and 5% Pen-Strep in Roswell Park Memorial Institute (RPMI) medium for one h at 37°C while stirring to aid in tissue digestion. The digest was strained through a 70 μm filter and centrifuged at 300 × g for 10 min. The cell pellet was reconstituted in 3 mL RPMI, layered carefully over 3 mL of Histopaque solution (Sigma-Aldrich, St. Louis, MO, USA), and centrifuged at 300 × g for 30 min with no brake at room temperature. The lymphocyte layer was visualized, aspirated from the Histopaque, and placed in a clean tube before adding 10 mL of RPMI. Lymphocytes were centrifuged at 300 × g for 5 min at room temperature to remove any residual Histopaque. Cells that were not immediately used for studies were placed in a freezing media consisting of 15% RPMI, 70% fetal bovine serum (FBS), and 15% dimethyl sulfoxide (DMSO) at a cell density no greater than 5 × 106 cells/mL and stored at −80°C or liquid nitrogen for long-term storage.
Following isolation of placental leukocytes, cells were centrifuged at 300 × g at 4°C for 10 min and resuspended in RPMI. To remove all B lymphocytes, CD19 MicroBeads (Miltenyi Biotec, San Diego, CA, USA) were added and the cells were incubated at 4°C for 15 min. The cell-MicroBead mixture was washed, centrifuged at 300 × g at 4°C for 10 min, and resuspended in RPMI. The cell-MicroBead mixture was filtered through a 30 μm pre-separation filter and then placed in LS Column in the MACS Separator (Miltenyi BiotecA). The unlabeled cells/effluent were centrifuged at 300 × g at 4°C for 10 min and resuspended in RPMI. CD4+ T cell isolation biotin-antibody cocktail (Miltenyi Biotec, San Diego, CA, USA) was added and the cells were incubated at 4°C for 5 min. CD4+ T cell MicroBead cocktail (Miltenyi Biotec, San Diego, CA, USA) was added and the cells were incubated at 4°C for 10 min. The cell-MicroBead mixture was placed in a new LS Column in the MACS Separator. The effluent in the collection tube consisted of untouched CD4+ T cells, of which a small sample was utilized for cell counting and flow cytometry to verify cell purity and the presence of human B cells by CD19+CD20+ staining. The absence of B cells supported the purity of the cell preparation.
Following isolation, CD4+ T cell purity was validated using flow cytometry. Isolated cells were then incubated for 10 min at 4°C with antibodies against rat CD4 (Miltenyi Biotec, San Diego, CA, USA), CD19 (Miltenyi Biotec, San Diego, CA, USA), and CD20 (Miltenyi Biotec, San Diego, CA, USA). Flow cytometry was performed on the Miltenyi MACSQuant Analyzer 10 (Miltenyi Biotec, San Diego, CA, USA) and analyzed using FlowLogic software (Innovai, Sidney, Australia). Lymphocytes were gated on forward- and side-scatter plot. After doublets were excluded, additional gates were set using fluorescence-minus-one (FMO) controls. Cells that were not immediately used for adoptive transfer were placed in a freezing media consisting of 15% RPMI, 70% FBS, and 15% DMSO at a cell density no greater than 5 × 106 cells/mL and stored at −80°C or liquid nitrogen for long-term storage.
Cells that had been previously stored in freezing media were washed with RMPI and then centrifuged at 300 × g at 4°C for 10 min to dilute and remove any residual DMSO prior to resuspending in TexMACS medium (Miltenyi, San Diego, CA, USA), IL-2 (1.022 ng/mL), and 5% Pen-Strep. A bead-antibody cocktail was prepared by combining CD2-biotin, CD3-biotin, and CD28-biotin with anti-biotin MACSiBead particles (Miltenyi Biotec, San Diego, CA, USA) and added to the leukocyte suspension. The cell-bead mixture was plated in 6-well tissue culture-treated plates at a cell density of 1.2 × 105 cells/mL and incubated at 37°C for 24 h. Following incubation, cells were removed from the 6-well plates using trypsin and a cell scraper, and 20 μL of cells were stained with Acridine Orange and Propidium Iodide (AOPI) and counted using as Cellometer cell counter (Nexcelom Bioscience, Lawrence, MA, USA). Cells were centrifuged at 300 × g at 4°C for 10 min and resuspended at a density of 2 × 107 cells/mL before being placed in the MACSiMAG Separator (Miltenyi Biotec, San Diego, CA, USA) to remove the MACSiBead particles from the cell suspension prior to adoptive transfer.
CD4+ T cells were resuspended in sterile saline at a concentration of 2 × 107 cells/mL saline. 100 μL of cells were injected intraperitoneally into pregnant nude-athymic rats under anesthesia on gestational day (GD) 12. A subset of rats that received PE CD4+ T cells also received mini-osmotic pumps infusing rituximab (250 mg/kg) on GD 14. For studies investigating T cell dependent B cell activation, a subset of isolated CD4+ T cells were incubated overnight in anti-CD40 T cell media [RPMI, 10% FBS, 1% 4-(2-hydroxyethyl)-1- piperazineethanesulfonic acid (HEPES), 5% penicillin/Streptomycin, 1.022 ng/mL IL-2, 4 ng/mL IL-12, and 60 ng/mL anti-CD40 ligand (anti-CD40L)] at 5% CO2, 37°C in a humidified atmosphere. After culture, cells were resuspended at a concentration of 2 × 107 cells/mL saline. 100 μL of cells were injected intraperitoneally into pregnant nude-athymic rats under anesthesia on GD 12. All adoptive transfer procedures were carried out in the barrier suite RUPP under aseptic conditions.
On GD 18, carotid arterial catheters were implanted for blood pressure measurement in all groups of rats. The catheters were V3 tubing (Scientific Commodities, Inc., Lake Havasu City, AZ, USA), which were tunneled to the back of the neck and exteriorized. On GD 19, the rats were placed in individual restraining cages and after a 30 min stabilization period, arterial blood pressures were recorded. Arterial pressures were monitored with a pressure transducer (Cobe III Transducer CDX Sema) for one hour and recorded as MAP. Litter size and pup weights were recorded, blood and urine samples were collected, and kidneys, placentas, and spleens were harvested under anesthesia.
Circulating APRIL was measured as a marker of B cell survival and potential transition into plasma cells. Sera collected on GD 19 was used in a commercially available anti-rat sandwich APRIL enzyme-linked immunoassay (ELISA) kit (MyBioSource, San Diego, CA, USA) and was performed as per manufacturer’s instructions. Sensitivity of the APRIL ELISA was reported as 0.1 ng/mL and a detection range of 0–50 ng/mL. The intra-assay and inter-assay variability is reported as < 10%.
Immunoglobulin was isolated from 1 mL of serum collected on GD 19 by epitope binding using a protein G column. This IgG fraction was used in a bioassay. AT1-AA was isolated from serum and analyzed using isolated cardiomyocytes as previously described [21]. AT1-AA was quantified by the chronotropic responses to AT1 receptor-mediated stimulation of cultured neonatal rat cardiomyocytes coupled with receptor-specific antagonists. Chronotropic responses were measured and are expressed in beats per minute (bpm). The results express the difference between the basal beating rate of the cardiomyocytes and the beating rate measured after the addition of the AT1-AA (increase in the number of bpm or Δ bpm).
Human and rat placental ACE-2 activity was assessed using a commercially available Abcam ACE-2 Activity Assay Kit (Cambridge, UK) designed to measure mammalian ACE-2. The assay sensitivity range is 0.4–400 μU/mL. The assay was performed as per manufacturer’s instructions and measures the ability of ACE-2 to cleave a synthetic Methyl Cumaryl Amide (MCA)-based peptide.
All data are expressed as means ± standard error of the mean and were performed with GraphPad Prism 7.02 software (GraphPad Software, San Diego, CA, USA). A student’s t-test or a one-way analysis of variant (ANOVA) with Bonferroni multiple comparisons test as post-hoc analysis was conducted for comparison of normally distributed variables. The Mann–Whitney U test or Kruskal–Wallis one-way ANOVA with Dunn’s multiple comparison post-hoc test was used for comparison of non-normally distributed variables. Results were reported as means ± standard error of the mean (SEM). Results with values of P < 0.05 were considered statistically significant.
MAP was increased in nude-athymic rats receiving PE CD4+ T cells (NP nude + PE CD4+ T cells, n = 9) compared to rats receiving NP CD4+ T cells (NP nude + NP CD4+ T cells, n = 7; 114 ± 1 mmHg vs. 98 ± 2 mmHg, P < 0.05; Figure 1). Administration of rituximab (103 ± 2 mmHg, n = 3) or anti-CD40L (102 ± 1 mmHg, n = 3) lowered MAP compared to PE CD4+ T cells alone (114 ± 1 mmHg, n = 9, P < 0.05 vs. NP CD4+ T cells, P < 0.05 vs. PE CD4+ T cells) as shown in Figure 1.
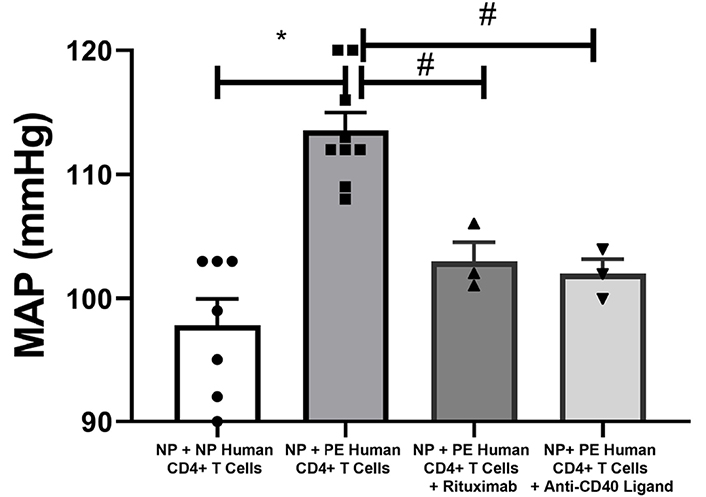
Blood pressure is increased in recipients of placental CD4+ T cells from PE patients compared to recipients of NP CD4+ T cells. MAP was increased in nude-athymic rats receiving PE CD4+ T cells compared to rats receiving NP CD4+ T cells (P < 0.05), but was significantly lowered when T cells–B cell communication was blocked with anti-CD40L or when endogenous B cells are depleted with Rituximab. Statistical differences were established using the Mann–Whitney U test or Kruskal–Wallis one-way ANOVA with Dunn’s multiple comparison post-hoc test. Results were reported as means ± SEM and considered statistically significant when P < 0.05 (* P < 0.05 vs. NP CD4+ T cells, Kruskal–Wallis, # P < 0.05 vs. PE CD4+ T cells, Mann–Whitney U test)
There were no changes in pup weight after administration of rituximab (1.265 ± 0.08 g, n = 3) or anti-CD40L (1.38 ± 0.13 g, n = 3) compared to NP nude + PE CD4+ T cells (1.23 ± 0.08 g, n = 9) or NP + NP CD4+ T cells (1.51 ± 0.14 g, n = 7) as shown in Figure 2A. Placental weight was increased in NP + NP CD4+ T cells (0.55 ± 0.02 g, n = 7) compared to PE CD4+ T cells (Figure 2B). Furthermore, administration of rituximab (0.65 ± 0.08 g, n = 3) or anti-CD40L (0.67 ± 0.04 g, n = 3) increased placenta weight compared to PE CD4+ T cells alone (0.50 ± 0.02 g, n = 9, P < 0.05 vs. PE CD4+ T cells; Figure 2B).
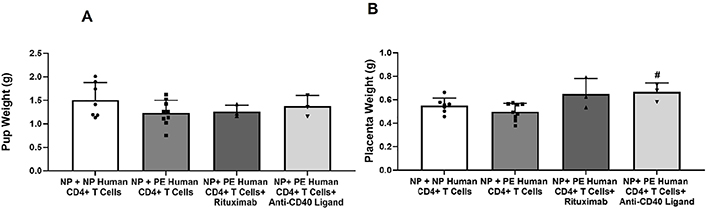
Recipients of PE CD4+ T cells have reduced fetal weight compared to recipients of NP CD4+ T cells. (A) Pup weights were less but not significantly decreased in rats receiving PE CD4+ T cells compared to NP CD4+ T cells. Also, administration of rituximab or anti-CD40L did not change pup weight in recipients of PE CD4+ T cells; (B) placenta weights were not significantly decreased in rats receiving PE CD4+ T cells compared to NP CD4+ T cells. Administration of rituximab or anti-CD40L increased placenta weight compared to PE CD4+ T cells alone. Statistical differences were established using the Kruskal–Wallis one-way ANOVA with Dunn’s multiple comparison post-hoc test. Results were reported as means ± SEM and considered statistically significant when P < 0.05 (# P < 0.05 vs. PE CD4+ T cells, Kruskal–Wallis)
After CD4+ T cell isolation, cell suspensions were stained with antibodies against CD19 and CD20. Prior to adoptive transfer, flow cytometry was performed on stained cells to confirm the absence of human B cells in the adoptive transfer preparation (Figure 3A). Circulating AT1-AA was increased in NP nude + PE CD4+ T cells (n = 4) compared to NP nude + NP CD4+ T cells (n = 5; 16.8 ± 0.32 bpm vs. 1.5 ± 0.9 bpm, P < 0.05; Figure 3B). NP nude + PE CD4+ T cells (n = 6) had elevated circulating APRIL (1.276 ± 0.0.13 ng/mL) compared to recipients of NP nude + NP CD4+ T cells (n = 5; 0.434 ± 0.11 ng/mL, P < 0.01) as shown in Figure 3C.
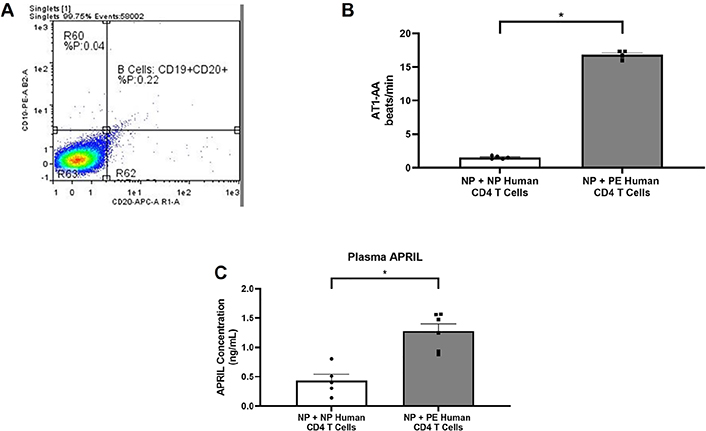
Human B cells are not present in CD4+ T cells preparation used for adoptive transfer. (A) Histogram showing forward and side scatter for CD19 and CD20 showing no human B cells in the adoptive transfer prep; (B) agonistic autoantibodies to the AT1-AA expression increased in rats receiving PE CD4+ T cells compared to rats receiving NP CD4+ T cells (P < 0.05); (C) ELISA data showing increased circulating APRIL in response to PE CD4+ T cells. Statistical differences were established using the Mann–Whitney U test. Results were reported as means ± SEM and considered statistically significant when P < 0.05 (* P < 0.05 vs. NP CD4+ T cells, Mann–Whitney U test)
Placentas from women with PE had increased placental ACE-2 activity (74.48 ± 10.29 mmol/min, n = 3) compared to NP (33.75 ± 8.81mmol/min, n = 4, P < 0.05) in Figure 4A. ACE-2 activity was elevated in recipient NP nude + PE CD4+ T cells and reduced with B cell inhibition (Figure 4B). NP nude + PE CD4+ T cells had elevated placental ACE-2 activity (13.35 ± 4.58 mmol/min, n = 4) compared to NP nude + NP CD4+ T cells (7.056 ± 3.315 mmol/min, n = 5, P = 0.14) in Figure 4B. Administration of rituximab (2.88 ± 2.86 mmol/min, n = 3) or anti-CD40L (1.44 ± 1.44 mmol/min, n = 3) had reduced placental ACE-2 activity compared to PE CD4+ T cells (Figure 4B).
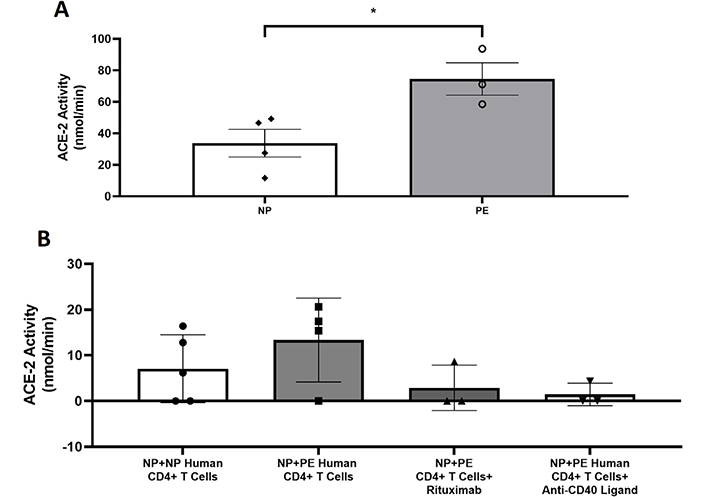
Placental ACE-2 is increased in PE placentas. (A) Placental ACE-2 activity is increased in PE placentas compared to NP placentas (P < 0.05); (B) ACE-2 activity is higher, albeit not significantly, in response to PE CD4+ T cells and is decreased with B cell depletion or inhibition of T cell–B cell communication. Statistical differences were established using the Mann–Whitney U test. Results were reported as means ± SEM and considered statistically significant when P < 0.05 (* P < 0.05 vs. NP CD4+ T cells, Mann–Whitney U test)
We have previously demonstrated that CD4+ T cells are implicated in the pathophysiology of PE through the adoptive transfer of CD4+ T cells from women with PE [13]. The data from this current study expound upon the initial results published by Harmon et al. [13], by showing additional downstream effects of preeclamptic CD4+ T cells such as elevated cytokines and AT1-AA. To our knowledge, the effect of CD4+ T cells to cause factors such as inflammatory cytokines, B cells secreting AT1-AA and sFlt-1 in models of PE is extremely limited thereby highlighting the importance of knowledge gained from these types of translational studies. In addition to elevated MAP, our new results show that adoptive transfer of CD4+ T cells from women with PE into nude-athymic rats leads to increased circulating AT1-AA expression when compared to adoptive transfer of CD4+ T cells from women without PE. Importantly, B cells were not carried over in the adoptive transfer preparation as shown by flow cytometry of CD4+ T cells prior to adoptive transfer. To determine CD4+ T cells activation of endogenous B cells, we measured APRIL, a B cell essential cytokine in response to CD4+ T cells. Our data demonstrated that APRIL was indeed elevated indicating that it was stimulated to engage in B cells growth. Unfortunately, there are no published studies examining a role for APRIL in PE, which will be a focus of further studies in our laboratory. Furthermore, inhibition of T cell to B cell communication by CD40-CD40L blockade and/or depletion of endogenous B cells lowered hypertension in response to PE CD4+ T cells, and therefore illustrated the importance of B cell activation, proliferation and secretion of the AT1-AA in causing hypertension in response to PE CD4+ T cells.
Early onset PE is classically defined as PE that manifests before 34 weeks gestation, and late onset PE is defined as PE that manifests after 34 weeks gestation [22]. Clinically, this is relevant because PE that manifests after 34 + 0 gestational age in women is not managed conservatively based on current guideline recommendations, and immediate delivery is recommended [1]. In fact, it has been proposed that early onset and late onset PE may represent two distinct disease processes [30]. After our results were stratified based on whether the CD4+ T cells were derived from women with early versus late onset PE, our results did not show a significant difference in MAP, pup weight, or placenta weight between the two groups of PE patients. Therefore, all data was analyzed and presented as PE compared to NP.
One of the functions of CD4+ T cells is to mediate B cell maturation [19]. CD40L is expressed on CD4+ T cells and promotes long-term B cell activation (maturation) by binding to CD40 on B cells [8]. CD20 is expressed on the surface of B cells, and stimulation of this protein leads to B cells entering circulation and producing antibodies [8]. Inhibition or disruption of CD4+ T cell–B cell communication at the CD40-CD40L prevents maturation of the B cells and blocks creation or proliferation of antibodies [8, 19]. B cells depletion with rituximab (anti-CD20) leads to B cell death and lowers or attenuates production of antibodies [19]. Our results demonstrated that inhibition of B cell maturation with anti-CD40L or depletion of B cells with rituximab led to significantly attenuated hypertensive response to PE CD4+ T cells in nude-athymic rats that received either of these agents. Furthermore, the addition of B cell-inhibiting agents significantly increased placental weight, thus some factor released in response to B cells was lowered possibly allowing better placental circulation.
Pregnant women naturally upregulate ACE-2 early in gestation in order to counteract the increased activity in the RAAS, which is necessary to manage the extracellular volume load that occurs early in pregnancy [22]. After examining if an increase in AT1-AA in PE women could lead to increased activation of the RAAS and upregulation of ACE-2, our data demonstrates that ACE-2 activity is increased in the PE placenta compared to NP placenta at delivery. One possibility that this occurs is that AT1-AA activation of AT1R leads to increases in ACE-2 [22]. Future studies will examine in greater detail the importance of the AT1-AA to cause endogenous changes in the AT1R expression or activation during pregnancy and how this causes pathophysiological changes in pregnancy. Importantly, recipient rats of PE CD4+ T cells exhibited higher placental ACE-2 activity in association with AT1-AA. With the depletion of B cells or inhibition of T cell–B cell communication, ACE-2 activity and hypertension were lowered, which further provides additional evidence supporting the importance of CD4+ T cells in the pathology of PE and the potential role of the AT1-AA to upregulate ACE-2, which may be a maternal compensatory response. Additional studies to examine the effect of AT1-AA on other factors of RAAS such as AT2 expression will be important for better understanding the complexity of RAAS alterations during pregnancy with and without AT1-AA. Nevertheless, these data present interesting associations between PE CD4+ T cells, AT1-AA, and placental ACE-2, which is an important area that will be further investigated by our group.
The collective results from this study support our previous data by further demonstrating that placental CD4+ T cells play an important role in the pathophysiology of PE by activating multiple inflammatory factors. One important factor is the production of AT1-AA in response to PE CD4+ T cells. We have previously shown that AT1-AA induced hypertension leads to increases in sFlt-1 and endothelin 1 (ET-1), which were both shown to be elevated in this model of PE [7]. Therefore, we believe the AT1-AA could be the mechanism for increases in downstream vasoactive pathways and a mechanism for further upregulating ACE-2 as a potential compensatory response. In this study, we show that reduction in B cell activation prevented ACE-2 activation and AT1-AA production and the resulting hypertensive response. Furthermore, rats receiving adoptively transferred NP CD4+ T cells or rats receiving PE CD4+ T cells in combination with B cell inhibiting agents did not have an increase in blood pressure compared to those receiving PE CD4+ T cells alone.
As the importance of B cells in either physiological or pathophysiological states is uncovered, new therapeutic options and targets will become available. The delicate nature of human pregnancy warrants careful treatment plans in order not to harm the mother and the fetus. Modulation of B cell function has the potential to be an impactful therapeutic route necessary to suppress autoantibodies known to cause pathophysiology in pregnancy and to normalize ACE-2 activity and thus should be ventured into with pure intentions of moving the field forward to developing better treatment possibilities for women with PE.
ACE-2: angiotensin-converting enzyme 2
ANOVA: analysis of variant
anti-CD40L: anti-CD40 ligand
APRIL: a proliferation-inducing ligand
AT1-AA: autoantibodies to angiotensin II type 1 receptor
AT1R: angiotensin II type 1 receptor
DMSO: dimethyl sulfoxide
FBS: fetal bovine serum
GD: gestational day
IL-17: interleukin-17
MAP: mean arterial pressure
NP: normal pregnant
PE: preeclampsia
Pen-Strep: penicillin-streptomycin
RAAS: renin-angiotensin-aldosterone system
RPMI: Roswell Park Memorial Institute
RUPP: reduced uterine perfusion pressure
SEM: standard error of the mean
sFlt-1: soluble fms-like tyrosine kinase-1
TNF-α: tumor necrosis factor alpha
UMMC: University of Mississippi Medical Center
KER bred all pregnant rats, collected placental T cells from patients, collected samples for molecular analysis and writing of the manuscript. ED aided in breeding rats, adoptive transfers and writing and editing of the manuscript. LMA collected patient placental T cells, performed adoptive transfer techniques and wrote and edited final version of the manuscript. DCC performed flow cytometry of collected T cells for adoptive transfer and aided in writing and editing of the manuscript. OH performed ace-2 assays and APRIL ELISA, measure blood pressure and collected samples for molecular analysis and edited final version of the manuscript. ACH performed adoptive transfer techniques, measured blood pressure and collected samples for molecular analysis and edited final version of the manuscript. NC performed adoptive transfer techniques, measured blood pressure and collected samples for molecular analysis and edited final version of the manuscript. SF bred all pregnant rats. TI performed adoptive transfer techniques, measured blood pressure and collected samples for molecular analysis and edited final version of the manuscript. GW measured AT1-AA in sera from recipient rats and edited final version of the manuscript. RD supervised measurements of AT1-AA in sera from recipient rats and edited final version of the manuscript. BL conceptualized all aspects of the study and supported the work financial through funding agencies listed below, edited versions of the manuscript. All authors have read and agreed to the published version of the manuscript.
The authors declare they have no conflict of interest.
This study was reviewed and approved by the University of Mississippi Medical Center Institutional Review Board (UMMC IRB). The study was conducted according to the guidelines of the University of Mississippi Medical Center Institutional Animal Care and Use Committee (UMMC IACUC), and the animals were handled according to the guiding principles published in the National Institutes of Health Guide for the Care of Animals and the Institutional Animal Care and Use Committee. All experimental procedures were approved by the IACUC at UMMC.
Informed consent to participate in the study was obtained from all participants.
Not applicable.
Not applicable.
The animals and assays performed in this study were completely supported by NIH grant RO1HD067541-11 (BL). 1U54GM115428 (LMA) and P20GM121334 (LMA BL), American heart association (AHA) early career award 19CDA34670055 (LMA), and the T32 Trainee Grant T32-HL105324 (ED) supported salaries of the authors initials listed by the grant that performed the work outlined in this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Rafael Luzes ... Adalberto Vieyra
Natalia L. Rukavina Mikusic ... Mariela M. Gironacci
Gian Paolo Rossi ... Teresa Maria Seccia
Sukhwinder K. Bhullar ... Naranjan S. Dhalla
Eran S. Zacks ... Richard B. Devereux
Casper N. Bang ... Peter M. Okin
Casper N. Bang ... Peter M. Okin
Marcello Chinali ... Richard B. Devereux
