 Review
Review

Affiliation:
1Specialized Service of Epidemiology and Molecular Biology, Santo Espírito Hospital of Terceira Island (SEEBMO/HSEIT), Canada do Briado, 9700-049 Angra do Heroísmo, Terceira-Azores, Portugal
2Comprehensive Health Research Centre, Nova Medical School, Campo Mártires da Pátria, 1169-056 Lisbon, Portugal
ORCID: https://orcid.org/0000-0003-3705-9121
Affiliation:
1Specialized Service of Epidemiology and Molecular Biology, Santo Espírito Hospital of Terceira Island (SEEBMO/HSEIT), Canada do Briado, 9700-049 Angra do Heroísmo, Terceira-Azores, Portugal
2Comprehensive Health Research Centre, Nova Medical School, Campo Mártires da Pátria, 1169-056 Lisbon, Portugal
ORCID: https://orcid.org/0000-0002-3600-2812
Affiliation:
3Santo Espírito Hospital of Terceira Island, Orthopedics Service, Canada do Briado, 9700-049 Angra do Heroísmo, Terceira-Azores, Portugal
ORCID: https://orcid.org/0000-0001-5527-527X
Affiliation:
1Specialized Service of Epidemiology and Molecular Biology, Santo Espírito Hospital of Terceira Island (SEEBMO/HSEIT), Canada do Briado, 9700-049 Angra do Heroísmo, Terceira-Azores, Portugal
2Comprehensive Health Research Centre, Nova Medical School, Campo Mártires da Pátria, 1169-056 Lisbon, Portugal
Email: brugesarmas@gmail.com
ORCID: https://orcid.org/0000-0001-7671-6521
Explor Musculoskeletal Dis. 2023;1:143–170 DOI: https://doi.org/10.37349/emd.2023.00018
Received: May 06, 2023 Accepted: August 29, 2023 Published: October 23, 2023
Academic Editor: Blanka Stiburkova, Charles University and General University Hospital, Czech Republic
The article belongs to the special issue Diffuse Idiopathic Skeletal Hyperostosis- A common but neglected disease
Bone formation is a complex process that occurs throughout life, and is normally limited to the skeletal system. In bone formation, osteoprogenitor cells follow several developmental stages, including differentiation in osteoblasts, proliferation, matrix maturation, and mineralization. The mechanisms involved in the mineralization process of bone, such as in the new bone formation, are extremely complex and have been under intense investigation for many years. Bone formation follows two distinct processes, intramembranous and endochondral ossification; both are regulated by signaling pathways involving numerous genes. Disturbance of these signaling pathways may cause a large spectrum of skeletal diseases characterized by new bone formation and bone growth anomalies. This review will only focus on the key genetic pathways involved in heterotopic bone formation. Wingless/integrated (Wnt), hedgehog (HH), and transforming growth factor beta (TGFβ)/bone morphogenetic protein (BMP) signaling pathways are described and illustrated; their relation with new bone formation is demonstrated through their involvement in bone formation disorders.
Bone is a specialized connective tissue that is comprised of cells and an extracellular matrix (ECM), which becomes mineralized by the deposition of calcium (Ca) hydroxyapatite (HA), providing strength, rigidity, and resistance [1]. Bone tissue has several key functions in the body; in addition to mechanical support and protection of vital organs, it also serves as a storage site of Ca, phosphate, and other minerals, which can be mobilized to the blood, as needed, to maintain homeostasis levels in body fluids. Bone tissue also provides an area for marrow as a place for hematopoiesis [2]. To fulfill its functions, bone is constantly remodeling. This requires a tightly controlled regulatory system that involves several specialized cells, which communicate with each other. These cells are constantly responding to many different internal and external signals, which can be systemic or local [3]. Bone homeostasis is maintained in adult life with bone resorption undertaken by osteoclasts and bone formation by osteoblasts. When bone resorption and bone formation occur on separate surfaces and are not combined, the process is called bone modeling [4]. Bone modeling occurs during birth to adulthood and is responsible for gaining bone mass and alterations in the skeletal form [5].
Bone formation follows two distinct processes, intramembranous and endochondral ossification; both are regulated by signaling pathways involving numerous genes. Variants in these genes and dysfunctions in their encoded proteins may have a substantial influence on bone homeostasis, leading to a variety of bone diseases [6]. Abnormal bone formation can occur as a result of mechanical, inflammatory, or metabolic stimuli and may affect bone (orthotropic bone formation) or soft tissue (heterotopic bone formation) [7].
In the past few years, the molecular understanding of bone formation has greatly increased, mainly due to the rapid advances in new biomedical technologies. This review, after providing a brief overview of bone formation biology, will focus on the fundamental subset of genes and pathways that are involved in the ossification process. The aim of this paper to summarize and discuss the current state of knowledge in the field of the molecular genetics of new bone formation.
Bone formation is a complex process occurring throughout life, normally limited to the skeletal system. Bone formation follows two distinct processes—intramembranous and endochondral ossification. The difference between them rests on whether the bone is formed by a direct process or by the intervention of a cartilage model; in several bones, both processes may occur [7]. Although there are two different ossification processes, in the end, the mature bone is the same, regardless of the process that produces it [1].
This type of ossification is mostly restricted to the flat bones and to some of the irregular bones [8]. It also contributes to the growth of short bones and the increase in thickness of long bones. Intramembranous ossification is initiated by mesenchymal tissue membranes during intrauterine life and by connective membranes in postnatal life [9]. The cells of the progenitor membranes migrate and aggregate in specific areas, forming ossification centers. In the ossification center, these cells elongate and differentiate into osteoprogenitor cells, which express runt-related transcription factor 2 (RUNX2)/core-binding factor alpha 1 (Cbfa1), crucial to osteoblast differentiation [1]. It has already been demonstrated, that Cbfa1 knockout mice die after birth and their skeletons completely lack ossification, having only a skeletal made of cartilage. In these mice, both intramembranous and endochondral ossification were blocked, since osteoblasts were in arrest state and neither osteocalcin (OCN) nor osteopontin (OPN) were expressed [10]. Mice heterozygous for Cbfa1 showed bone defects similar to the human syndrome called cleidocranial dysplasia, a rare condition that effects teeth and bones, such as the face, skull, clavicles, spine, and legs, in which ossification is often absent or deformed [11].
It is believed, that bone morphogenetic proteins (BMPs) activate the RUNX2 gene. Once activated, the RUNX2/Cbfa1 transcription factor activates OCN, OPN, osterix transcription factor, and other bone-specific ECM proteins [12, 13]. Osteoblasts begin to secrete around themselves the osteoid, an un-mineralized collagen-proteoglycan matrix, that is able to join Ca salts [13]. The presence of Ca in osteoid, results in the hardening of the matrix, and the entrapped osteoblasts, in the calcified matrix, become osteocytes. Osteocytes separate from one another as more matrix is produced, but they are interconnected with each other through canaliculi [1]. As calcification proceeds, bone spicules are released out of the ossification center and are surrounded by compact mesenchymal cells that form a membrane that surrounds the bone, the periosteum. The cells in the periosteum also become osteoblasts and deposit osteoid matrix parallel, and many layers of bones are formed [13].
Endochondral ossification is the main mechanism of ossification in mammals and is essential for the formation of axial and appendicular bones. This type of ossification involves the formation of a hyaline cartilage template and the subsequent replacement by bone [14]. Endochondral ossification can be divided into several steps:
Some of the condensed mesenchymal cells differentiate into chondroblasts that proliferate and secrete an ECM to form the cartilage template, composed of hyaluronic acid, collagen fibers, chondroitin sulfate, and water. The cartilage model is essential to provide a mechanically stable model for bone formation and also as a source of longitudinal bone growth [14]. As the matrix surrounds chondroblasts, they may be designated as chondrocytes. Several transcription factors are involved in this stage. The SRY-box transcription factor 9 (SOX9) gene encodes the main transcription factor involved in chondrocyte differentiation [15, 16]. SOX9, together with SOX5 and SOX6, are essential to cartilage formation and proliferation by promoting the expression of cartilage genes, such as collagen type II alpha 1 chain (COL2A1) and aggrecan (ACAN), a cartilage-specific proteoglycan [17].
The chondrocytes stop dividing and increase their volume by 10-fold to 20-fold, which they are known as “hypertrophic chondrocytes” [18]. The hypertrophic chondrocytes have the ability to alter the ECM by adding collagen type X and more fibronectin, allowing for calcification. Hypertrophic chondrocytes secrete several small membrane-bound vesicle organelles into the extracellular space to initiate the mineralization process within the cartilaginous matrix [13]. The membrane-bound vesicle organelles provide the nucleation site for cartilage matrix mineralization and are composed of a specific combination of enzymes involved in the generation of Ca and inorganic phosphate (Pi) ions, including annexins, Pi transporters, and phosphatases [19–21]. Chondrocyte hypertrophy process is regulated by an upregulation of RUNX2 and COL10A1 gene expression with simultaneous downregulation of activating transcription factor 4 (ATF4) gene [22–24]. Collagen type X seems to have a key role in the mineralization process in hypertrophic chondrocytes [25].
The calcification of the ECM by hypertrophic chondrocytes prevents nutrients from reaching them, causing its death by apoptosis.
The apoptosis of the hypertrophic chondrocytes favours the production of angiogenic factors, such as the vascular endothelial growth factor (VEGF) which stimulates the invasion of blood vessels [13]. The blood infiltration facilitates the recruitment of osteoclasts, which destroy the mineralized cartilage, and osteogenic cells derived by perichondrium to be differentiated into osteoblasts, the bone-forming cells [26].
Osteoblasts will deposit bone matrix on the degraded cartilage, by producing the extracellular components collagen type I and OPN, and alkaline phosphatase (ALP), essential to the mineralization process and the deposition of HA crystals [27, 28]. The replacement of cartilage cells by bone cells is deeply dependent on ECM mineralization [13]. Osteocytes derived from osteoblasts are surrounded by bone matrix, thus appearing where there was previously cartilaginous tissue [9].
The mechanisms involved in the mineralization process of bone and new bone formation are maintained through four distinct cell types: (A) osteoblasts, the bone-forming cells; (B) osteoclasts, the cells that resorb bone; (C) bone lining cells, which differentiate into osteoblasts; and (D) osteocytes, that coordinate bone remodeling and act as mechanosensors, under the control of both local and systemic factors [29].
Chondrocytes are the foundation of articular cartilage joint homeostasis but can also serve as a base for bone formation and chondral repair [30] These cells maintain the ECM and produce a multi-component matrix containing water, proteoglycans, and collagen. Chondrocytes begin to distance from each other due to the presence of the matrix [31]. Instability or imbalance in this process can result in abnormal architecture or function, leading to inadequate, excessive, or ectopic calcification, and consequential serious health consequences [32].
Minerals are formed by organisms of all biological kingdoms for the most diverse and important purposes. Biomineralization is thus a fundamental biological process by which organisms produce hard inorganic matter, required for crucial functions of the body. The process by which the mineral deposition occurs can be briefly explained as a result of: (A) ions reaching the tissue; (B) combining with their counterions; (C) achieving supersaturation of the salt solute; and (D) precipitating as a solid phase (i.e. crystal) [33]. The predominant mineral of the vertebrate skeleton is Ca HA, the main constituent of bone.
Ca and Pi are two key players in the biomineralization process, contributing directly to the formation of HA crystals. Homeostasis regulators maintain the extracellular concentrations and equilibrium of these ions as constant as possible, in order to protect the organism against deficiency or overload [33]. Ionized Ca2+ and inorganic Pi, are maintained by a dynamic equilibrium through fluctuations occurring at the level of: (A) the intestine (absorption through cellular and paracellular mechanisms); (B) the kidney (leading role in the extracellular concentration regulation of both ions); and (C) bone (interchange of mineral by cellular bone resorption and possible diffusion through bone cell envelope). Ca2+ is involved in paramount biological functions such as muscle function, neural transmission, cell signaling, blood coagulation, and biomineralization. Ca2+ levels have to be particularly controlled in intra- and extracellular compartments in order to be maintained in a fine range. Extracellular control key players include parathyroid hormone (PTH), 1,25-dihydroxy-vitamin D, and calcitonin [34]. Inorganic pyrophosphate (PPi), which consists of two molecules of Pi connected by a hydrolysable high-energy ester bond, is also a critical part of the regulation of crystal deposition [35]. Pi is essential for the physiological functioning of osteogenic cells, not only because it is an integral component of HA crystal but also because it may affect the production rate of the bone matrix. The transport of Pi is the main factor leading to the accumulation of minerals inside cells and is fundamental for the calcification process [36]. For normal mineral deposition to proceed, the ratio of extracellular Pi and PPi is required to be in balance. Extracellular PPi is required to induce calcification and is a potent inhibitor of the crystallization of Pi with Ca thereby suppressing HA crystal propagation [37]. PPi is produced, degraded, and transported by specialized mechanisms. Dysregulation of any of these processes has been associated with ectopic mineralization [35]. PPi is produced either: (A) directly by ectoenzymes that generate Pi (pyrophosphatases and adenosine triphosphatases) and PPi [nucleoside triphosphate pyrophosphohydrolase (NTPPPH)] on apoptotic particles and matrix vesicles (MVs) membrane surfaces or (B) indirectly as a metabolic byproduct of multiple biochemical and biosynthetic reactions during synthesis and secretion of matrix proteins [38]. The three major regulators of PPi homeostasis are: (A) ectonucleotide pyrophosphatase/phosphodiesterase (PDE) 1 (ENPP1), an ubiquitous type II transmembrane glycoprotein with enzymatic activity, which produces PPi from ATP, inhibiting PPi related calcification and mineralization [39]; (B) tissue non-specific ALP (TNAP), an isozyme of a family of four homologous human ALP genes, presented in the MVs and with the ability to hydrolyze PPi [40]; and (C) ankylosis progressive homolog (ANKH), a multiple-pass transmembrane protein which is involved in the transport of PPi across plasma membrane to the extra cellular compartment. Mutations in ANKH can be the cause of the abnormal deposition of Ca PPi crystals in the cartilage [41].
Bone is mineralized in two phases; primary mineralization is achieved when MVs are secreted by osteoblasts. Secondary mineralization corresponds to the gradual increase of bone mineral density. MVs consist of extracellular membrane-invested organelles with 20–200 nm in diameter [42]. These key organelles are secreted by hypertrophic chondrocytes and mature osteoblasts and are the place where the first Ca/Pi crystals are formed. MVs are budded off from the plasma membrane of the osteoblasts/chondrocytes to the extracellular space, where they act as a reservoir of mineralization materials and as mineralization niduses [43]. The first crystals of Ca HA mineral are generated inside MVs at some point of biomineralization of growth plate cartilage, newly formed bone, tendons, and the predentine of teeth [43, 44]. The calcification process, initiated through MVs, begins with the absorption of Ca2+ determined by the high concentration of Ca-phospholipids and proteins. Meanwhile, Pi accumulates within the MVs, as a result of Pi transport by its specialized transporters, plus enzymatic activity of phosphatases. When sufficient Ca2+ and Pi have accumulated in the interior of the MVs, amorphous apatite crystals start to precipitate. This non-crystalline form will be converted to octacalcium Pi, which is then transformed into highly insoluble HA [21]. MVs potential for therapeutic use in bone regeneration or treatment of bone pathologies is preliminary but it will surely become noteworthy in the future [45]. The composition of MVs depends on the mineralizing nature of their precursor cell but mostly includes:
Lipids—plasma membrane phospholipids (e.g. cholesterol and sphingomyelin) and an unusually high concentration of acidic phospholipids such as phosphatidylserine [46]; this anionic phospholipid shows a selective ability to create complexes with Ca2+ and Pi [47]. These complexes, in vitro, are very effective inducing HA precipitation [48].
Enzymes—TNAP and nucleotide pyrophosphatase/PDE 1 (NPP1)—where TNAP is the major phosphatase of these vesicles, being typically enriched on the surface of MVs [49]; NPP1 can act as a backup phosphatase in the absence of TNAP [50]. TNAP also modulates the phosphorylation status of OPN. OPN is an important non-collagenous bone protein, which inhibits the nucleation and growth of HA, through binding to nascent crystals by means of the phosphorylated residues of the protein [51].
Proteins—small integrin-binding ligand N-linked glycoproteins (SIBLINGs)—include OPN, bone sialoprotein, matrix extracellular phosphoglycoprotein, dentin matrix protein, and dentin sialoprotein. These proteins share a conserved arginine-glycine-aspartic acid motif mediating their cell attachment and signaling functions [52]. The conserved acid serine and aspartate-rich motif peptide region within this family of proteins appears to be the key determinant of their role in mineralization [53].
Pi transporters—two related type III sodium/Pi pituitary-specific Pi co-transporters, encoded by solute carrier family 20 type A1 [SLC20A1; Pi transporter 1 (PiT1)] and SLC20A2 are both expressed in chondrocytes and osteoblasts; however, PiT1 is the major mediator of Pi influx in these cell types [54].
Another phosphatase—phosphoethanolamine/phosphocholine phosphatase 1 (PHOSPHO1)—a phosphatase that is crucial for the initiation of skeletal mineralization, for its central role in Pi generation, was shown to be highly expressed in calcifiable MVs from osteoblasts, chondrocytes and odontoblasts [55, 56]. Studies with animal models suggests that PHOSPHO1 is involved in MVs biogenesis and shows that PiT1 is involved in skeletal mineralization [57].
microRNAs (miRNAs)—a single miRNA may affect the transcription of multiple genes in different pathways. Studies with miRNAs were carried out in vascular mineralization; a number of dysregulated miRNAs in MVs were identified from chronic kidney disease rats showing aortic mineralization, including miR-667, -702, -3562, -3568, and -3584 [58].
New bone is synthesized by osteoblasts, which synthesize and deposit organic bone matrix (osteoid) proteins that will mineralize in developing skeletons during the process of bone remodeling [59]. These cells participate in both membranous and endochondral bone formation. A number of signaling pathways that converge on specific transcription factors, regulate both of these ossification processes. Disturbance of these signaling pathways may cause a large spectrum of skeletal diseases characterized by new bone formation and bone growth anomalies [60].
Wingless/integrated (Wnt) signaling is a key pathway involved in numerous cellular activities, including bone development, homeostasis, and remodeling [61]. In the context of new bone formation, Wnt is critical for chondrocyte development and osteoblast differentiation [62]. When a secreted extracellular Wnt glycoprotein binds a frizzled receptor in the cell membrane, the pathway is activated by inducing bone formation [63]. In humans, at least 19 Wnt glycoproteins and 10 frizzled receptors have been described and are involved in the activation of at least three distinct intracellular Wnt signaling pathways: the canonical Wnt, the non-canonical planar cell polarity (PCP), and the non-canonical Wnt/Ca pathway [61, 64]. It is known that the three Wnt pathways are involved in the regulation of bone biology [65–67]; however, the canonical Wnt is best described in the context of bone formation [68].
The canonical Wnt pathway (or Wnt/β-catenin) mediates signaling through the stabilization of β-catenin in the cytoplasm upon activation. In the presence of Wnt glycoproteins, occur the binding to the complex frizzled receptor to one of the co-receptors low density lipoprotein receptor-related protein (LRP), LRP5 or LRP6, causing the phosphorylation of the Dishevelled (Dsh) protein, which inhibits glycogen synthase kinase-3beta (GSK3β) from phosphorylating β-catenin in the cytoplasm. This causes the accumulation of dephosphorylated β-catenin, which, upon reaching a certain concentration level, translocates into the nucleus (Figure 1). In the nucleus, β-catenin acts as a transcriptional coactivator with lymphoid-enhancer binding factor (LEF)/T-cell specific transcription factor (TCF) to activate the expression of Wnt target genes, such as CYCLIN D1, AXIS inhibition protein 2 (Axin2), c-Myc and peroxisome proliferator-activated receptor [68–72]. In osteoblasts, the activation of the canonical Wnt also activates the expression of osteoprotegerin (OPG, encoded by TNFRSF11B gene), an inhibitor of receptor activator of nuclear factor kappa beta (RANK) ligand (RANKL) [73]. OPG inhibits osteoclastogenesis by blocking the interaction between RANK-RANKL, thus preventing bone resorption [74, 75]. On the other side, in the absence of a Wnt ligand, β-catenin is constantly degraded by the action of the tetramer degradation complex composed of GSK3β, adenomatous poliposis coli (APC), and Axin, which tags β-catenin for ubiquitination and degradation by the proteasomal machinery (Figure 1) [68, 69, 72].
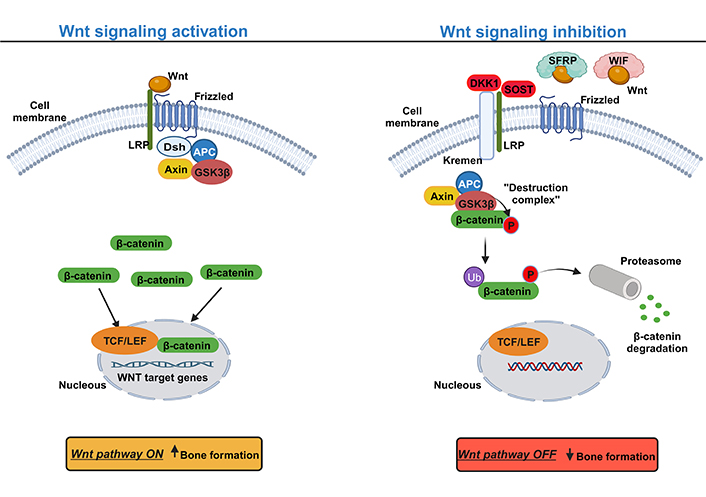
Canonical Wnt signaling pathway: a simplified schematic illustration. Activation of the canonical Wnt signaling pathway (left) with Wnt ligands interacting with the transmembrane frizzled protein and the co-receptor LRP to activate the pathway and hence the inhibition of the degradation complex composed by GSK3β, APC, and Axin. The suppression of the degradation complex induces the accumulation of β-catenin at the cytoplasm level and consequently its translocation into the nucleus to trigger the expression of Wnt target genes, inducing bone formation. Inhibition of the canonical Wnt signaling pathway (right) with the presence of extracellular factors, such as dickkopf-related protein 1 (DKK1), sclerostin (SOST) that with the cooperation of Kremen protein occurs the binding to the LRP co-receptor, thereby competing with Wnt ligands. In addition, Wnt signaling can also be inhibited by Wnt inhibitor factor (WIF) and secreted frizzled-related protein (SFRP) by sequestering Wnt ligands, preventing its binding to the Frizzled and LRP. Without the binding of Wnt ligands to LRP and frizzled, β-catenin is phosphorylated by the degradation complex and then tagged for degradation by the proteasome. Upward arrow indicates an increase; downward arrow indicates a decrease. Ub: ubiquitin; P: phosphate. This figure was created with BioRender.com
Given the importance of the Wnt pathway, in different cellular processes, several regulatory systems are involved in its control. Based on their sites of action, the regulators may be divided into extracellular or intracellular antagonists. Most extracellular antagonists are secreted proteins that can interact with either Wnt ligands or Wnt receptors. Examples of extracellular antagonists are DKK1, SOST, WIF-1/2, and SFRPs. DKK1 binds to LRP and the transmembrane protein Kremen 1/2, leading to the internalization and degradation of LRP, thus decreasing the amount of LRP for the binding of the Wnt ligand [76, 77]. Similarly, SOST binds to the co-receptor LRP competing with the binding Wnt protein [78, 79]. However, the WIF-1/2 and SFRPs directly bind and neutralize Wnt proteins, preventing them from binding to the frizzled and LRP complex (Figure 1) [80, 81]. At the intracellular level, the most predominant inhibitors of the Wnt pathway are the β-catenin phosphorylation complex composed of GSK-3β, Axin, and APC. It is believed that APC membrane recruitment protein 1 (AMER1), also called WTX is also involved in the β-catenin destruction complex, however, the exact molecular function is under debate [82]. Finally, at the nucleus, the β-catenin/TCF complex could also be a subject of inhibition [83].
In the “non-canonical” Wnt pathway, the signaling response to Wnt proteins is independent of β-catenin and LRPs. The Wnt PCP pathway uses the receptor tyrosine kinase-like orphan receptor 2 (ROR2), neurotrophin receptor homolog 1 (NRH1), receptor like tyrosine kinase (RYK), or tyrosine-protein kinase-like 7 (PTK7) as co-receptor, instead of LRP5/6 (Figure 2). The binding of the non-canonical Wnt protein to their frizzled receptor acts directly on Dsh to form a complex with Dsh associated activator of morphogenesis 1 (DAAM1), which activates the rhodopsin (Rho, the small G protein) and Rac family small GTPase 1 (RAC1). Rho activates the Rho associated protein kinase (ROCK) involved in the modulation of the cytoskeleton and RAC1 stimulates c-Jun N-terminal kinase (JNK) to activate target gene expression and remodel cytoskeletal actin (Figure 2) [68, 84, 85].
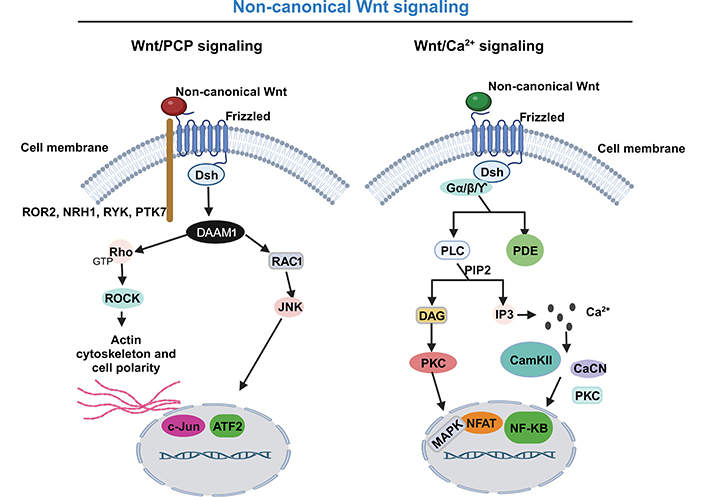
Non-canonical Wnt signaling pathway: a simplified schematic illustration. Activation of the non-canonical Wnt/PCP signaling pathway (left), with non-canonical Wnt ligands interacting with the transmembrane Frizzled protein and uses as co-receptor the ROR2, NRH1, RYK, or PTK7 to recruit the Dsh. They form a complex with DAAM1, which activates both the Rho and RAC1. Rho activates the ROCK and RAC1 to remodel cytoskeletal actin and stimulate JNK to activate target gene expression. Activation of the non-canonical Wnt/Ca pathway (right), with non-canonical Wnt protein binding to their frizzled receptor and acting directly on Dsh and trimeric G protein (Gα/Gβ/GƳ) leading to the activation of phospholipase C (PLC) and PDE. PLC, by phosphatidylinositol 4,5-bisphosphate (PIP2) cleavage, generates inositol triphosphate (IP3) and diacyglycerol (DAG). IP3 stimulates the release of Ca from intracellular stores. Ca release activates several Ca-sensitive enzymes, such as Ca/calmodulin-dependent protein kinase II (CamKII), calcineurin (CaCN), and protein kinase C (PKC), with subsequent activation of cyclic adenosine monophosphate (cAMP) response element binding protein, nuclear factor-kappa B (NF-κB) and nuclear factor of activated T cell (NAFT). Activated NAFT translocate to the nucleus to regulate the expression of target genes. On the other hand, DAG activates PKC and subsequently mitogen-activated protein kinase (MAPK). GTP: guanosine-5’- triphosphate. This figure was created with BioRender.com
The non-canonical Wnt/Ca2+ pathway is the most complex of the three Wnt pathways. This pathway regulates intracellular Ca levels, by releasing intracellular Ca by the endoplasmic reticulum and consequent activation of Ca-sensitive enzymes, which causes the activation of transcription factors. More specifically, the non-canonical Wnt protein binding to their frizzled receptor, and acting directly on Dsh and trimeric Gα/Gβ/GƳ, leads to the activation of PLC and PDE. PLC, by PIP2 cleavage, generates IP3 and DAG. IP3 binds to its receptor in the endoplasmic reticulum and releases Ca from intracellular stores. Ca release activates several Ca-sensitive enzymes, such as CamKII, CaCN, and PKC, and subsequently the activation of cAMP response element binding protein, NF-κB, and NAFT (Figure 2). Activated NAFT translocates into the nucleus to regulate the expression of target genes involved in cell adhesion, migration, and other functional aspects. On the other hand, DAG activates PKC and subsequently MAPKs involved in a variety of physiological processes [68, 72, 84, 86, 87].
The association of the Wnt pathway with bone biology was found through the identification of variants in genes related to the pathway, causing bone diseases (Table 1) [88–98]. Also, several studies using mouse models confirm the importance of the Wnt pathway in bone, in which its activation or inhibition leads to increased or decreased bone formation, respectively [99]. The first evidence of the association of the Wnt pathway with osteoblastogenesis came from the identification of gain or loss of function variants in the LRP5 gene causing high or low bone density, respectively [88, 100]. The gain of function variants in the LRP5 gene affects the ligation of the inhibitors DKK1 and SOST to LRP5 decreasing the inhibition of the Wnt signaling pathway, leading to bone formation [101]. Contrariwise, loss of function variants in the LRP5 gene leads to osteoporosis Pseudoglioma syndrome [online mendelian inheritance in man (OMIM)#259770] with an excessively low bone mass and an excessive risk of developing skeletal fractures [88].
Overview of several genes related in the Wnt signaling pathway associated with diseases marked by bone overgrowth
| Protein activity | Gene | Chr | Protein function in Wnt signaling | Disease (manifestation) | References |
|---|---|---|---|---|---|
| Transmembrane co-receptors | LRP5 | 11 | Co-receptor that works with frizzled protein in the membrane | Endosteal hyperostosis (Worth disease; OMIM#144750) and steosclerosis (high bone mass) | [75, 102] |
| LRP4 | 11 | Related to LRP5/6 in modulation of Wnt/β-catenin pathway. Bind to SOSTDC1, also known as WISE, a family of Wnt antagonists [103] | Sclerosteosis type 2 (OMIM#614305) | [97] | |
| Inhibitors | DKK1 | 10 | Inhibitor of Wnt signaling pathway | DISH (OMIM#106400), OPLL (OMIM%602475), AS (OMIM#106300), and OYL (low levels of DKK1 in humans) | [104] |
| SOST | 17 | SOST by osteocytes is responsible for inhibiting bone formation | Sclerosteosis type 1 (OMIM#269500), Van Buchem disease (OMIM#239100), and Craniodiaphyseal dysplasia (OMIM#122860; high bone mass) | [94, 96, 105] | |
| SFRP4 | 7 | Secreted protein which binds to Wnt ligands and inhibits Wnt signaling | Pyle’s disease (OMIM#265900) | [106–108] | |
| WTX/AMER1 | X | Encodes an intracellular repressor of canonical Wnt signaling | Osteopathia striata (OMIM#300373; high bone mass) | [91] |
Chr: chromosome; SOSTDC1: SOST domain-containing protein 1; DISH: diffuse idiopathic skeletal hyperostosis; OPLL: ossification of the posterior longitudinal ligament; AS: ankylosing spondylitis; OYL: ossification of yellow ligament
Another player of the Wnt pathway is LRP4 which, unlike LRP5/6, is involved in the inhibition of Wnt through interaction with SOST protein. Variants in the LRP4 gene may cause sclerosteosis type 2 (OMIM#614305), a severe disease characterized by progressive bone overgrowth (Table 1). Due to the compromised binding of LRP4 and SOST in this disease, serum levels of SOST are elevated as well as the activity of the Wnt pathway in osteoblasts and consequently increased bone formation [97].
Several evidences show that Wnt signaling is involved in various diseases characterized by ectopic ossification. Niu et al. [104] found that higher levels of OCN and SOST and lower levels of DKK1 are correlated to the ectopic ossification observed in OPLL (OMIM%602475), AS (OMIM#106300), DISH (OMIM#106400), and OYL diseases. According to the authors, the high levels of SOST are counterbalanced by underproduction of DKK1. An interesting study using a mice model shows that DKK1 blockade stimulates the expression of collagen type X, induces the formation of hypertrophic chondrocytes and promotes ankyloses of sacroiliac joints. In these animals was a subsequent enhancement of the Wnt pathway with new bone formation and ankyloses, suggesting that Wnt signaling has a role in new ectopic bone formation in the axial skeleton [109]. To support this hypothesis, SOST and DKK1 gene expression were found to be downregulated in the spine of a mouse model of AS [110]. Another study investigated a positive relationship between DKK1 inhibitor and excessive ossification in DISH and the authors showed that lower levels of DKK1 were associated with more severe spinal involvement [111]. Diarra et al. [112] reported that low expression of DKK1 leads to bone formation and high levels of bone loss. According to the authors, DKK1 inhibitor plays an important function in bone remodeling. According to previous studies, DKK1 inhibitors play important roles in the control of the Wnt pathway; however, variants in the DKK1 gene are not described in the literature as associated with monogenetic bone disorders [101]. Several evidences point to the fact that the alterations in DKK1 levels, found in some skeletal diseases, are not genetically determined, but probably caused by posttranscriptional changes [113]. In fact, one study performed by Daoussis et al. [114], found that functional levels of DKK1 are decreased in AS patients, suggesting that DKK1 in these patients is dysfunctional and may contribute to ectopic bone formation. The SOST gene is a crucial inhibitor of the Wnt pathway, and its production in bone is performed by mature osteocytes. Loss of function variants in the SOST gene are found in patients with sclerosteosis type 1 (OMIM#269500), a severe disease with progressive skeletal overgrowth [94]. In addition, deletion of 52 kilobase in the same gene results in reduced transcription of SOST [115], responsible for causing a very similar and related disease, the van Buchem disease (OMIM#239100) [96]. Both diseases, sclerosteosis, and van Buchem disease, result from osteoblasts hyperactivity and both show autosomal recessive inheritance [116]. In both diseases, in order to compensate for the loss of SOST, an upregulated of DKK1 expression is verified, which is not sufficient to prevent enhanced bone formation, but, it may prevent ectopic calcification [117–119]. Another disease caused by SOST variants is craniodiaphyseal dysplasia (OMIM#122860), a rare autosomal dominant inherited disease, marked by generalized hyperostosis and sclerosis. Kim et al. [105] found in two children with craniodiaphyseal dysplasia two severe variants affecting the secretion signal of the SOST gene. Variants in the SFRP4 gene, another inhibitor of the Wnt pathway, have also been shown to be disease-causing in Pyle’s disease (OMIM#265900), a metaphyseal dysplasia with autosomal recessive inheritance pattern [106–108]. As already mentioned, SFRP4 binds to Wnt ligands and inhibits Wnt signaling activation. Kiper et al. [108], showed that the knockout mice of SFRP4 manifest the same phenotype observed in Pyle’s disease. Osteopathia striata (OMIM#300373) is another disease caused by inactivating mutations in another mediator of the Wnt pathway, the AMER1 or WTX gene, an intracellular inhibitor of Wnt. The disease is X-linked and is virtually lethal in male patients [91].
Several studies have investigated the role of epigenetics in modulating Wnt signaling, suggesting that miRNAs have a role in osteoblast differentiation and bone formation. miRNA are non-coding small RNA molecules responsible for regulating post-transcriptionally the expression of several genes. miR-29a seems to activate bone formation by enhancing Wnt signaling [120], maybe by downregulating the expression of the DKK1 gene [121]. Similarly, Zhang et al. [122] found that the DKK1 gene was downregulated via a 3’ untranslated region (3’ UTR) by the miR-335-5p. As a result of decreased DKK1 protein level, by miR-335-5p, the Wnt pathway is enhanced and promotes osteogenic differentiation and bone formation. In the AS mice model, the miR-96 is increased and seems to promote osteoblast differentiation and bone formation, via Wnt signaling activation, through downregulation of the SOST gene [123]. Another miRNA that interacts with Wnt signaling is miR-27, which inhibits the expression of APC, a player in the degradation complex of β-catenin. This inhibition favors the activation of Wnt signaling and bone formation [124]. In addition, the non-canonical Wnt through Rho could be able to mediate osteogenic differentiation induced by mechanical loading in tendon-derived stem cells, which might contribute to tendon ossification [125].
DNA methylation is also an important epigenetic mechanism that plays a role in the regulation of bone formation. This mechanism is characterized by the introduction of a heritable epigenetic mark involving a reversible modification of cytosines in the context of CpG dinucleotides by DNA methyltransferases [126]. It was shown that DNA methylation modulates SOST gene expression, a potent inhibitor of the Wnt pathway. A decreased methylation of the SOST gene promoter is verified during the differentiation of osteoblasts to osteocytes, thus allowing osteocyte-specific expression of SOST [127]. Another evidence of the involvement of DNA methylation in regulating bone formation comes from a study with osteoporosis patients which showed increased methylation of the SOST gene promoter accompanied by a decreased level of serum SOST [128].
Hedgehog (HH) signaling pathway is another pathway that has been implicated in the process of new bone formation [113]. In vertebrates, the HH family consists of three ligands: Indian HH (IHH), sonic HH (SHH), and desert HH (DHH). Despite that all three ligands function through the same pathway and sometimes with overlapping functions, IHH seems to be more implicated in chondrocyte differentiation and SHH is more responsible to induce the formation of osteoblasts [129–131]. IHH seems to be involved in both endochondral and intramembranous ossification. In endochondral ossification, IHH is essential for osteoblast differentiation and survival in the perichondrium [90], by inducing the expression of RUNX2, essential for osteoblastogenesis [91]. IHH ligands are produced by prehypertrophic and hypertrophic chondrocytes and are considered the main players in endochondral ossification [92, 93].
The HH signaling pathway includes HH ligands, the patched (PTCH) receptor, the signal sensor smoothened (SMO) receptor, the suppressor of fused (SUFU), and transcription factors glioma-associated oncogenes homologs (GLIs). The HH signaling pathway can be in active or inactive form, and its control is mediated by the presence or absence of HH ligands, respectively. Normally, the responsive transcription in HH signaling is performed downstream by GLI-mediated transcription factors (GLI1, GLI2, and GLI3), which act as terminal transcription factors of the HH signaling pathway. GLI1 and GLI2 are transcriptional activators of the HH pathway and GLI3 acts as a repressor [89]. GLI1 is a critical transcriptional activator that acts on the HH pathway and is one of the HH target genes [89, 90]. Specifically, in the presence of HH ligands, such as SHH, DHH, or IHH, occurs the binding to the PTCH transmembrane receptor and consequently, the SMO inhibition by PTCH is abolished [132]. Active SMO is released, and in the primary cilium, recruits the GLI/SUFU/Kinesin family member 7 (KIF7) complex resulting in the detachment of GLI from SUFU and KIF7. The detachment of active GLI permits its entrance to the nucleus to promote the transcription of HH target genes, crucial to cellular differentiation, migration, and proliferation (Figure 3) [133–135]. One of the main target genes in HH signaling is PTH-related protein, which is essential to promote the proliferation of chondrocytes during endochondral ossification [136]. On the other hand, in the absence of HH ligands, the PTCH represses the activity of SMO, which is located in the intracellular endosome. The GLI/SUFU/KIF7 complex recruits kinases, such as casein kinase I (CKI), PKA, and GSK3, leading to GLI phosphorylation and consequently promoting the processing of the repressor form of GLI (GLIR). GLIR translocates to the nucleus to inhibit the transcription of HH target genes, and in this case, the pathway remains in its inactive form (Figure 3) [134, 137, 138].
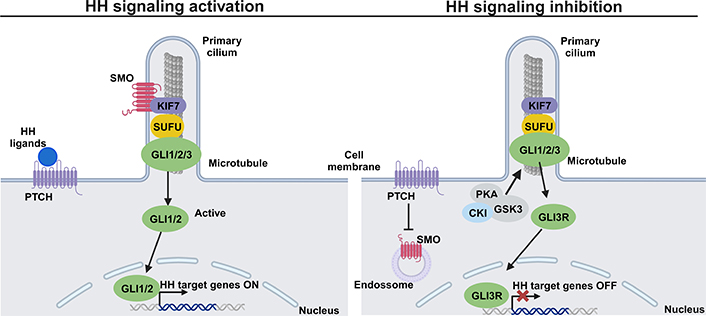
HH signaling pathway: a simplified schematic illustration. Activation of the HH signaling pathway (left) with binding of HH ligands to the transmembrane PTCH at the plasma membrane. This binding abolishes SMO inhibition by PTCH and allows the accumulation of SMO in the primary cilium. Active SMO recruits the GLI/SUFU/KIF7 complex resulting in the detachment of GLI1/2 from KIF7 and SUFU. GLI1/2 translocates into the nucleus and goes on to activate the transcription of HH target genes. Inhibition of the HH signaling pathway (right) by PTCH inhibiting SMO, prevents its entry into the primary cilium. Some kinases, such as PKA, GSK3, and CKI are recruited by the complex GLI/SUFU/KIF7 and phosphorylates GLI, and consequently, the repressor GLI3R is formed. GLI3R translocates to the nucleus to block the transcription of HH target genes. This figure was created with BioRender.com
HH pathway is essential in cartilage and bone growth, and its disturbance has been confirmed in several bone disorders, such as osteoarthritis, heterotopic ossification, bone tumors, and others [139]. Specifically, the local aberrant activation of HH signaling participates in the pathophysiology of heterotopic ossification [140]. In a study using a mice model of osteoarthritis, the authors demonstrated that the increased HH signaling is positively related to the severity of osteoarthritis. Importantly, pharmacological inhibition of HH signaling in this model led to attenuation of osteophyte formation and osteoarthritis severity. In human osteoarthritic samples, the authors also showed an enhanced activation of HH signaling [141]. Another evidence of the importance of the HH signaling pathway in pathological bone formation comes from a mouse model of inflammatory arthritis. In these mice, the authors used a blockage of SMO, a key component of HH signaling, and found a reduction of osteophyte formation in arthritis compared with mice without blockage of SMO. In addition, the authors found that the SMO blockage did not affect inflammation, showing that HH signaling is responsible for new bone formation in the context of inflammatory arthritis [142]. One more evidence, showing the importance of the HH signaling pathway in new bone formation, comes from a mouse model genetically modified to have increased HH signaling pathway activity at spinal chondrocytes. These mice developed a fusion of their spine (ankylosing spine) without any signs of inflammation, showing once again that new bone formation and inflammation are two independent processes [143]. Daoussis et al. [144], reported that patients with AS have higher serum levels of IHH secreted factor compared with healthy controls, suggesting that IHH may be involved in new bone formation in these patients. Also, in human cervical OPLL, the overexpression of HH signaling (IHH) promotes atypical chondrocyte differentiation and enhances new bone formation [145].
In progressive osseous heteroplasia (POH; OMIM#166350), a human disease characterized by extensive heterotopic ossification in soft tissues (Table 2), the HH pathway is also dysregulated by loss of function variants in the guanine nucleotide binding protein (G Protein) gene (GNAS). GNAS encodes the alpha-subunit of the stimulatory G protein (Gαs), an activator of kinase PKA, which is an inhibitor of HH signaling [146–148]. Regard et al. [148] in a study using a GNAS-/- mice model showed that HH signaling was up-regulated in ectopic osteoblasts and progenitor cells, while Wnt-β-catenin was decreased [148]. However, gain of function variants in GNAS upregulated Wnt-β-catenin signaling in fibrous dysplasia (OMIM#174800), showing that Gαs is a critical regulator of osteoblasts differentiation by maintaining the balance between the Wnt-β-catenin and HH signaling pathways [149]. In normal soft tissues, Gαs is the main responsible for maintaining HH signaling suppressed to prevent bone formation [150].
Overview of several genes related in HH signaling pathway associated with diseases marked by bone overgrowth
Another bone disease associated with the HH signaling pathway is Gorlin syndrome (OMIM#109400), characterized by multiple tumor formation and skeletal abnormalities, including ectopic ossification, polydactyly, and rib anomalies. The disease is caused by variants in the PTCH1 gene, which encodes the transmembrane protein PTCH, directly involved in the inhibition of the HH signaling pathway (Table 2) [152–154]. Ohba et al. [155], demonstrated that deficient PTCH+/- mice showed an accelerated osteoblast differentiation with consequently high bone mass due to an increased bone formation. Additionally, the authors verified a reduction of the repressor transcription factor GLI3 in osteoblast precursor cells.
As verified in the Wnt signaling pathway, a variety of miRNAs could also be involved in the osteogenesis process mediated by HH signaling. In one study using human umbilical mesenchymal stem cells, the miR-342-3 was reported to down-regulate SUFU to activate the SHH pathway and consequently accelerate osteogenic differentiation [156]. It is also reported that miR-342-3 overexpression, by activating the HH signaling, upregulates the expression levels of ALP, RUNX2, and OPG, all of them important in the osteogenic process [157]. Otherwise, the miR-467g seems to be an inhibitor for osteoblast differentiation that could downregulate the osteogenesis progression by targeting IHH/RUNX2 signaling. The authors also found that miR-467g silencing led to a significant increase in new bone formation [158].
A third important pathway is the transforming growth factor beta (TGFβ) signaling pathway which is involved in several cellular processes and plays important roles throughout life, including the regulation of bone homeostasis [159]. It is well known that TGFβ has essential roles in coordinating the bone remodeling process [2, 160]. TGFβ superfamily has several members, such as TGFβs, nodal, activin, and BMPs [161]. TGFβ and BMP members have several functions in bone development through regulating the maintenance of postnatal bone and cartilage. Due to the great complexity, this paper will only summarize the general process involving the TGFβ and BMP members in bone, with occasional references to the other members.
TGFβ secreted proteins are synthesized as a latent form and are composed of mature TGFβ and latency-associated protein (LAP) [162]. The latent TGFβ ligand is stored in the ECM and its activation depends on osteoclast bone resorption [160]. At the cell surface, the secreted active ligand binds to the complex of two signaling receptors (type II and I) and transduce signals to both the canonical mothers against decapentaplegic homolog (SMAD)-dependent signaling pathway and the non-canonical SMAD independent signaling pathway (Figure 4). In human cells, at least five type II and seven type I receptors can be expressed and generate receptor complexes that function with all the ligands involved in the TGFβ family [163]. Specifically, in the canonical SMAD-dependent signaling pathway, following a TGFβ binding (or other family members such as activins and nodal), the type II receptor phosphorylates and activates the type I receptor, which in turn activates both SMAD2 and SMAD3 proteins by phosphorylation events. In the case of BMPs, three other SMAD proteins are activated, SMAD1, SMAD5, and SMAD8 [164]. The group of SMAD proteins, which are activated by a TGFβ receptor, are usually designated by receptor activated SMADs (R-SMADs). Activated R-SMADs dissociates from the receptor type I and form a trimeric complex with the mediator SMAD4 in cytoplasm. The trimeric complex enters into the nucleus, where it associates with transcription factors and chromatin modifiers proteins, to regulate the transcription of TGFβ/BMP target genes (Figure 4). BMP signaling mediated by SMADS regulates the expression of transcription factors (RUNX2, osterix, Msh homeobox 2, distal-less homeobox5/6, and SOX9) [165], which are essential for chondrogenesis and osteoblastogenesis [166]. Several mediators are implicated in the regulation of TGFβ/BMP signaling. The SMAD6 and SMAD7 proteins are intracellular mediators with the ability to prevent R-SMAD activation and consequently the SMAD signaling propagation. SMAD6/7 recruits the specific E3 ubiquitin ligases 1 (SMURF1) and SMURF2 to the type I receptor, promoting its ubiquitin/proteasome degradation [167–170]. Into the nucleus, SMAD7 could also inhibit the transcriptional activity of the SMAD complex, by binding on the regulatory sequences of TGFβ target genes [161]. TGFβ signaling could also be inhibited or modulated by BMP and activin membrane-bound inhibitor (BAMBI) which is a transmembrane pseudo-receptor that resembles the TGFβ type I receptors, but with an absent intracellular kinase domain, which is required for signaling activation. Specifically, BAMBI can associate with TGFβ family receptors blocking its function by preventing phosphorylation of the receptor and consequent inhibition of BMP and TGFβ ligands [171]. BAMBI can also form a complex to the intracellular inhibitor SMAD7 to inhibit the interaction between type I receptor and R-SMADs blocking the activation of the SMAD signal transduction pathway [172].
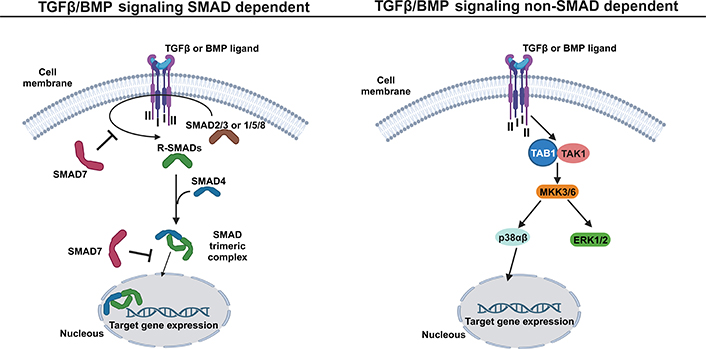
TGFβ/BMP signaling pathway: a simplified schematic illustration. In SMAD dependent pathway (left), the TGFβ family members, such as TGFβ and BMP, bind to the tetrameric receptor complex formed by two type II and two type I receptors, allowing the activation of SMAD 2 and 3 (in the case of TGFβ ligand) or SMAD 1/5 and 8 (in case of BMP ligand). The active R-SMADs then interact with SMAD4, forming an SMAD trimeric complex that enters into the nucleus to activate the expression of TGFβ or BMP target genes. The mediator SMAD7 has the ability to prevent the R-SMAD activation at the cytoplasm level and inhibit the transcriptional activity of the SMAD trimeric complex in the nucleus. In the non-SMAD dependent pathway (right), the binding of TGFβ family members, such as TGFβ and BMP, to the tetrameric receptor complex allows the direct activation of TGFβ-activated kinase 1-binding protein 1 (TAB1) and TGFβ-activated kinase 1 (TAK1), which initiates the MAP kinase kinases (MKK)-p38 or MKK- extracellular signal-regulated kinase 1/2 (ERK1/2) MAPK cascade to induce the expression of TGFβ or BMP target genes. This figure was created with BioRender.com
Since TGFβ and BMP have important roles in cartilage and bone homeostasis, dysregulation in these signaling pathways is frequently associated with multiple diseases. A number of human genetic variants in genes related to the TGFβ/BMP signaling pathway have been identified in several inheritable bone diseases [173–179]. Gain of function variants in the TGFβ1 gene, especially located in the LPA region of TGFβ1, are causative of Camurati-Engelmann disease (CED; OMIM#131300), an autosomal dominant disease characterized by generalized hyperostosis affecting the skull and the long bones of the arms and legs [180, 181]. Normally, the non-covalent binding of LAP to the active TGFβ1 is important to keep TGFβ1 inactive in the bone matrix. However, in this disease, the release of active TGFβ1 from the bone matrix is increased. Importantly, it was found that after bone resorption, the TGFβ1 is essential for the migration and proliferation of osteogenic cells to the bone region. This shows that due to CED variants, the release of active TGFβ1 from the bone matrix results in disturbed bone remodeling with increased bone turnover [2]. Since the importance of the TGFβ pathway in regulating bone metabolism, several players of this pathway have been extensively studied in several other ectopic bone diseases, such as OPLL. It was verified, in vivo, that TGFβ is present in the ossified matrix and chondrogenic areas of the posterior longitudinal ligament, but it is absent in the non-ossified ligament [179, 182]. The single nucleotide polymorphism (SNP) with the reference SNP identification rs1982073, located in exon 1 of the TGFβ1 gene, is considered a risk factor for genetic susceptibility to OPLL [183]. However, one study found no association between this variant and the occurrence of OPLL [184], and another study found that the C allele seems to be related to the area of the ossified lesion [185]. TGFβ3 gene is another gene presenting two intronic variants (rs226862 and rs22847) with significant association with OPLL disease [184]. In the same way, variants in TGFβ receptors seem to be associated with OPLL disease. Three variants in the TGFBR2 gene seem to be associated with OPLL; however, the exact mechanism is unknown [186]. Variants in genes involved in the inhibition of the TGFβ pathway were also found. LEM domain containing 3 (LEMD3, also called MAN1) gene encodes a nuclear membrane protein with the ability to bind to SMAD proteins and consequently promote inhibition of the TGFβ signaling pathway [1, 9]. Loss of function variants in LEMD3 is the cause of osteopoikilosis (OMIM#166700), an autosomal dominant inherited disease, characterized by increased bone density with sclerotic lesions in one or several bones [187]. In some cases, other tissues, such as skin and muscles, are also affected [1, 9]. One study performed, based on linkage analysis, tried to find an association between LEMD3 variants with both DISH and chondrocalcinosis, but none of the variants were segregated within the studied families [188].
BMP growth factors stimulate chondrocyte and osteoblast differentiation, resulting in cartilage and bone formation [189]. Variants in the BMP2 gene could induce thoracic ossification of the ligamentum flavum in Han Chinese populations. Subsequently, the same authors showed, by functional analysis, that the BMP2 variants upregulate the BMP2 gene expression and promote osteogenic differentiation [175]. Also, a significant expression of BMP ligands and their receptors in ossified and chondrogenic regions has been noted in OPLL and thoracic ossification of the ligament flavum [178, 179].
In fibrodysplasia ossificans progressiva (FOP; OMIM#135100), the gain of function variants in the activin receptor A type 1 (ACVR1)/activin receptor-like kinase-2 (ALK2) gene, which encodes a BMP receptor type I, designated as ACVR1, causes dysregulation of BMP signaling with resulting ectopic endochondral ossification (Table 3) [177]. In FOP disease, the BMP signaling is dysregulated, keeping BMP signaling activated when it should be inactivated [176].
Overview of several genes related to TGFβ/BMP signaling pathway associated with diseases marked by bone overgrowth
| Gene | Chr | Protein function in the TGFβ/BMP pathway | Disease (manifestation) | References |
|---|---|---|---|---|
| TGFβ1 | 19 | Encodes a ligand involved in the activation of the TGFβ pathway | CED (OMIM#131300) | [180] |
| ALK2/ACVR1 | 2 | Encodes the type I BMP receptor | FOP (OMIM#135100) | [177] |
| LEMD3 | 12 | Encodes a nuclear membrane protein that can inhibit the TGFβ pathway | Osteopoikilosis; Buschke-Ollendorff syndrome (OMIM#166700) | [190] |
Several evidences show that epigenetic modifications could also control TGFβ and BMP signaling pathways. For example, miR-133 targets RUNX2 and SMAD5 genes to inhibit osteogenesis induced by BMP2 [191]. RUNX2 is one of the main early BMP response genes essential for bone formation. Also, miR-30 family members target RUNX2 and SMAD1 genes to negatively regulate osteoblast differentiation induced by BMP2. Interestingly, the overexpression of miR-30 leads to a decrease in ALP activity, and the opposite increases its activity, showing that miR-30 could be an important regulator during the biomineralization process [192]. The binding of miR-542-3p to the BMP7 gene inhibits bone formation by suppressing osteoblast proliferation and differentiation induced by BMP7. However, the inhibition of miR-542-3p seems to promote the expression of osteoblast-specific genes and increase the activity of ALP and matrix mineralization [193]. On the contrary, the miR-20a is involved in promoting osteogenic differentiation by up-regulating BMP and RUNX2. The miR-20a targets negative regulators of BMP signaling, such as BAMBI, peroxisome proliferator activated receptor gamma (PPARγ), and cysteine rich transmembrane BMP regulator 1 (CRIM1) [194].
In bone, canonical Wnt/β-catenin functions as a mediator of two other important pathways involved in bone formation, the HH [129], and BMP signaling pathway [195]. Wnt/β-catenin signaling increases BMP2 expression levels, promoting BMP signaling [196]. Wnt/β-catenin and BMP pathways work together to regulate osteoblast differentiation and bone formation. Both pathways contribute to chondrocyte hypertrophy by regulation of the co-receptor LRP5 [197]. The Wnt and HH signaling, control the proliferation and differentiation of osteoblasts/chondrocytes. The Wnt signaling is enhanced in mature osteoblasts and HH is enhanced in immature osteoblasts [129]. It is well known that the cooperation of Wnt and HH signaling induces osteogenesis in POH disease, causing heterotopic ossification [148]. In bone, HH signaling could function both individually as well as cooperating with other signaling pathways, such as Wnt, BMP, and PTH-related protein [198, 199].
The current knowledge of new bone formation is improving; however, much is yet to be disclosed. It is assumed that heterotopic bone formation is triggered by the same mechanisms occurring in normal bone formation. Due to the great complexity of signaling pathways involved in bone formation, only the three main pathways involved in osteogenesis were described in this review paper. It is now well established that Wnt, HH, and TGFβ/BMP signaling pathways are connected and regulate each other, in the early stages of osteogenesis and osteoblast maturation. Rare and severe genetic bone disorders have contributed enormously to the knowledge of the already-mentioned signaling pathways. Besides disruptions or interferences in these pathways, the study of other biological mechanisms involved, such as inflammation, hypoxia, and mechanical factors, seem fundamental. Animal models have also been a valuable resource and will probably keep its importance in the future. Concerning the existing therapeutic approaches, disorders characterized by a decrease in bone density are clearly benefited from several options in use. The use of MVs for therapeutic purposes, for bone regeneration or treatment of bone pathologies, is still embryonic but it is already encouraging. Disorders characterized by “milder” heterotopic ossification/calcification are still far from being disclosed. In this group of disorders, we can include OPLL, DISH chondrocalcinosis, and even AS, all without any clear conclusions besides the complex nature of their genetic backgrounds. Collaborative efforts and a large number of samples are necessary to overcome the obstacles. In the future, it is also very important to explore the factors that trigger osteogenesis in the context of new bone formation. For instance, some pathways are mechanosensitive and can be activated by mechanical loading leading to new bone formation.
ACVR1: activin receptor A type 1
ALP: alkaline phosphatase
AMER1/WTX: adenomatous poliposis coli membrane recruitment protein 1
APC: adenomatous poliposis coli
AS: ankylosing spondylitis
Axin2: AXIS inhibition protein 2
BAMBI: bone morphogenetic protein and activin membrane-bound inhibitor
BMP: bone morphogenetic protein
Ca: calcium
CaCN: calcineurin
CamKII: calcium/calmodulin-dependent protein kinase II
cAMP: cyclic adenosine monophosphate
Cbfa1: core-binding factor alpha 1
CED: Camurati-Engelmann disease
CKI: casein kinase I
DAAM1: Dishevelled associated activator of morphogenesis 1
DAG: diacyglycerol
DISH: diffuse idiopathic skeletal hyperostosis
DKK1: dickkopf-related protein 1
Dsh: Dishevelled
ECM: extracellular matrix
FOP: fibrodysplasia ossificans progressiva
GLIR: repressor form of glioma-associated oncogenes homolog
GLIs: glioma-associated oncogenes homologs
GNAS: guanine nucleotide binding protein (G protein) gene
GSK3β: glycogen synthase kinase-3beta
Gαs: alpha-subunit of the stimulatory G protein
HA: hydroxyapatite
HH: hedgehog
IHH: Indian hedgehog
IP3: inositol triphosphate
JNK: c-Jun N-terminal kinase
KIF7: Kinesin family member 7
LEF: lymphoid-enhancer binding factor
LEMD3: LEM domain containing 3
LRP5: low-density lipoprotein receptor-related protein
MAPK: mitogen-activated protein kinase
miRNAs: microRNAs
MKK: MAP kinase kinases
MVs: matrix vesicles
NAFT: nuclear factor of activated T cell
NF-κB: nuclear factor-kappa B
NRH1: neurotrophin receptor homolog 1
OCN: osteocalcin
OMIM: online mendelian inheritance in man
OPG: osteoprotegerin
OPLL: ossification of the posterior longitudinal ligament
OPN: osteopontin
PCP: planar cell polarity
PDE: phosphodiesterase
PHOSPHO1: phosphoethanolamine/phosphocholine phosphatase 1
Pi: inorganic phosphate
PIP2: phosphatidylinositol 4,5-bisphosphate
PiT1: inorganic phosphate transporter 1
PKC: protein kinase C
PLC: phospholipase C
POH: progressive osseous heteroplasia
PPi: inorganic pyrophosphate
PTCH: patched
PTH: parathyroid hormone
PTK7: protein kinase-like 7
RAC1: Rac family small GTPase 1
Rho: rhodopsin
ROCK: rhodopsin associated protein kinase
ROR2: receptor tyrosine kinase-like orphan receptor 2
R-SMADs: receptor activated mothers against decapentaplegic homologs
RUNX2: runt-related transcription factor 2
RYK: receptor like tyrosine kinase
SFRP: secreted frizzled-related protein
SHH: sonic hedgehog
SMAD: mothers against decapentaplegic homolog
SMO: smoothened
SOST: sclerostin
SOX9: SRY-box transcription factor 9
SUFU: suppressor of fused
TCF: T-cell specific transcription factor
TGFβ: transforming growth factor beta
TNAP: tissue non-specific alkaline phosphatase
WIF: wingless/integrated inhibitor factor
WNT: wingless/integrated
BP: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. ARC and LP: Writing—review & editing. JBA: Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Iris Eshed
Netanja I. Harlianto ... Jorrit-Jan Verlaan
Fernando Pérez-Ruiz ... Ana María Herrero-Beites
David Kiefer ... Xenofon Baraliakos
Fabiola Atzeni ... Reuven Mader
Greta Pellegrino ... Piercarlo Sarzi-Puttini
