
Abstract
Soft tissue sarcoma (STS) is a rare malignancy with a high incidence. Early diagnosis can reduce the rate of amputations and increase survival, however, this is typically delayed. The diagnosis and treatment of smaller lesions have a better prognosis; nonetheless, patients present to physicians when the soft tissue mass is large with obvious signs of red flags. In addition, the symptoms of this disease are highly non-specific and overlap greatly with benign conditions, resulting in a lack of clinical suspicion and low awareness among practitioners and the general public. Thusly, it is entitled as “the loneliest cancer”. This can make an accurate diagnosis difficult, with a great proportion of misdiagnoses leading subsequent inadvertent to incomplete STS excision, affecting the overall prognosis of the disease and devastating consequences in the disease process. A timely and precise diagnosis is essential because half of people with STS progress toward quietly aggressive illness. The purpose of this review is to raise awareness of STSs so that early recognition, accurate work-up, overview of conventional treatment plans, and appropriate referral to a tumor center can be achieved, avoiding whoop situations, and improving patient outcomes. In addition, insight into the advances in immunotherapy, nanotechnology, and artificial intelligence (AI) can lead to STS diagnosis and treatment prognosis.
Keywords
Soft tissue sarcoma, soft tissue lump, referral system, biopsy, diagnosis, treatmentsIntroduction
Sarcomas are a rare malignancy that is derived from the mesenchymal origin and is broadly divided into soft tissue sarcomas (STSs) and bone sarcomas [1]. STSs are cancers that develop in the connective and supporting tissues of the body such as fats, muscles, blood vessels, nerves, cartilage. The most common subtype of STSs is liposarcomas and leiomyosarcomas [2].
The World Health Organization (WHO) 5th edition of soft tissue and bone tumors recognizes more than 100 histological types that are named according to the tissue they most closely resemble (Table 1) [3, 4]. In an analysis of patients with STS it was found that altogether the extremities constitute approximately 50–70% of all STS, with most cases occurring in the thighs due to its volume of soft tissue [4, 5], followed by trunk wall (10%), and retroperitoneum (10%; Figure 1) [6]. STS in the extremities occurs in 6–15% paediatric (< 15 years) and 5% approximately in the adult and young population (15–29 years).
Common STSs and benign tumors
| Common STSs | Common benign tumors that are not cancers, but can start in soft tissue |
|---|---|
|
|
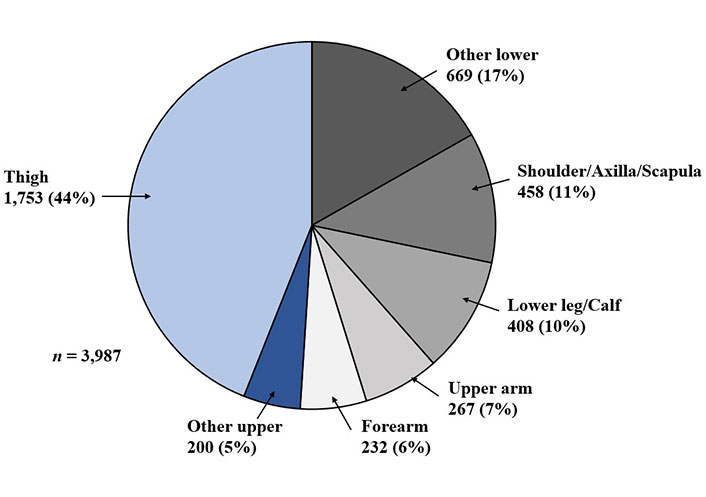
In addition, it was found that the peak age of STS was generally 40–60 years [7]. The aetiology of STS is mostly unknown but is thought to be sporadic in nature with a minor contribution from environmental influences, irradiation, viral infections, immunodeficiency and genetic susceptibility. The largest environmental contributor is exposure to ionizing radiation accounting for only 3–6% of all sarcomas [8].
Epidemiology and incidence of STSs around the world
STSs are rare neoplasms that make up less than 1% of all malignancies [9, 10]. When describing disease burden, incidence is a more appropriate indicator than prevalence as it describes the number of new cases of STS.
In Europe, rare cancers account for approximately 24% of all cancers [8]. Although STS represents less than 1% of all cancers, it is still one of the rare cancers with the highest incidence rate. In Italy, there was a total of 4,072 new cases of STS estimated for the entire country in 2015, with STS accounting for approximately 80% of all sarcomas (4,072 of 4,957). In addition, the incidence of STS in women was found to be slightly greater than that of men because of gynaecological uterine sarcomas and breast sarcomas [8]. In the United Kingdom (UK), approximately 3,300 cases of STS have been reported in 2021 with 13,500 new cases being reported in the United States at this time [11]. Furthermore, China had approximately 39,900 new STS cases nationwide in 2014, accounting for 1.05% of their overall cancer incidence [12].
The most common anatomical sites for STS are extremities 50–70%, followed by trunk wall and retroperitoneum with other sites representing less than 5%. Malignant tumors of the retroperitoneum are approximately four times more frequent than benign lesions. Retroperitoneal tumors are often much larger before they become symptomatic and tend to present at a later stage, resulting in a lower survival rate than tumors of the extremities. Retroperitoneal sarcoma (RPS) tends to be more locally aggressive, with high rates of local and metastatic recurrence, and therefore, poor overall long-term survival. As surgical resection is the only curative therapy for RPS, the grade and extent of surgical resection with a negative margin are the most important prognostic factors [13].
Clinical features of soft tissue masses that require urgent investigation
The clinical symptoms observed in patients with STS are nonspecific, with the most common finding being a painless, gradually enlarging mass with site-dependent symptoms of increased pressure, such as paraesthesia and distal edema. “Red flag symptoms” are the clinical features of soft tissue masses that require urgent investigation. A greater risk of malignancy is associated with a greater number of clinical features [11]. UK guidelines suggest that any lump greater than 5 cm is growing, is located deep in the body, is painful, and should be considered malignant until proven otherwise [14]. This is because 86% of tumors that meet these criteria are malignant [15].
Red flags symptoms for soft tissue masses:
Increasing in size.
Mass > 5 cm.
Deep lying mass.
New onset pain.
Mass recurrence.
Firmer than surrounding tissues.
Signs and symptoms of local infiltration.
Challenges to clinical recognition and acknowledgment to people
The rare and heterogeneous nature of STS is the cause of the poor prognosis of which there is little knowledge to the general people. There is extensive overlap in presentations between benign and malignant tissue masses, as well as a wide range of lesion sizes, presentations, and ages of affected individuals. Being rare, not often recognized and far from fully understood, it is referred to as “the loneliest cancer”. Patients with STS were more likely to be treated for another condition or counselled that their symptoms were not concerning [16].
Benign soft-tissue masses outnumber malignant tumors by approximately 150:1, with 20 malignant soft-tissue masses per 1 million people in the United States [16]. Although both kinds of lesions are frequently painless, it is important to distinguish between benign or malignant and determine whether it requires strategic treatment plans. Often benign lesions can also give rise to considerable morbidity that requires surgical interventional.
The best individual indicator of increased malignancy risk is an increase in tumor size. An accurate differential diagnosis can be obtained after a comprehensive history and physical examination, including examination of the site, size, shape, contour, consistency, tenderness and tethering [17]. Appropriate imaging and histological analysis will further assist in confirming this diagnosis.
Multidisciplinary and collaborative approach
Treatment for STS is best conducted in a multidisciplinary setting as this allows for optimal patient management, including consideration of preoperative induction treatment, discussion of possible reconstructive strategies, neo-adjuvant/adjuvant measures, and planning of rehabilitation. This will require adequate communication and collaboration of interdisciplinary decisions by all members, including primary physicians or surgeons, orthopaedic surgical oncologists, pathologists experienced in soft tissue and bone tumors, onco-radiologists, pediatric oncologists, radiation experts, rehabilitation specialists, nurse specialists, and social workers.
As STS is broad in nature and can be located anywhere in the body, general practitioners or surgeons from any specialty may be the first line of contact for patients presenting with STS, and are essential for their accurate identification and diagnosis (Figure 2). Experienced radiologists and pathologists are required to accurately diagnose STS features through imaging and histopathology and to provide suggestions for further evaluation. After the diagnosis of STS is confirmed, the patient must be referred to a center providing interdisciplinary multimodal care. They will undergo appropriate staging by a trained oncologist and discuss a continuing multimodal treatment plan. The multimodal approach to treatment begins with sufficient surgical resection of the tumor performed by an experienced surgeon (Figure 3). Reconstruction with the goal of unimpaired wound healing, general rehabilitation, and early initiation of postoperative radiation treatment will follow.
Illustrative figure showing appearances of suspicious lumps on different parts of the body
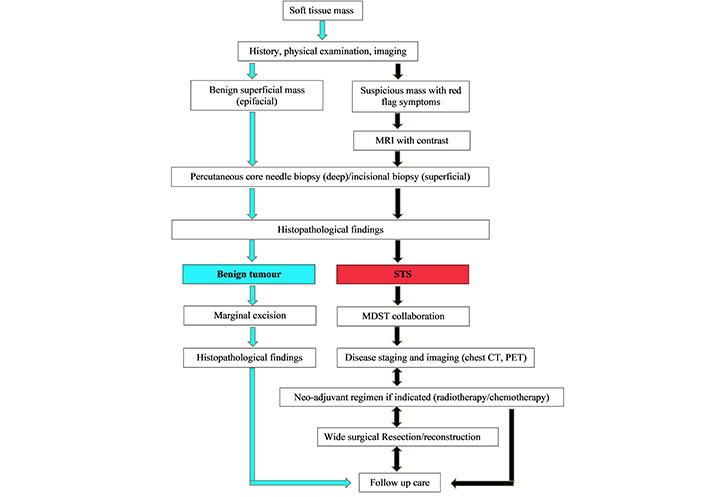
The algorithm for diagnosis of soft tissue tumors and overview of multimodal treatment strategy. MRI: magnetic resonance imaging; MDST: multidisciplinary sarcoma team; CT: computed tomography; PET: positron emission tomography
Referral centres
Timely referral to a specialized center is essential, allowing for early treatment that can improve the survival rate and lower the amputation rate. Currently, patients are initially managed in institutions with little exposure to STS cases, with fewer than 3 cases per year [18]. Many physicians, general surgeons, and even orthopaedic surgeons see soft tissue lumps in their career practice, but may not see and diagnose sarcoma because it represents low evidence, despite various raising awareness of sarcoma, educational strategies in public places, medical institutes, and social media. There are problems with the practice pattern of referring to specific oncologic orthopaedic surgeons. Consequently, inadvertent and incomplete STS excision occurs in a larger percentage of initial operations, accounting for up to 24–60% [19–21]. Despite strong evidence of lower post-surgery complication rates, mortality rates, and good outcomes after resection, all these initial procedures have taken place in low-volume sarcoma institutions, which eventually lacked an MDST.
In 2006, the UK introduced the National Institute for Health and Clinical Excellence (NICE) guidelines. The guideline highlighted any “red flag” symptoms should immediately trigger a referral to an MDST hospital before any interventional. As a result, more than 100 cases of STS were referred per year [22]. This effort has improved both survival and functional outcomes, and standardized protocols led to improved outcome efficacy.
Acute and late diagnosis and prognosis
It is essential to diagnose STS early as it is silently aggressive in nature, negligence and unawareness will result in the disease progression from acute to chronic. Acute disease presentation is defined as at least two months of initial presentation of symptoms and diagnosis. Whereas, chronic presentation is 6–12 months between initial symptoms and diagnosis. The course of disease progression depends upon both physician and patient related factors. Misdiagnosis, delay in the workup, lack of multi-disciplinary approach, lack of referral centres, are the attributes of physician related negligence, whereas, ignored symptoms, overlap or attributed symptoms of other cause, lack of awareness, rural located patients and poorer communities are patients related factors. In relation to the timeline it is found that long symptom duration is associated with low-grade sarcoma and high-graded sarcoma with red flags is with acute time duration.
Studies have found that there is a linear relationship between an increasing lesion size with a poorer prognosis, especially for lesions greater than 5 cm [23]. In the UK, it takes an average of 92 weeks from a patient noticing symptoms to their referral and investigation. By the time the tumor has been accurately diagnosed there is an increase in size by greater than 10 cm [23].
Diagnostic tools for investigation
Appropriate and effective imaging modalities are necessary for diagnosis, along with clinical indications. MRI is the imaging modality of choice for the diagnosis of the primary tumor, with CT, nuclear imaging, and ultrasonography (USG) as secondary options. For any lesions, an appropriate differential diagnosis can be made based on the patient’s age, location of lesions, and radiographic appearance. Conventional X-ray is the first-line imagining technique that gives at least two orthogonal views for any suspected soft tissue or bone sarcomas in any symptomatic patients. It provides information on calcifications, calcification patterns, bone destruction patterns, a zone of transition, cortical destruction, types of periosteal reaction, soft tissue, and joint involvement [24, 25].
However, USG is an effective initial investigation modality for patients suspected to have STS. It has a high negative predictive value for soft tissue masses and is proven to be highly cost-effective [11]. The favorable features of USG are that it measures the depth of the lesion, soft tissue internal echotexture, determining the relation to fascia, and vascularity (color Doppler). Often, it can differentiate benign lesions from pseudotumors, such as lipomas, ganglion cysts, and inflammatory conditions or hematomas that require further evaluation [24]. If the ultrasound finding raises suspicion of STS or is inconclusive, refer the patient to a sarcoma center [14].
MRI has become the X-ray of today. It is the cornerstone of musculoskeletal disease considering its non-invasive nature to assess the soft tissue, bone, and articular structures. MRI has excellent specificity for classifying different subtypes of periosteal reaction, the lesion’s true nature and aggressiveness are much more accurately determined in comparison with other imaging techniques [24, 26]. MRI is preferred in the confirmatory diagnosis of soft-tissue sarcomas of the limbs due to its superior anatomical definition and multiplanar approach allowing for precise biopsy planning. Additionally, contrast-enhanced MRI can provide further information such as characteristics of the soft tissue mass, relation to neurovascular structure, muscular compartment, adjacent joints, and fascia-tumor relationship. MRI has a very high negative predictive value (100%) for distinguishing a benign lipoma from a malignant lipoma [27]. Every tumor that takes up contrast medium should be considered malignant until proven otherwise.
CT has a limited role in diagnosing STS due to less contrast resolution compared to MRI, and concern for radiation [28]. CT cannot be totally reliable to distinguish lipoma or lipoma variant liposarcoma. It may be because CT attenuation of sarcoma is slightly less or similar to that of muscles [29]. Contrasted CT is the modality of choice for staging if MRI is contraindicated. CT angiography and three-dimensional (3D) reconstructed CT images can provide an even more sensitive evaluation of vascular invasion and visually display the anatomy for surgeons. CT is the modality of choice for distant metastasis assessment and surveillance.
Importance and role of biopsy
Imaging procedures alone do not permit the exact classification of a tumor as benign or malignant, therefore diagnosis is dependent on the histopathological investigation. Following confirmation of STS by biopsy a course of action can be determined depending on the histopathological grade and staging.
Percutaneous core needle biopsy is often used as it is safe and effective. It is considered a standard diagnostic approach and in most cases, multiple percutaneous core needle specimens are obtained under ultrasonographic guidance [14]. It can simply be performed in an outpatient setting under local anaesthesia for palpable tumors of the extremities. In 80% of core needle biopsies, the subtype and grade of the tumor can be determined and for experienced pathologists, there is a diagnostic accuracy of 95% to 99% [30]. Incisional biopsies are far less commonly used and if done by an inexperienced physician, they have a higher rate of complications than core needle biopsies. Therefore, it is advocated that such biopsies should be performed in specific circumstances by experienced tumor-trained orthopaedic surgeons who will perform subsequent operations in the future.
Regardless of how the tumor specimen is collected, this material should be analyzed by a pathologist specializing in soft-tissue disease for greater accuracy. Following the confirmation of STS, histopathology and staging are carried out so that the treatment plan can be determined by the corresponding surgeons.
Avoiding a “whoops” of STS
A “whoops procedure” is the process where a mass that was considered benign is resected in an unplanned manner without final diagnosis, preoperative imaging, or planning and is unexpectedly found to be a sarcoma when pathology results come back [11, 31]. Three-quarters of the referrals from a sarcoma center were from a “whoops procedure” occurring in a primary or secondary health setting by inexperienced surgeons [32]. Therefore, all the necessary steps must be taken before initiating a treatment plan.
The consequences of a “whoops procedure” harm the overall prognosis of the disease. These consequences include:
Increase in reoccurrence rate in the original and distant sites.
Residual tumorous cells in scar tissue due to inadequate margin of excision.
Breaching the tumor fascia can lead to wound complications and infection requiring flap coverage.
Increased rate of limb salvage and amputation.
Subsequent distant metastasis.
Poor functional outcome.
Financial and economic burden.
Mental and psychological impact.
There is a 20% to 25% local recurrence rate for visceral and extremity lesions at 10 years with 2nd/3rd of them occurring within the first 2 years [6]. Adequate follow-up post-treatment employing clinical examination imaging is necessary to detect treatable recurrence and metastasis, especially during the first 2 years.
Because of unplanned surgical procedures, surgeons are likely to excise inadequate margins of superficial and small sarcomas leading to re-occurrence and re-excision [33, 34]. During re-histological assessments, micro or macro tumor residuals are often present in the initial tissue scar and surgical margin [34–36]. In such scenarios, systematic re-excision is warranted within 3 months of the initial unplanned surgery despite any radiological or clinical evidence of local reoccurrences [37].
A compressive approach and awareness to the public and professionals, which may result in early diagnosis and avoidance of inappropriate treatment leading to whoop situations, are essential. The STS campaign contributes to spreading awareness among the public and advocates policymakers, conducting free outpatient services for screening for preventive measures in an educational manner.
Treatment therapy of soft-tissue sarcomas
Radiotherapy aims to preserve limb function, decrease the risk of reoccurrence and avoids amputations [38]. Radiotherapy combined with limb-sparing surgery was validated in a significant randomized controlled trial and has been the gold standard treatment since 1982 [39]. Since then, STS has been managed this way achieving local tumor control in 95% of cases [40]. Radiotherapy can be used either pre- and post-operatively in the management of STS. Preoperatively, a lower total dose is sufficient, which entails low toxicity and increased efficacy due to better oxygenation and vascularization of the tumor, and surgical resection becomes easier due to tumor shrinkage [41]. Radiation has also been effective in reducing post-operative wound complications and limiting acute reactions if performed before 4–8 weeks of surgery. Otherwise, there is the risk of developing oedema, joint stiffness, and fibrosis [42, 43]. In a randomized trial of pre- versus post-surgical radiation, local control rates were the same, but with a significantly lower number of wound complications in the post-operative treatment group [44]. Radiotherapy should be considered for high- and intermediate-grade tumors of the limbs. The most frequent post-operative radiation dose is 50–60 Gy doses sometimes boosted to 66 Gy [15]. If surgery is not appropriate or is refused by the patient, radiotherapy can be used alone; however, the local control rate is only 30–60% [30].
Chemotherapy is also widely used but is reserved for advanced disease stages and has a palliative role and systemic control. The sensitivity of chemotherapy (neoadjuvant, adjuvant or isolated) and survival is greatly influenced according to the type of tumor, grade of tumor, metastatic potential, case-by-case basis operability, individual characteristic and the likelihood of a response [45]. Ifosfamide and doxorubicin are the gold standard chemotherapies and have proven to be highly effective [46]. Doxorubicin as a single agent exhibits a limited dose response of 60 mh/m2 per 3-week cycle and appears to be effective, with toxicity above 75 mg/m3 [47]. It has clear antitumor activity in STS, with a response rate of 16–27% [48]. Ifosfamide can be effectively used as the first- or second-line treatment, and is often used in combination with doxorubicin [49]. However, the combination of both agents has been reported to have a good response rate in young patients with aggressive tumors [50]. There have been various studies on chemotherapy agents, such as taxanes, trabectidin, dactinomycin, etoposide, gemcitabine, docetaxel, trabectedin, pazopanib, and cyclophosphamide, and their various combinations in clinical trials. The advances in targeted therapy and immunotherapy are molecular level potential treatment options of major interest in STS.
Surgical management remains the standard therapy for non-metastatic diseases and offers the best chance of cure. The goal of treatment for STS of the limbs is function-preserving. Surgical resection involving wide margins by an experienced surgical team should be performed after a thorough study of the scans. Conservative limb-sparing surgery has been proven to be as effective as limb amputation. STS expands spherically along the tissue planes with outward growth that creates a pseudo capsule with the compressed surrounding tissue that malignant cells can penetrate [30, 51]. The removal of only visible tumors in this plane can result in 90% recurrence if there is no further treatment and subsequent death. With greater than 30% recurrence after further excision of the tumor bed and little difference with the addition of postoperative surgery [52]. Wide-margin excision surgery involves resection of a large proportion of surrounding healthy tissue, with safety margins of 4–5 cm to the sides and 1–2 cm deep to the tumor [53, 54]. Excision surgery, with planned postoperative radiotherapy, has a local recurrence rate of only 4%.
Primary amputation can only be considered in justified cases when the tumor is advanced, infiltrating major neurovascular-muscular compartments and in procedures such as downstaging with oncological therapy, marginal excision, and use of major reconstructive surgery [55, 56]. In 5–10% of cases, amputation may be necessary for patients with STS of the limbs, most commonly after previous limb-salvaging operations. Major amputation may be necessary in these cases because recurrence occurs proximally. These procedures provide good local control and are well tolerated by patients [56]. The extremity of STS can be minimized by the use of reconstructive procedures, particularly myocutaneous pedicled flaps to proximal limbs and limb girdles, and free vascularized flaps to more distal sites [57, 58].
Advances in STS and prospectives
Surgery is the mainstay of treatment, conventional chemotherapies and radiotherapies are minimal and do not lead to durable responses or cure, and patients may experience substantial toxicities. Immunotherapy has emerged as a novel approach that has revolutionized and rejuvenated the field of tumor immunology. Several types of immunotherapies, including adoptive cell transfer (ACT) and immune checkpoint inhibitors (ICIs), have yielded durable clinical responses. However, the challenges of efficacy in STS and bone sarcoma are limited owing to their distinct subtypes. Growing evidence on the pathophysiology and immune infiltrates in the tumor microenvironment of cancer cells impedes the effectiveness of immunotherapies. In recent years, nanotechnology has also shown potential in sarcoma treatment thanks to the development of smart materials and more effective drug delivery systems.
On the contrary, the use of AI in the pathological management of STS is a rapidly growing body of work, trying to use machine learning algorithms to improve diagnosis and derive other clinically important information from conventional histopathological slides and survival prediction in STS in near future. Finally, advances in biotechnology, the pharmaceutical industry, and collaboration can overcome the challenges of STS.
Conclusions
Overall, understanding of the “loneliest cancer” has greatly increased over the years but with very little progress. Standardized protocols have led to a slight improvement in outcome efficacy; however, there is currently a delay in the diagnosis of STS, although early treatment improves the survival rate and lowers the incidence of amputation. To improve this, a soft tissue mass presenting with red flag symptoms should be considered malignant until proven otherwise, and all necessary steps in management should be taken when there is clinical suspicion of STS. Treatment for STS is best conducted in a multidisciplinary setting, as it allows for optimal patient management, with surgery being the mainstay of treatment, and radiotherapy and chemotherapy being useful in selected cases. Despite the increasing awareness of STS, problems in the practice pattern still remain with regard to the timely referral of specific oncology-orthopaedic professionals.
Abbreviations
| CT: |
computed tomography |
| MDST: |
multidisciplinary sarcoma team |
| MRI: |
magnetic resonance imaging |
| STS: |
soft tissue sarcoma |
| USG: |
ultrasonography |
Declarations
Acknowledgments
The authors would like to acknowledge Ms. Simran Shakya for helping digitally draw the figures and performing English language editing of the manuscript.
Author contributions
SS: Conceptualization, Recourses, Writing—review & editing. ELB and SC: Writing—review & editing. JS: Writing—review & editing, Conceptualization. XZ: Conceptualization, Investigation, Validation.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2024.