 Review
Review

Affiliation:
1Department of Translational Medical Sciences, Section of Cardiology, University of Campania “Luigi Vanvitelli”, 80131 Naples, Italy
2Cardiology Unit, Azienda Ospedaliera Universitaria Luigi Vanvitelli, 80138 Naples, Italy
Email: giovanni.cimmino@unicampania.it
ORCID: https://orcid.org/0000-0002-3802-7052
Affiliation:
3Vanvitelli Cardiology Unit, Monaldi Hospital, 80131 Naples, Italy
ORCID: https://orcid.org/0000-0001-7824-6033
Affiliation:
1Department of Translational Medical Sciences, Section of Cardiology, University of Campania “Luigi Vanvitelli”, 80131 Naples, Italy
Affiliation:
1Department of Translational Medical Sciences, Section of Cardiology, University of Campania “Luigi Vanvitelli”, 80131 Naples, Italy
Affiliation:
1Department of Translational Medical Sciences, Section of Cardiology, University of Campania “Luigi Vanvitelli”, 80131 Naples, Italy
Affiliation:
1Department of Translational Medical Sciences, Section of Cardiology, University of Campania “Luigi Vanvitelli”, 80131 Naples, Italy
Affiliation:
4Department of Precision Medicine, University of Campania “Luigi Vanvitelli”, 80138 Naples, Italy
Affiliation:
1Department of Translational Medical Sciences, Section of Cardiology, University of Campania “Luigi Vanvitelli”, 80131 Naples, Italy
3Vanvitelli Cardiology Unit, Monaldi Hospital, 80131 Naples, Italy
ORCID: https://orcid.org/0000-0002-3590-6983
Affiliation:
5Department of Advanced Biomedical Sciences, Division of Cardiology, University of Naples “Federico II”, 80131 Naples, Italy
ORCID: https://orcid.org/0000-0001-7818-4952
Explor Musculoskeletal Dis. 2024;2:189–207 DOI: https://doi.org/10.37349/emd.2024.00048
Received: January 09, 2024 Accepted: February 22, 2024 Published: June 03, 2024
Academic Editor: Blanka Stiburkova, Charles University and General University Hospital, Czech Republic
Medical attention to uric acid (UA) has been increasing in recent years, mainly because this molecule has been shown to be associated with increased cardiovascular risk, both in the general population and in the hypertensive patients. A growing body of clinical and experimental data supports this view and prompts reconsideration of the role of UA in the development of atherosclerosis and the genesis of cardiovascular disease. It is known that this substance, in certain plasma concentrations, induces increased oxidative stress, a chronic inflammatory state, and a whole series of other modifications that are potentially deleterious at the cardiovascular level leading to hypertension, atherosclerosis, atrial fibrillation (AF), and other metabolic changes such as diabetes, metabolic syndrome, non-alcoholic fatty liver disease and kidney failure. Despite this epidemiologic and mechanistic evidence, the current guidelines from international cardiology scientific societies do not give precise indications in this regard, and some of them only suggest UA evaluation as part of an initial screening of the hypertensive patient. The purpose of this review is to briefly describe the main clinical and epidemiological evidence supporting the role of hyperuricemia as a possible emerging cardiovascular risk factor and to analyze the potential pathophysiological mechanisms through which elevated UA levels may exert a detrimental effect on the cardiovascular system.
Uric acid (UA) is the end product of purine catabolism in the humans [1]. There are basically two mechanisms underlying hyperuricemia: increased production or decreased elimination [1]. The first is mainly related to the increased consumption of fructose-sweetened beverages that correlates with increased risk of developing hyperuricemia (whose average values in the Western world have risen from 3.5 mg/dL in 1920 to 6.5 mg/dL) [2]. In 80% of cases, however, hyperuricemia is related to decreased excretion rather than excessive intake [1]. In cases of decreased excretion, UA plasma levels increases; given its relatively low solubility, a concentration above 6.5 mg/dL results in precipitation of its solid salt, i.e., monosodium urate, at the joint level. This results in the formation of deposits (tophi) responsible for the clinical symptoms of gout, a pathological condition whose effects were already described by the ancient fathers of medicine (e.g., Hippocrates, 5th century B.C.) and which until a few years ago was considered the only clinical manifestation of the accumulation of this substance [3]. Increased UA plasma levels in addition to being a possible cause of gouty arthritis may be responsible for nephro/urolithiasis. Other pathophysiological processes promoted by UA, such as inflammation and oxidative stress, also play an important role in the pathogenesis of many cardiovascular diseases (CVD) [4–8]. Several clinical studies have documented an association between elevated UA plasma levels and cardiovascular risk factors [9], such as high blood pressure [10], obesity [11], metabolic syndrome (MS) [11, 12], dyslipidemia [13, 14], and chronic kidney disease (CKD) [15, 16]. However, some other reports seem to not support its cardiolesive role [17]. The “rediscovery” of UA in the cardiovascular setting is relatively recent [18]. Changes in diet and lifestyle as well as pharmacological changes in recent decades [19], have led to a high prevalence of gout and hyperuricemia in the adult population (1–4% and up to 20% in the U.S. population, respectively) [20, 21].
Current evidence clearly indicate that high levels of UA lead to an increased risk of hypertension (HTN) [22, 23]. The first report of a possible link between gout (hyperuricemia), HTN, CVD, and renal failure dates back to the late 19th century [24]. However, this link remained poor studied until the late 1950s [25, 26]. Since then, several other epidemiological studies have controversly documented the significant correlation between hyperuricemia and a wide range of cardio-metabolic diseases, including HTN, obesity, dyslipidemia, MS, type 2 diabetes (T2DM), ischemic heart disease, vascular dementia, pre-eclampsia, and CKD [25–28]. Data from a mendelian randomization study seems to support this correlation reporting that each 1 mg/dL increase in genetically predicted UA level are significant for cardiovascular death and sudden cardiac death [29]. However, the possible role of hyperuricemia in the pathogenesis of CVD still remains the subject of intense scientific debate, as it is not completely clear whether increased uricemia simply represents a marker or a cardiovascular risk factor [30, 31]. The close association of hyperuricemia with classic cardiovascular risk factors (such as abdominal obesity, HTN, diabetes, dyslipidemia, and CKD), on the other hand, makes it extremely difficult to clarify the possible causal role of hyperuricemia in the development and progression of CVD [30, 31]. For that matter, it is evident that recognition of a possible causal role of hyperuricemia in the pathogenesis of CVD could be of great clinical importance given that hypouricemic drugs currently available in clinical practice are quite effective in reducing circulating levels of UA. This review aims to better define the importance of serum UA in cardiovascular setting with a focus on the atherosclerotic burden briefly describing the main clinical and epidemiological evidence supporting the role of hyperuricemia as a possible emerging cardiovascular risk factor and to analyze the potential pathophysiological mechanisms by which chronic hyperuricemia might exert its damaging effects on the cardiovascular system.
Hyperuricemia is defined by UA levels above 7 mg/dL in males and above 6 mg/dL in females [32]. Pathophysiological processes promoted by UA such as inflammation and oxidative stress [6] could play a causal role in the pathogenesis of many CVD such as HTN, atrial fibrillation (AF), heart failure (HF), and coronary artery disease (CAD) [31, 33]. Although an association between hyperuricemia and CVD is still matter of debate, UA remain a potential therapeutic target in clinical practice.
In recent years, numerous studies have linked hyperuricemia and HTN. Up to 25% of hypertensive patients are hyperuricemic [26], and the prevalence of hyperuricemia is higher in patients with severe HTN [34]. Furthermore, elevated UA levels seem to be associated with inadequate blood pressure control in pharmacologically treated patients [10]. Hyperuricemia appears to correlate with an increased risk of developing HTN-related organ damage, particularly left ventricular hypertrophy [35], subclinical myocardial damage [36], and worsening renal function [37]. The pathophysiological mechanisms underlying this association have not been fully elucidated, however, it has been hypothesized that some mechanisms play a frontline role regarding arteriolar vasoconstriction and increased renal sodium reabsorption [38]. Possible mechanisms are endothelial dysfunction (ED) [6], activation of the renin-angiotensin-aldosterone system (RAAS) [39], and hyperinsulinemia secondary to insulin resistance [40]. Consequently, the current European Hypertension Guidelines [41, 42] and the 2021 ESC Cardiovascular Disease Prevention Guidelines [43] include UA among the additional risk factors for CVD in hypertensive patients and recommend its serum levels be assessed in HTN screening.
Due to the considerable evidence linking hyperuricemia and HTN, studies have been conducted with the aim of analyzing the preventive role of hypouricemic treatment in the hypertensive population, with mixed results [41]. A randomized, double-blinded, placebo-controlled trial selected a group of pre-hypertensive obese adolescents aged 11 years to 17 years to receive either allopurinol, probenecid or placebo, showing that both the xanthine oxidase (XO) inhibitor (allopurinol) and the uricosuric agent (probenecid) resulted in a significant reduction in blood pressure levels [44]. Another study investigated the effects of hypouricemic treatment on blood pressure in the adult population older than 65 years. Using data from the UK Clinical Practice Research Datalink, 365 elderly patients with HTN and 6,678 controls were enrolled, and a significant reduction in both systolic and diastolic blood pressure was found in response to allopurinol [45]. On the other hand, a recent study of about 82 hypertensive patients aged 18–40 years did not demonstrate a reduction in systolic and diastolic blood pressure and C-reactive protein (CRP) in response to allopurinol compared with placebo [46].
In conclusion, there is a close association between hyperuricemia and the development of HTN, however, the role of hypouricemic treatment in blood pressure control is still debated and further clinical studies are needed.
The correlation between hyperuricemia and congestive HF has been widely studied over the years [47]. The main focus was on whether there was a causal relationship between hyperuricemia and HF and whether elevated serum UA levels could be an independent marker of HF development or predict unfavourable outcomes in the context of patients with HF.
With regard to the etiopathological correlation, several studies have hypothesized a role for UA as a determinant in the development of congestive HF: hyperuricemia could be an indicator of high metabolic oxidative stress [47–50].
Metabolic alterations as the normal coupling between the anaerobic threshold and insulin sensitivity with the consequence of insulin resistance occurring in congestive HF may cause hyperuricemia [48–50].
The enzyme XO catalyses the formation of UA from the purine bodies at the expense of the formation of a superoxide anion molecule. The upregulation of XO that occurs, for example, in conditions of hypoxia resulting from vascular and cardiac dysfunction, typically present in HF, would therefore result in increased production of reactive oxygen species (ROS) that contribute to the pathophysiology of congestive HF through increased oxidative stress [51].
ROS in turn promote not only cell apoptosis [52] but also the production of inflammatory cytokines [53] and together with them contribute to vascular dysfunction.
Moreover, a correlation between hyperuricemia and HF could also be explained through inflammation, as it has been shown that patients with HF and higher levels of circulating UA had higher levels of CRP, interleukin-6 (IL-6), and neutrophil count [47, 54].
Furthermore, hyperuricaemia, through increased ROS, would reduce the bioavailability of endothelial nitric oxide (NO) and lead to ED through a loss of NO-dependent vasodilation [55, 56].
A further contributor to the increase in UA levels in the HF setting is impaired renal function, which is frequent in this type of patient [15] and the use of high-dose diuretics, which would increase reabsorption [57]. About half of HF patients with preserved or reduced ejection fraction have been shown to have UA concentrations above the reference limit [58]. The meta-analysis by Huang et al. [59] showed that for every 1 mg/dL increase in UA, the risk of developing HF increases by 19%.
In the Framingham Offspring Cohort Study, patients in the highest quartile of UA (> 6.3 mg/dL) had a six-fold increased risk of developing HF compared to those in the lowest quartile (< 3.4 mg/dL) [47].
A linear correlation between elevated UA levels and the development of congestive HF was also demonstrated in the large AMORIS study [60]. Furthermore, in the context of patients with congestive HF, hyperuricemia appears to have an unfavourable prognostic role in terms of outcomes [61].
A previous meta-analysis has reported that elevated serum UA levels were independent predictors of all-cause mortality and cardiovascular mortality: indeed, for every 1 mg/dL increase in serum UA, all-cause mortality increased by 4% [62]. In another analysis by the Italian Group for the Study of Survival in Heart Failure-Heart Failure (GISSI-HF), it was found that serum UA levels > 7 mg/dL were directly related to increased all-cause mortality, poor long-term survival and risk of hospitalization for cardiovascular causes in people with HF [63]. Moreover, other studies have reported that hyperuricemia is associated to all-cause mortality and hospitalization in patients with advanced HF [64–66]. In a multivariate analysis serum UA levels emerged as a significant predictor of New York Heart Association (NYHA) functional class, independent of other confounding factors [67]. A more recent mendelian randomization study [68] further support the notion that elevated UA levels may heighten risks of HF. Based on the data accumulated over the years, hyperuricemia has been proposed as a parameter in several prognostic scores in HF, including the metabolic, functional and haemodynamic decompensation staging system [69]. Thus, on the basis of these clinical evidence, the use of uricosuric agents or XO inhibitors in patients with decompensation has been hypothesized.
Several studies, including randomized ones, have been carried out to explore this hypothesis, but unfortunately, they have not demonstrated a statistically significant improvement in clinical, left ventricular ejection fraction and exercise capacity in these patients [62, 64, 70].
New well-designed studies are needed to rule out the possibility of these drugs playing a key role in the treatment of congestive HF.
Recent studies have shown a correlation between hyperuricemia and the risk of developing AF [71]. In the Atherosclerosis Risk In Communities (ARIC) study it was shown that elevated serum UA levels increased the risk of AF by 1.16-fold [72]; the association was found to be stronger in the black population and in women [72]. In addition, an increased risk of AF was found in diabetic patients with hyperuricemia [73]. Hyperuricemia is also related to an increased risk of thrombus formation in the left atrial appendix (LAA) in patients with nonvalvular AF [74]. One study has also shown that the UA levels negatively correlate with LAA contractile function and could provide additional prognostic information about future thromboembolic events in patients with AF [75].
The pathophysiological mechanisms underlying this correlation have been only partially elucidated. Hyperuricemia-induced mechanisms such as inflammation, oxidative stress, ED, and activation of the RAAS, that regulates blood pressure, fluid and electrolyte balance, and systemic vascular resistance, could play a causal role in bringing about atrial remodeling and thus promote the formation of reentry circuits [76]. At the molecular level, it has been shown in mouse atrial myocytes (HL-1 cells) that UA penetrates myocytes through specific transporters and induces Kv1.5 protein expression. The final effect is enhanced ultra-rapid delayed-rectifier K+ channel currents and shortened action potential duration in HL-1 cells thus inducing AF [77]. Further clinical studies are needed to demonstrate whether hypouricemic preventive treatment is effective in reducing the incidence of AF.
In the last years several evidence have also linked hyperuricemia with atherosclerotic CAD. Some prospective studies have shown a correlation between elevated serum UA levels and the incidence of acute coronary syndrome (ACS) [78]. In the ACS patients, independent studies have shown a correlation between hyperuricemia at admission an increase in both in-hospital and long-term mortality, the incidence of adverse cardiovascular events [79], worse prognosis, expecially for patients with Killip class III at presentation [80–82], increased risk of acute plaque complications [83], as well as more complex percutaneous procedures [84]. A more recent observation by our group has shown that elevated UA levels ad hospital admission in patient presenting ACS is correlated with a higher inflammatory burden and myocardial damage, multivessel diseases and complex coronary stenosis (type C of Ellis classification) [85], further supporting the role of UA in the pathophysiology of acute coronary events.
By the cellular point of view, during acute myocardial ischemia tissue hypoxia occurs, especially when ACS results in reduced ejection fraction or cardiogenic shock, leading to an increased purine release and UA synthesis [86]. This UA blood level increase, is also favoured by the renal damage that often occurs in the clinical context of ACS leading to a reduced excretion of UA [86].
On the other hand, in patients with chronic coronary syndrome (CCS), hyperuricemia has been correlated with mortality from cardiovascular causes and increased incidence of cardiovascular events [87]. As for our observation in the acute setting [85] also in the CCS elevated UA levels were correlated with the presence and severity of coronary atherosclerosis and multivessel involvement [88], higher Syntax score and higher long-term mortality [89]. However, other studies, evaluating patients with previous acute myocardial infarction and already revascularized have failed to confirm this association [90–92]. Finally, the ALL-HEART study [93], a large randomized clinical trial, have failed to show benefits on major cardiovascular outcomes from allopurinol treatment in patients with documented CAD without gout. However, it should be taken into account that baseline UA levels in ALL-HEART study were quite normal with a mean of 0.35 mmol/L serum concentrations.
Despite the controversy still ongoing, current data seem to support the role of UA in the pathogenesis of coronary atherosclerosis, expecially in the early rather than advanced stages of CAD. Further and well-designed studies are needed to better define how and at which stage of coronary atherosclerosis development, UA levels might contribute.
In the pathophysiology of stroke, UA could play an important role. Despite some evidence support a negative relationship between hyperuricemia and acute ischemic stroke [78, 94–96], other observations reports opposite findings [97, 98]. These controversial results might be explained partially for the presence of other cardiovascular risk. In the REGARDS study, severe HTN has been considered the main interplay between hyperuricemia and stroke [99]. To better define the primary role of UA/stroke relationship, another study was conducted in 3,243 elderly Chinese patients without comorbidities showing a significant correlation between asymptomatic hyperuricemia and incident stroke with a up to 2-fold increased risk [100]. Further supporting evidence come from the studies using uric lowering agents. Different independent reports have shown that allopurinol administration in hyperuricemic patients was associated to a decreased risk of major cardiovascular events including stroke [101] with greater benefits for longer use [102].
A recent meta-analysis of 19 prospective cohort studies including up to 37,386 males and 31,163 females indicates that elevated UA concentrations are associated to a significant risk of ischemic and hemorrhagic stroke in adults, mainly females [95].
A summary of the association between UA and CVD is reported in Figure 1.
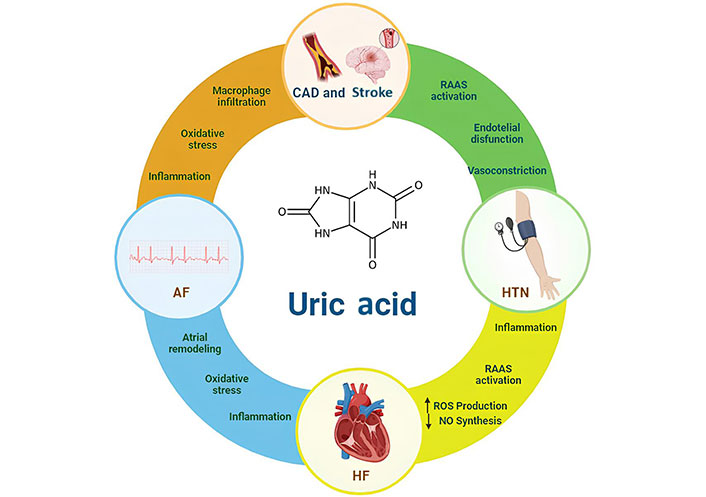
CVDs linked to uric acid and putative pathways involved. The pathophysiological mechanisms underlying these associations have been hypothesized. The possible mechanisms that related hypertension (HTN) to hyperuricemia include activation of the RAAS and increased renal sodium reabsorption, endothelial dysfunction (ED), with consequent arteriolar vasoconstriction. Same mechanisms together with oxidative stress and inflammation could play a causal role in bringing about atrial remodeling and thus promote the formation of reentry circuits in the setting of AF. The inflammation itself, the oxidative stress with the activation of macrophage, the infiltration of them in the vessel involved in atherosclerotic cascade could be mechanisms implicated in developing CAD and stroke. The RAAS and increased renal sodium reabsorption, the inflammation, oxidative stress, increased production of NO are all possibly involved in HF. AF: atrial fibrillation; HF: heart failure; ROS: reactive oxygen species; NO: nitric oxide; RAAS: renin-angiotensin-aldosterone system; CAD: coronary artery disease; CVD: cardiovascular diseases
UA is the end product of a exogenous (food) and endogenous (damaged, dying and dead cells) pool of purines that occurs mainly in the liver, intestines and the vascular endothelium [103]. Beyond the well-established role as etiological agent of the severe, acute and chronic inflammatory arthritis, gout [19] and its implication also in metabolic and non-metabolic disorders [38, 40, 104, 105], it is important to recognize that UA may exerts some protective functions in healing and defense [103]. It has antioxidant properties as ROS and peroxynitrite (ONOO-) scavenger [106], favour endothelial function at normal levels [107, 108], help tissue repair [109]. Furthermore, UA is a potent mediator of type 2 immune responses [110], it is involved in the resistance to parasites [103] and ultimately it represent a defense against neurological and autoimmune diseases [103, 111, 112].
In the paragraphs below the molecular pathways linking UA to atherosclerosis will be analyzed.
Endothelial activation represents a state where endothelial cells become both proinflammatory and procoagulant due to the influence of oxidized lipoproteins and cytokines released during inflammatory conditions [113]. This activation involves the heightened expression of adhesion molecules and the recruitment of inflammatory cells [113]. Oxidative stress is crucial in orchestrating synthesis and release of cytokines, thereby linking ROS with inflammation and endothelial activation, as well as dysfunction [113]. It is well known that UA is linked to oxidative stress, inflammation, and ED [114]. When XO reacts with xanthine to produce UA and superoxide anion, ROS are released [6]. Nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase, XO, lipoxygenase, myeloperoxidase, mitochondrial enzymes, and uncoupled NO synthase are the primary producers of ROS [115]. Excessively generated O2—concomitant with increased UA production in the process of purine metabolism interacts directly with NO with high affinity, resulting not only in decreased NO bioavailability by degrading and inactivating it, but also leading to an increase in the formation of ONOO-, a powerful oxidant that causes DNA damage, cell death, and lipid peroxidation [116]. ONOO- could cause the oxidation of tetrahydrobiopterin, an essential cofactor of endothelial NO synthetase (eNOS) in its inactive form, causing a deficiency. If inadequate concentrations of tetrahydrobiopterin occurrs, eNOS is converted from an NO-producing enzyme into an O2-generating enzyme. This process is referred to as eNOS uncoupling [116]. The increased production of O2 further impairs endothelial function through a vicious cycle of increased oxidative stress, decreased NO bioavailability, increased ONOO- production, and eNOS uncoupling [116]. Therefore, it is hypothesized that the bioavailability of NO is reduced by the production of O2, which together with UA production in the process of purine metabolism, catalyzed by XO, leads to ED [6]. XO is present not only inside the cytoplasm but also on the external surface of the endothelial cell membrane. Experimental studies have shown that, in pathophysiological conditions, the circulating enzyme released by XO-rich organs is bound to glycosaminoglycans on the surface of endothelial cells and can then be endocytosed into intracellular compartments, inhibiting the NO-dependent relaxation of vascular smooth muscle cells [117]. These observations indicate that endothelial endogenous XO as well as the inducible circulating XO released from XO-rich organs are important source of ROS, thus contributing to ED [118].
XO has been shown to be involved in the transformation of macrophages into foam cells via acceleration of low-density lipoprotein (LDL) intake [119], a process that is directly involved in the atherosclerosis development [113].
It has been reported that UA can induce the endothelial-to-mesenchymal transition (EndMT) in human umbilical vein endothelial cells (HUVECs) and hyperuricemic rats by promoting oxidative stress and glycocalyx shedding [120]. Disruption to the endothelial glycocalyx, which is associated with inflammation, can increase vascular permeability and promote leukocyte and platelet adhesion to endothelial cells [121], thus participating to atherosclerosis development. Overall, high UA levels might drive the EndMT-mediated loss of endothelial function [122].
UA is transported through the plasma membrane via transport proteins, urate transporter 1 (URAT1) and URATv1, which act in tandem for urate reabsorption in renal tubules [1]. Recent experimental studies revealed UA transporters are also expressed in other types of cells, including vascular cells [123, 124]. This is of great importance since activation of these transporters in endothelial cells is associated to ED via reduction of endothelial NO bioavailability and ROS generation [6, 125, 126]. It has been reported that reduced NO release in HUVECs by UA is also associated to increased CRP production, through the activation of p38 and extracellular signal-regulated kinase 44/42 mitogen-activated protein kinases (MAPK) pathways [127], and inhibited migration and proliferation of smooth muscle cells [128]. A summary of the mechanisms involved in ED is provided in Figure 2.
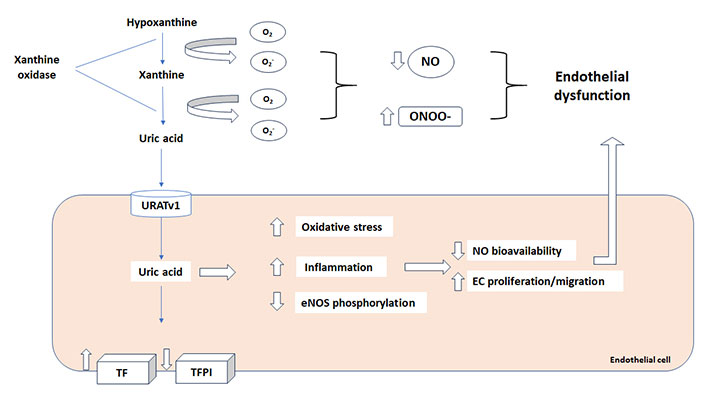
Uric acid and endothelial dysfunction. Reactive oxygen species (ROS), produced by xanthine oxidase (XO) during the uric acid synthesis, decrease NO bioavailability and increase peroxynitrite (ONOO-) impairing endothelial function. Moreover, uric acid increase tissue factor (TF) and procoagulant activity with a concomitant decrease in TF pathway inhibitor (TFPI) expression. URATv1: urate transporter v1; NO: nitric oxide; eNOS: endothelial NO synthetase
It is interesting to note that these UA-induced alterations are reversed in vitro by pretreatment with probenecid, a UA transporter inhibitor [129]. Activation of HUVEC renin-angiotensin system by UA has been also documented, leading to ROS production, apoptosis and senescence [130] and also this effect is suppressed by probenecid [130]. Moreover, it has been reported that angiotensin receptor blockers, such as irbesartan and losartan, exert uricosuric properties [131, 132] improving NO bioavailability in HUVEC [129] and this effect is of great importance by the therapeutical point of view.
These data suggest that UA absorbed into endothelial cells through UA transporters causes inflammation, oxidative stress, and dephosphorylation of eNOS, leading to ED through decreased NO bioavailability and that uric lowering agents might be of help in improving endothelial function.
The final effect of UA in inducing ED and inflammation, through all the pathways already indicated above, is a procoagulant state that might result in vascular thrombosis [133]. The link between ED, inflammation and thrombosis is well known [113, 134]. A recent study clearly shows that UA induce expression of functional tissue factor (TF), the key initiator of coagulation cascade, and decreases the level of TF pathway inhibitor (TFPI), the only physiological inhibitor of TF, in cultured endothelial cells in a dose-dependent manner expression [135] as schematically summarized in Figure 2. On this regard, different studies have linked high UA levels with the risk of venous thrombosis [136–139].
Existing clinical evidence support the association between UA levels and different inflammatory diseases like atherosclerosis that is a chronic immuno-inflammatory CVD [33, 140] and other conditions such as MS, diabetes mellitus [141], CKD [141], Hashimoto’s thyroiditis (HT) [142], and non-alcoholic fatty liver disease (NAFLD) [143]. High intracellular UA concentrations promote intracellular signaling pathways and the expression of inflammatory markers including nuclear factor κB (NF-κB), growth factors, vasoconstrictive substances (angiotensin II, thromboxane, and endothelin-1), and chemokines via activating MAPK [7, 16]. Furthermore, UA can activate NOD-like receptor protein 3 (NLRP3) inflammasomes [144]. The inflammasome is a multimeric protein complex that acts activating caspase-1 and inflammatory cytokines such as IL-1β [145].
The NF-κB regulates NLRP3 gene transcription in the nucleus and release it into the cytoplasm [146]. Then, the NLRP3 inflammasome assembles and forms a complex with an adaptor protein and procaspase-1. Afterwards procaspase-1 cleaved to active caspase-1 [146]. The active caspase-1 processes pro-IL-1β and pro-IL-18 expressed in the nucleus into the mature inflammatory cytokines IL-1β and IL-18 and also guide the mature processing of the gasdermin D protein, which can causes cell lysis and pyroptosis [147]. It has been reported that UA promotes the activation of NLRP3 inflammasomes via AMP-activated protein kinase (AMPK)-mammalian target of rapamycin (mTOR)-mitochondrial ROS pathway [148] and suppresses the activity of AMPK, that involves the activation of the mTOR [149].
The alterations of these pathways result in the generation of mitochondrial ROS and activation of hypoxia-inducible factor 1-alpha (HIF-1α) that promotes the secretion of IL-1β, increase inflammation and damages the cardiovascular system [148, 149]. A schematic view is reported in Figure 3.
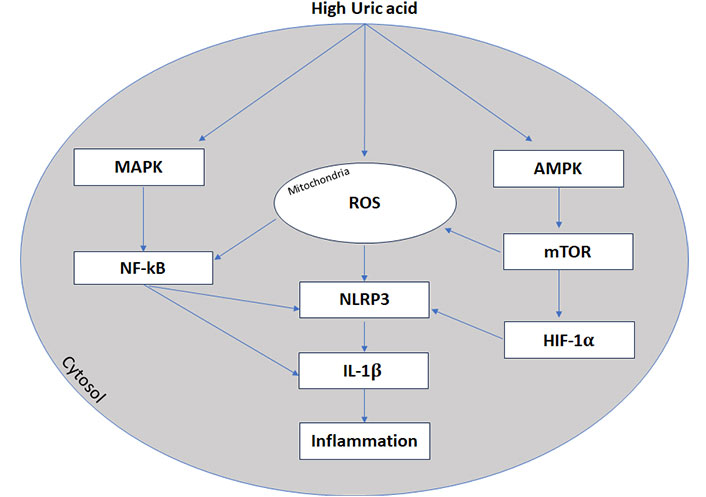
High uric acid and inflammation. Inflammasone (NLRP3) transcription is promoted by nuclear factor κB (NF-κB) activation and AMPK-mTOR-mitochondrial ROS pathway. MAPK: mitogen-activated protein kinases; ROS: reactive oxygen species; NLRP3: NOD-like receptor protein 3; IL-1β: interleukin-1β; AMPK: AMP-activated protein kinase; mTOR: mammalian target of rapamycin; HIF-1α: hypoxia-inducible factor 1-alpha
Recently, different reports indicate that UA/high density lipoprotein (HDL) ratio (UHR) is promising index of inflammation associated with CVD, such as HTN [150] and non CVD. Hyperuricemia is significantly associated with the development of MS and T2DM [11–13] and UHR seems to be correlated with the risk of MS and T2DM development [151, 152], new onset diabetes [153] and even prediabetes [154]. Moreover, UHR seems to be inversely correlated with glomerular filtration rate (GFR) [155] but positively associated with waist circumference, body weight, body mass index, serum creatinine, fasting plasma glucose (FPG), and glycated hemoglobin (HbA1c) levels [152]. Consequently, UHR could be used with other indices such as HbA1c and fasting glucose as a predictor of diabetic control in the diabetic population [12, 153]. UHR has been also correlated with uncontrolled systemic HTN [150]. Additionally, UHR is significantly increased in subjects with NAFLD and it could potentially serve as novel marker for hepatic steatosis [143]. Finally, high levels of UHR have been also found in patient with HT [142].
The important lifestyle changes in the last decades have generated newer risk factor for CVD. UA seems one of these new contributors to the development of atherosclerosis and CVD. The evidence to date seems to support the primary role of UA in this context showing that UA might be both an innocent bystander as well as a ruthless killer based on the diseases and comorbidities. UA might be either a marker of more severe manifestation of CVD as well as a key actor in CVD development. Novel biomarker, such as UHR, are supported by different evidence as promising tool to use in clinical practice to improve the predictive role of UA. However, the current CVD guidelines still fail to give clear recommendations in the evaluation of UA levels. UHR is a very simple parameter to get and as UA should be included in the SCORE system and became a true biomarker for cardiovascular risk evaluation. Several studies support this role, thus it’s time for act.
ACS: acute coronary syndrome
AF: atrial fibrillation
AMPK: AMP-activated protein kinase
CAD: coronary artery disease
CKD: chronic kidney disease
CRP: C-reactive protein
CVD: cardiovascular diseases
ED: endothelial dysfunction
eNOS: endothelial nitric oxide synthetase
HF: heart failure
HIF-1α: hypoxia-inducible factor 1-alpha
HTN: hypertension
HUVECs: human umbilical vein endothelial cells
IL-6: interleukin-6
MAPK: mitogen-activated protein kinases
MS: metabolic syndrome
mTOR: mammalian target of rapamycin
NF-κB: nuclear factor κB
NLRP3: NOD-like receptor protein 3
NO: nitric oxide
ONOO-: peroxynitrite
RAAS: renin-angiotensin-aldosterone system
ROS: reactive oxygen species
T2DM: type 2 diabetes
TF: tissue factor
UA: uric acid
UHR: uric acid/high density lipoprotein ratio
URAT1: urate transporter 1
XO: xanthine oxidase
GC, FN, and PC: Conceptualization. RF, MM, GT, VMC, and NM: Investigation, Writing—original draft. GC, PG, and PC: Writing—review & editing. PG and PC: Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 7979
Download: 123
Times Cited: 0
