 Review
Review

Affiliation:
1Arthrosi Therapeutics, Inc., San Diego, CA 92121, United States
Email: rkeenan@arthrosi.com
ORCID: https://orcid.org/0000-0002-2341-7314
Affiliation:
1Arthrosi Therapeutics, Inc., San Diego, CA 92121, United States
Affiliation:
1Arthrosi Therapeutics, Inc., San Diego, CA 92121, United States
Affiliation:
1Arthrosi Therapeutics, Inc., San Diego, CA 92121, United States
Affiliation:
2VA New York Harbor Health Care System, New York, NY 10010, United States
3Department of Medicine, Biochemistry and Molecular Pharmacology, New York University Grossman School of Medicine, New York, NY 10016, United States
ORCID: https://orcid.org/0000-0003-3168-1542
Explor Musculoskeletal Dis. 2024;2:529–554 DOI: https://doi.org/10.37349/emd.2024.00077
Received: September 16, 2024 Accepted: November 05, 2024 Published: December 12, 2024
Academic Editor: Blanka Stiburkova, Charles University and General University Hospital, Czech Republic
The article belongs to the special issue Pharmacological and Non-Pharmacological Management of Gout
Gout is a common inflammatory arthritis preceded by chronically elevated levels of serum urate. In addition to leading to gouty flares, hyperuricemia can result in stone-like deposits of monosodium urate crystals (tophi) being deposited in joints and soft tissue, where they cause severe pain and damage. Although gout is an ancient disease with a well-characterized etiology, its treatment landscape has not kept pace with that of other rheumatic conditions. Therapy centers on lowering serum urate concentrations, with urate-lowering drugs falling into three categories: xanthine oxidase inhibitors (e.g., allopurinol, febuxostat) that reduce urate production by blocking the conversion of hypoxanthine to uric acid; uricosurics [primarily urate transporter-1 (URAT1) inhibitors, including probenecid, lesinurad] that promote the renal excretion of urate; and recombinant uricases (e.g., pegloticase) that convert uric acid to allantoin (a water-soluble compound that is more readily excreted). Some treatments have been available for decades, but are often limited by toxicities, primarily relating to the liver and kidneys. Recent research has focused on developing more potent and specific URAT1 inhibitors in the hope that these safety concerns can be overcome, and that better tolerated, more effective therapies can be made available. Newer uricosurics have different chemical structures from their predecessors, resulting in greater URAT1 selectivity in order to reduce off-target effects. Several of these have shown promising results in clinical trials and could prove to be viable alternatives to suboptimal existing therapies. Indeed, newer generation uricosurics may have the potential to become viable therapies in indications other than gout, such as some metabolic diseases. In this narrative review, we discuss the position of uricosurics (primarily URAT1 inhibitors) in the landscape of chronic gout treatment of the past, present, and future.
Gout is a common and potentially debilitating inflammatory arthritis with a well characterized etiology [1, 2]. In gout, chronically elevated serum urate (hyperuricemia) eventually causes monosodium urate crystals to be deposited in the joints and other tissues, triggering recurrent disease flares [2, 3]. If inadequately treated, palpable tophi (stone-like deposits of monosodium urate surrounded by inflammatory coronas) can develop in or near joints, damaging them and causing severe pain [4, 5].
The persistent hyperuricemia that leads to gout is a consequence of overproduction or, more commonly, underexcretion of urate (90% of cases) [6, 7]. Uric acid is the end-product of the human catabolism of purines (adenosine and guanosine), which are found in many foods including shellfish and legumes. Under normal physiological conditions, for example in plasma, uric acid exists primarily as its ionized form, urate [5, 8, 9].
Ordinarily, around two-thirds of urate is excreted renally, and the remainder through the gastrointestinal tract [10]. Plasma urate is freely filtered by renal glomeruli, but most of it is then reabsorbed as it travels through the nephron. This reabsorption is mediated by various urate-anion exchangers, most importantly urate transporter-1 (URAT1) [8]. URAT1 employs an active transport process to resorb uric acid from renal ultrafiltrates passing through the proximal tubule [7]. In addition to tubular reabsorption, uric acid can also be actively transported into the tubular lumen by other transporter proteins [8]. The balance between filtration and tubular secretion on the one hand, and reabsorption (mainly by URAT1) on the other, determines the outcome of renal handling of uric acid.
Urate homeostasis is tightly regulated; however, hyperuricemia can result if the balance between production and excretion of uric acid is disrupted [5]. For example, in patients with chronic kidney disease, where kidney filtration is impaired, the gastrointestinal tract may clear a greater fraction of plasma urate than it would otherwise. However, gastrointestinal urate excretion may be insufficient to restore homeostasis of serum urate levels, raising the likelihood of developing hyperuricemia and gout.
Reducing serum urate concentrations to restore uric acid homeostasis is currently the cornerstone of gout management. Treat-to-target urate-lowering therapy is recommended in guidelines published by the American College of Rheumatology (ACR) and the European Alliance of Associations for Rheumatology (EULAR) [11, 12]. Both recommend reducing serum urate levels to ≤ 6 mg/dL when treating with urate-lowering therapy, with the EULAR guidelines suggesting an even lower target of ≤ 5 mg/dL for severe tophaceous disease [11, 12].
Currently recommended urate-lowering therapies include xanthine oxidase inhibitors (XOIs), uricosurics, and recombinant uricases, each with a different mechanism of action. XOIs (e.g., allopurinol, febuxostat) reduce urate production by blocking the conversion of hypoxanthine to uric acid [13]. Uricosurics (e.g., URAT1 inhibitors including probenecid and lesinurad) promote renal urate excretion by blocking renal uric acid reabsorption, the body’s normal method of maintaining uric acid homeostasis. Recombinant uricases (e.g., pegloticase) catalyze the conversion of uric acid to water-soluble allantoin, a more soluble and easily excreted metabolite [12].
The XOI allopurinol is the current first-line urate-lowering therapy in the US and Europe [11, 12, 14, 15]. However, allopurinol has several drawbacks: it is not always adequately effective, may require a long up-titration period to safely reach an effective dose, and associates with a rare, but potentially fatal, hypersensitivity syndrome [15, 16]. Therefore, effective alternatives are needed.
Currently used uricosurics inhibit renal transporters, particularly URAT1 [17, 18], whose inhibition prevents uric acid from being reabsorbed from the proximal tubule, therefore increasing renal excretion [8, 17, 18]. Other renal uric acid transporters include (but are not limited to) organic anion transporter 1 (OAT1), OAT3, glucose transporter 9 (GLUT9), ATP-binding cassette super-family G2 (ABCG2), and multidrug resistance protein 4 (MRP4), which all work to regulate uric acid homeostasis [19, 20]. ABCG2 is also expressed in the intestine and becomes important in renal disease for the intestinal excretion of urate [19, 20]. Historically, modulation of uric acid excretion has been achieved using non-selective inhibitors, but owing to advances in transporter characterization, the newer medications in development are more selective in their transporter interaction [19].
Uricosurics have been marketed for several decades [21], but their use remains infrequent, partially due to efficacy and safety concerns [6, 11, 22] and variations in availability depending on geographical area. For example, only probenecid (a relatively weak uricosuric) is available in the US and used in only 2–3% of cases [23–25]. Conversely, benzbromarone, which is not approved in the US, is commonly used in patients with gout in some Asian countries, even above allopurinol [6, 26, 27].
To overcome some of the limitations of existing urate-lowering therapies, drugs specifically and more potently targeting URAT1 are in development and show promising potential for the treatment of gout. In this review, we discuss the position of uricosurics (specifically URAT1 inhibitors) in the chronic gout treatment landscape of the past, present, and future. Other drugs approved for non-gout indications have demonstrated secondary uricosuric properties, including the antihypertensives captopril, amlodipine, and losartan [28–30]; the statin atorvastatin [31]; the lipid-lowering agent fenofibrate [31]; and sodium-glucose cotransporter-2 (SGLT2) inhibitors, including dapagliflozin, canagliflozin, and empagliflozin, used in the treatment of type 2 diabetes mellitus (T2DM) [32]; however, detailed discussion of these is beyond the scope of this article.
Uricosurics for gout have a long and controversial history, with several early agents causing safety concerns that led to the withdrawal of some drugs. The use of uricosuric agents goes back as far as the 19th century, when high doses of salicylates were first used to promote renal clearance of uric acid [33]. However, such high-dose therapy was both impractical and toxic, and lower doses of salicylate actually promote uric acid retention [33]. In the mid-20th century, salicylates were superseded by the second-generation uricosurics probenecid, sulfinpyrazone, and benzbromarone [33].
Five uricosurics are currently approved [6]. These are probenecid in the US, Europe, and Japan [6, 34]; benzbromarone in Japan and China, other southeast Asian countries or regions, and Europe (although largely withdrawn by the manufacturer in Europe due to hepatotoxicity concerns) [6, 7, 35–37]; lesinurad in the US (but discontinued by the manufacturer who stated commercial reasons for this withdrawal) [21, 38]; sulfinpyrazone in the US and Europe, although also largely discontinued by the manufacturer) [6, 21, 39]; and dotinurad in Japan [40]. Individual uricosurics are described in more detail below Table 1 and the receptors they interact with are further summarized in Table 2.
Overview of uricosurics: past, present, and future
| Time period | Agent | Clinical experience to date | Mechanism of action | Key safety signals |
|---|---|---|---|---|
| Past | Probenecid [6, 21, 34, 41, 42, 109] |
| Non-specific: inhibits anion transporters URAT1, OAT1, OAT3, and GLUT9 |
|
| Benzbromarone [6, 21, 36, 110] |
| Non-specific: acts mainly on URAT1, but also on OAT1, OAT3, and GLUT9 |
| |
| Sulfinpyrazone [6, 7, 21, 39, 43, 111] |
| Thought to inhibit URAT1 and MRP4 |
| |
| Phenylbutazone [21, 112, 113] |
| NSAID, small study demonstrated mild uricosuric properties | Blood dyscrasias | |
| Zoxazolamine [21] |
| Muscle relaxant (calcium channel blocker) | Fatal hepatotoxicity | |
| Benziodarone [21, 49] |
| Vasodilator | Jaundice | |
| Present | Lesinurad [6, 17, 21, 38, 55, 57] |
| URAT1, OAT1, OAT3, and OAT4 inhibitor | Raised creatinine and nephrolithiasis when used as monotherapy |
| Verinurad (RDEA3170) [6, 21, 65, 66] |
| Selective URAT1 inhibitor | Renal-related adverse events and elevated creatinine were common in clinical trials | |
| Dotinurad [21, 71] |
| Selective URAT1 inhibitor |
| |
| Future | Ruzinurad (SHR4640) [73, 74] |
| Selective URAT1 inhibitor | No serious hepatic or safety signals have been observed [76, 77]; yet acute kidney injury has occurred rarely in clinical trials [77] |
| Pozdeutinurad (AR882) [75, 77, 78] |
| Selective URAT1 inhibitor; optimized from benzbromarone metabolite scaffold | No serious adverse events and no renal or hepatic safety signals have been reported in clinical trials | |
| Epaminurad (URC102/UR-1102) [40, 79] |
| URAT1 inhibitor; derived from benzbromarone | No serious adverse events have been reported in clinical trials | |
| ABP-671 [82] |
| URAT1 inhibitor | Nephrolithiasis reported in clinical trials | |
| Xininurad (XNW-3009) |
| URAT1 inhibitor | No trial data are yet available |
* Only trials in the indications of gout or hyperuricemia are listed. FDA: US Food and Drug Administration; GI: gastrointestinal; GLUT9: glucose transporter 9; MRP4: multidrug resistance protein 4; NSAID: nonsteroidal anti-inflammatory drug; OAT1: organic ion transporter 1; URAT1: urate transporter 1; XOI: xanthine oxidase inhibitor
Receptors inhibited by non-selective and selective uricosurics
| Uricosuric | URAT1 | OAT1 | OAT3 | OAT4 | GLUT9 | ABCG2 | MRP4 |
|---|---|---|---|---|---|---|---|
| Probenecid [6, 43] | √ | √ | √ | – | √ | – | – |
| Benzbromarone [6, 43] | √ | √ | √ | – | √ | – | √ |
| Sulfinpyrazone [6, 43] | √ | – | – | – | – | – | √ |
| Lesinurad [6, 43, 55, 56] | √ | √ | √ | √ | – | – | – |
| Dotinurad [6, 43, 71] | √ | – | √ | √ | – | √ | – |
| Ruzinurad* [73, 74] | √ | – | – | – | – | – | – |
| Pozdeutinurad (AR882)* [6] | √ | – | – | – | – | – | – |
| Verinurad (RDEA3170)* [43, 65] | √ | √ | – | √ | – | – | – |
| Epaminurad (URC102/UR-1102)* [6, 40] | √ | No data | No data | No data | No data | No data | No data |
| ABP-671* [6] | √ | No data | No data | No data | No data | No data | No data |
| Xininurad (XNW-3009)* [6] | √ | No data | No data | No data | No data | No data | No data |
* URAT-1 inhibitors under clinical development. – indicates that the drug is not an inhibitor of the receptor. No data indicates no published data available. √ indicates target inhibited. ABCG: ATP-binding cassette super-family G; GLUT: glucose transporter; MRP4: multidrug resistance protein 4; OAT: organic anion transporter; URAT: urate transporter
Probenecid is a non-specific uricosuric that inhibits multiple anion transporters (URAT1, OAT1, OAT3, and GLUT9) [6]. First developed in 1949, probenecid was initially used to reduce the renal clearance of penicillin, and was the first urate-lowering therapy to be commercialized and widely used. Today, it is rarely used for either purpose, its popularity dwindled with the introduction of allopurinol in the 1960s [7, 41, 42], and it has only moderate efficacy as a urate-lowering agent in patients with gout [24].
Although generally well tolerated, probenecid carries a risk of nephrolithiasis, and there is concern over multiple drug-drug interactions, namely with penicillin, furosemide, and methotrexate [6]. Some physicians advocate alkalinizing the urine of patients on probenecid to protect against nephrolithiasis, but the ACR guidelines recommend against this approach [11]. The risk of nephrolithiasis means that probenecid is not recommended in patients with a history of this condition, or in those with stage ≥ 3 chronic kidney diseases, in whom it lacks efficacy [6, 11, 17].
Sulfinpyrazone is thought to inhibit both URAT1 and MRP4 [43]. Although US Food and Drug Administration (FDA) approved, sulfinpyrazone has been discontinued by the manufacturer and is not generally available; as such, it has less available efficacy and safety data than probenecid and benzbromarone [6, 7].
Gastrointestinal intolerance, skin rashes, platelet aggregation impairment, and rare bone marrow toxicity are potential side effects of sulfinpyrazone [7]. Additionally, it is thought to cause renal failure through inhibition of prostaglandin synthesis in the kidney and platelets [44]. Various case reports from the 1980s and 1990s describe acute renal failure attributed to sulfinpyrazone [45–47].
Phenylbutazone is a nonsteroidal anti-inflammatory drug (NSAID) that was first approved as an analgesic in 1952 (particularly for arthritic pain) and was widely used for around 30 years. It has subsequently been withdrawn for use in humans owing to severe side effects including blood dyscrasias, although it remains in widespread use as an equine analgesic [48]. Similarly, after it was discovered that the muscle relaxant zoxazolamine had potent uricosuric activity, this was used briefly during the 1950s for treating gout. However, zoxazolamine was removed from the market shortly afterward amid reports of fatal hepatotoxicity [21]. Benziodarone is another historical uricosuric that was withdrawn due to reports of jaundice [49]. None of these agents are relevant in today’s gout treatment landscape.
Compared with probenecid, benzbromarone is a more potent uricosuric [12], with efficacy comparable to that of allopurinol [36, 50, 51]. Like probenecid, it is non-selective, acting mainly on URAT1, but also on GLUT9, OAT1, and OAT3 [6], although it has higher URAT1 specificity compared with probenecid [43].
Benzbromarone has been in use for treating gout for over 30 years [36]. Despite its efficacy, its clinical use has been hampered by real-world reports of severe liver toxicity, the incidence of which is estimated at 1 in 17,000 [37]. Furthermore, benzbromarone has never been approved by the FDA and has been withdrawn by its original manufacturer on account of its reported hepatotoxicity [6, 36]. Still marketed by other manufacturers, it is readily available in some Asian countries including China and Japan. Benzbromarone also has limited availability in Brazil, New Zealand, and some European countries [36].
Several hypotheses relating to hepatotoxicity caused by benzbromarone have been proposed [36]. Firstly, benzbromarone may cause hepatotoxicity in a similar manner to amiodarone, by inhibiting the mitochondrial respiratory chain and beta-oxidation. The resultant uncoupling of oxidative phosphorylation and generation of reactive oxygen species is hypothesized to lead to mitochondrial edema, apoptosis, and cellular necrosis [52]. Secondly, there is an unanticipated accumulation of 6-OH benzbromarone in poor cytochrome P450 2C9 metabolizers, which has been attributed to the sequential metabolic pathway through cytochrome P450 2C9 [53]. Thirdly, several metabolites of benzbromarone, such as orthoquinine and epoxide, have been implicated in its toxicity [36]. Benzbromarone can undergo sequential hydroxylation steps that can ultimately result in the formation of a reactive quinone intermediate, or metabolism by cytochrome P450 3A to epoxide intermediates and other reactive species; all of which are capable of adducting proteins to cause hepatotoxicity [53, 54].
Unlike probenecid, benzbromarone is considered suitable for use in patients with chronic kidney disease because it is predominantly metabolized by the liver [12, 36]. Indeed, while not considered at all in the ACR treatment guidelines, benzbromarone is recommended in the EULAR guidelines for patients with renal impairment, to be given with or without allopurinol [12].
In 2015, lesinurad became the first relatively selective URAT1, OAT1, OAT3, and OAT4 inhibitor to be approved in the US [55–57]. It was approved for use only after other therapies had failed [7], and in combination with an XOI, as evidence from clinical trials suggested that lesinurad monotherapy was associated with elevated creatinine levels [17]. A fixed-dose combination of lesinurad plus allopurinol was later approved in 2018 [58].
Lesinurad is more potent than probenecid [17], and more selective. Unlike probenecid, it does not alter the function of OAT1 and OAT3 in vivo and therefore these receptors are not clinically relevant to the uricosuric action of lesinurad [6, 55, 56]. Phase 3 trials demonstrated that lesinurad combined with allopurinol has superior efficacy (in terms of serum urate-lowering) to either allopurinol alone or febuxostat alone [59, 60].
As with several past uricosurics, patients treated with lesinurad required renal monitoring on account of potential renal toxicities [61]. Lesinurad nephrotoxicity or acute kidney injury is dose-dependent and thought to be related to uric acid crystalluria, although direct kidney toxicity cannot be excluded. Tubular cell death, with fatal outcomes, has been seen with high doses in rat studies, although tubular dilation and biochemical changes have been seen even at lower doses [61]. Lesinurad has a short half-life (around 5 hours) so it increases uric acid concentrations in urine transiently, but quite rapidly, after administration. It was hypothesized that the rapid spike in lesinurad effect could flood the tubule with uric acid and result in microcrystallization in the renal tubules and the urinary system, leading to acute injury; however, despite these concerns, no evidence of increased incidence of kidney stones was seen with lesinurad in clinical trials [59, 61].
Lesinurad was withdrawn by its manufacturer in 2019, and the fixed-dose combination of lesinurad plus allopurinol was withdrawn the following year [62]. It has been suggested this was due partly to renal safety concerns, and also for commercial reasons owing to its limited activity as a monotherapy and the need for dosing alongside allopurinol [17, 63]. However, the FDA database states that lesinurad discontinuation was unrelated to either drug safety or efficacy.
Verinurad is one of the most potent and specific URAT1 inhibitors yet discovered. It requires the presence of URAT1 residue Cys-32, a unique feature relevant to its potency [64].
Two phase 2 trials of verinurad in both Western (Study 1) and Japanese patients (Study 2) with gout have been completed [65]. Verinurad, compared with placebo, significantly reduced serum urate levels over 12 weeks in Study 1 and 16 weeks in Study 2. The least squares mean (± standard error) estimate of percentage change in serum urate levels from baseline in Study 1 was 1.2 ± 2.9 for placebo, –17.5 ± 2.8, –29.1 ± 2.8, and –34.4 ± 2.9 for verinurad 5, 10, and 12.5 mg, respectively. For Study 2, results were –2.4 ± 2.5 for placebo, and –31.7 ± 2.5, –51.7 ± 2.6, and –55.8 ± 2.5 for verinurad 5, 10, and 12.5 mg, respectively. The difference versus placebo was significant for each verinurad dose in both trials (p < 0.0001). However, renal-related treatment-emergent adverse events, including elevation in creatinine levels, occurred with a higher frequency in verinurad-treated patients.
Indeed, similar to lesinurad, verinurad increases the fractional excretion of uric acid within the first six hours of dosing, causing a “dumping” of uric acid through the tubules, potentially causing microcrystallization of uric acid in the renal tubules and consequent acute kidney injury. It was concluded that verinurad, like lesinurad, would be unsuitable as monotherapy in gout, but potentially could be combined with an XOI in hope of minimizing this “dumping” effect [65].
Several trials evaluating verinurad in combination with XOIs have been carried out, demonstrating superior urate-lowering effects relative to XOIs alone [6]. For example, when combined with allopurinol or febuxostat, verinurad dose-dependently reduced serum urate, and was generally well tolerated [66, 67].
Notwithstanding these early findings, no phase 3 trials of verinurad in gout have been registered on ClinicalTrials.gov to date, and it appears that clinical development of verinurad for this indication has been terminated [68].
Dotinurad is another selective URAT1 inhibitor with only minimal effects on other receptors (ABCG2, OAT1, and OAT3) [40]. It has been approved for treating hyperuricemia and gout in Japan since 2020 [40].
Two phase 3 trials [69, 70] have demonstrated that dotinurad is non-inferior to febuxostat or benzbromarone in terms of serum urate-lowering activity; notably, these trials identified no renal or hepatic safety signals [71]. Further, mild to moderate renal impairment did not adversely affect dotinurad’s serum urate-lowering effect.
Uricosurics, particularly those with a URAT1-specific mechanism of action, have been shown to be effective in lowering serum urate and treating patients with chronic gout [72]. However, as we have described, their use has been limited by various factors, not least renal and other safety concerns. Much recent interest has focused on developing URAT1 inhibitors that are more effective, but also safer than their predecessors. To that end, several experimental URAT1 inhibitors are currently in advanced clinical development (Table 3), most of which work specifically to inhibit URAT1 (Table 2).
Ruzinurad is an investigational agent that has been evaluated in two phase 2 trials, both conducted in China. In the first (a dose-ranging study), 198 Chinese patients were randomized to receive once-daily ruzinurad, benzbromarone, or placebo [73]. Ruzinurad was superior to placebo for all doses studied. Proportions of patients who achieved target serum urate of ≤ 360 μmol/L at week 5 were 32.5%, 72.5%, and 61.5% in the ruzinurad 5 mg, 10 mg, and benzbromarone groups, respectively; significantly higher than placebo (0%; p < 0.05 for ruzinurad 5 mg and 10 mg groups). The incidences of gout flares requiring intervention were similar across all groups, no serious adverse events occurred in any group, and no renal or hepatic safety signals were observed. In the second, a randomized trial in which 93 patients received one of three dose combinations of ruzinurad plus febuxostat, the combination was effective, with target serum urate levels achieved in 75–97% of patients across the groups [74]. One patient (1.1%) had a serious treatment-emergent adverse event of acute kidney injury, which resolved with treatment.
Following on from these phase 2 studies, it appears that a 36-week, phase 3, randomized, allopurinol-controlled trial has concluded (NCT04052932), but no results have yet been published.
Pozdeutinurad is a potent and selective URAT1 inhibitor that was designed by optimizing a benzbromarone metabolite scaffold. In three phase 2 trials, pozdeutinurad has been studied as monotherapy versus allopurinol or placebo, as well as combination therapy with allopurinol or febuxostat [75–77].
In one trial, pozdeutinurad showed promising efficacy for reducing serum urate, as well as monosodium urate crystal volume in tophi, assessed by photography, caliper measurement, and dual-energy computed tomography (DECT). After 3 months’ treatment, serum urate levels were reduced by nearly half for the patients who received pozdeutinurad either alone or with allopurinol, compared with around a 30% reduction for patients on allopurinol alone. Serum urate levels of < 6 and < 5 mg/dL were achieved by 86% and 64% of patients on pozdeutinurad, respectively, after 3 months’ treatment. Further, four patients (29%) experienced a complete resolution of one or more tophi after 6 months of treatment. There were no serious adverse events, no elevation of serum creatinine observed, and no other renal or hepatic safety signals related to pozdeutinurad treatment reported, indicating that this drug could provide a safer alternative to existing treatments [75].
In further support of this conclusion, a separate phase 2 trial showed that the serum urate-lowering effect of pozdeutinurad in patients with mild or moderate renal impairment was comparable to that seen in patients with normal renal function. In a phase 1 study of subjects with mild or moderate renal impairment, only mild increases in plasma pozdeutinurad exposure were noted after a single oral 100 mg dose. There were 50–60% reductions in serum urate, in line with the findings in patients with normal renal function. No clinically relevant serum creatinine elevations were observed in these patients [78].
A 12-month, global, phase 3, placebo-controlled trial of pozdeutinurad in patients with gout is currently recruiting (NCT06439602), with an estimated completion date in late 2026.
Epaminurad is a URAT1 inhibitor, also derived from benzbromarone [40]. Three phase 2 trials have been completed, and results of two of these have been published: in these, 140 Korean patients with gout were treated for 14 days with either epaminurad or placebo. The mean (standard deviation) reduction in serum urate was 54.3% (± 12.4) for patients receiving the highest tested epaminurad dose (10 mg), and 1.2% (± 7.75) for patients receiving placebo in the same part of the study [79]. A multicenter phase 3 trial to evaluate the efficacy and safety of epaminurad in combination with febuxostat is currently recruiting (NCT05815901), and investigational new drug applications have been submitted in Malaysia and South Korea, in anticipation of further studies in these regions.
Although they are derivatives of benzbromarone, epaminurad, and pozdeutinurad are more selective and have superior safety profiles [40, 75]. For example, in vitro, epaminurad shows less potential for hepatotoxicity compared with benzbromarone, owing to it having a lower potential for reactive metabolites to form in the liver, as well as reduced mitochondrial toxicity [80]. Similarly, in vitro, metabolic stability has been demonstrated for pozdeutinurad [81]. Unlike benzbromarone, pozdeutinurad does not undergo a sequential metabolic pathway through cytochrome P450 2C9 and the accumulation of 6-OH benzbromarone is avoided.
ABP-671 has been evaluated in a randomized, placebo-controlled, ascending dose phase 2a study involving 59 patients with gout and one patient with hyperuricemia. After 4 weeks, serum urate levels at all doses tested were significantly decreased from baseline compared with the placebo group, and ABP-671 was well tolerated. Overall, 3 (5.0%) participants experienced nephrolithiasis: 1 (2.1%) who received ABP 671 and 2 (16.7%) who received placebo [82]. A global phase 2b/3 study is currently active, but not recruiting (NCT05818085).
Xininurad is another URAT1 inhibitor under development [6]. No clinical trial data are currently available, but a phase 2/3 trial is underway in China (CTR20222360).
Other urate-lowering therapies in varying stages of clinical development include SAP-001 (NCT04040816), AC201 (NCT02287818), YL-90148, ACQT-1127, HP-501, D-0120 (NCT03923868), FCN-207 (NCT04622124); CDER167 and NC-2700 are in preclinical development [6].
Summary of clinical development to date for pipeline uricosurics
| Uricosuric | Design | N | Population | Intervention | Key efficacy findings | Key safety findings |
|---|---|---|---|---|---|---|
| Ruzinurad (SHR4640) | Phase 2; randomized; multicenter; double-blind; placebo/active-controlled (NCT03185793) [73] | 198 | Chinese participants with:
| Ruzinurad 2.5, 5, or 10 mg QD, 50 mg benzbromarone QD, or matched placebo, once-daily for 5 weeks |
|
|
| Phase 2; randomized; multicenter; double-blind; parallel-controlled (CTR 20192429) [74] | 93 | Chinese participants with:
| Ruzinurad plus febuxostat in one of three regimens:
|
|
| |
| Phase 3; randomized; multicenter; double-blind; active-controlled (NCT04052932) [114] | 594 | Chinese participants with sUA ≥ 480 μmol/L with gout and BMI ≥ 18 and ≤ 35 kg/m2 | Ruzinurad (2 dose levels, not published), allopurinol 300 mg, or placebo orally QD for 36 weeks | Unavailable (study completed in July 2021, but results so far unpublished) | Unavailable (study completed in July 2021, but results so far unpublished) | |
| Pozdeutinurad (AR882) | Phase 2; global; proof-of-concept (NCT05253833) [75] | 42 |
| Pozdeutinurad 75 mg, pozdeutinurad 50 mg + allopurinol, or allopurinol alone at doses up to 300 mg, all orally QD for 6 months | After 3 months (primary endpoint):
After 6 months:
|
|
| Phase 1 and 2a [78] | 28 | Patients with gout and either mild or moderate impairment, or normal renal function | Oral pozdeutinurad up to 75 mg as multiple doses over 1 week, or up to 150 mg as a single dose | In participants with mild or moderate renal impairment, after a single 100 mg dose:
In participants with mild or moderate renal impairment, after multiple 50 mg doses:
| No clinically relevant serum creatinine elevation | |
| Phase 2b; randomized; double-blind; placebo-controlled (NCT05119686) [77] | 140 | Patients with gout and eGFR > 30 mL/min | Pozdeutinurad 50 or 75 mg, or placebo, orally QD for 12 weeks |
|
| |
| Phase 3; randomized; multicenter; double-blind; placebo-controlled (NCT06439602) | Approximately 750 | Patients with gout and ≥ 2 flares in the last year; sUA ≥ 7 mg/dL if on urate-lowering therapy, or > 6 mg if not on urate-lowering therapy; serum creatinine < 3.0 mg/dL and estimated creatinine clearance ≥ 30 mL/min | Pozdeutinurad 50 or 75 mg, or placebo, orally QD for 12 months | Unavailable: estimated study completion November 2026 | Unavailable: estimated study completion November 2026 | |
| Verinurad (RDEA3170) | Two phase 2; randomized; multicenter; double-blind; placebo-controlled (NCT01927198; NCT02078219) [65] | Study 1: 172; Study 2: 204 | Western (Study 1) patients with gout and sUA > 6.5 and ≤ 10 mg/dL; Japanese (Study 2) patients with gout sUA ≤ 10 mg/dL, and either:
|
| Least squares mean (± SE) percentage change in sUA from baseline:
|
|
| Phase 2a; open-label; randomized; multicenter (NCT02246673) [67] | 64 | Patients with gout, with estimated creatinine clearance ≥ 60 mL/min | Complex treatment arms: verinurad at doses 2.5–20 mg given in combination with febuxostat either 40 or 80 mg, in one of four different sequences, all oral, QD, and for 4 weeks |
|
| |
| Phase 2a; open-label; randomized; multicenter (NCT02498652) [66] | 41 | Patients with gout, sUA ≥ 8 mg/dL, and estimated creatinine clearance ≥ 60 mL/min |
|
|
| |
| Epaminurad (URC102) | Two phase 2; randomized; multicenter; placebo-controlled (NCT02290210 and NCT02557126) [79] | 64 (Study 1); 76 (Study 2) | Korean patients with gout and sUA ≥ 7 mg/dL and ≤ 10 mg/dL |
| Mean (± SD) sUA changes were dose-dependent, ranging from:
|
|
| Phase 3; randomized; multicenter; double blind; active-controlled (NCT05815901) [115] | Approximately 588 | Korean patients with gout, ACR/EULAR 2015 score ≥ 8, sUA ≥ 7 mg/dL |
| Unavailable: estimated study completion August 2025 | Unavailable: estimated study completion August 2025 | |
| ABP-671 | Phase 2a; randomized; multicenter; double-blind; placebo-controlled (NCT04638543) [82] | 60 | Patients with gout or hyperuricemia |
|
| 3 (5.0%) participants reported nephrolithiasis: 1 (2.1 %) in the ABP-671 groups and 2 (16.7%) in the placebo groups |
| Phase 2b/3; multicenter, randomized, double-blind (NCT05818085) [116] | Estimated 580 | Patients with gout; sUA ≥ 7.0 mg/dL |
| Unavailable: estimated primary completion of August 2024, and study completion expected August 2025. However, no results have yet been posted | Unavailable: estimated primary completion of August 2024, and study completion expected August 2025. However, no results have yet been posted | |
| Xininurad (XNW-3009) | Phase 3; multicenter, randomized, double-blind; active-controlled (CTR20222360) [117] | Unknown | Patients with gout (details unpublished) | Febuxostat or XNW-3009 (doses unpublished) | Unavailable: study recruiting, but primary completion date unknown | Unavailable: study recruiting, but primary completion date unknown |
ACR: American College of Rheumatology; AEs: adverse events; BID: twice-daily dosing; BMI: body mass index; EULAR: European Alliance of Associations for Rheumatology; QD: once-daily dosing; SD: standard deviation; SE: standard error; sUA: serum uric acid; TEAE: treatment-emergent adverse event; Cmax: maximum concentration; AUClast: area under the curve from time of dosing to the last measurable concentration; eGFR: estimated glomerular filtration rate
URAT1 inhibitors all bind to a common site in the core of this transporter molecule, sterically hindering the transit of uric acid through its substrate channel. Different inhibitors have vastly different potencies and interact with different amino acids within the transporter. Those with the highest inhibitory potency all interact with URAT1 residues Ser-35, Phe-365 (which are important for uric acid transport kinetics), and Ile-481 [64].
Based on early-stage clinical trials of the novel, selective URAT1 inhibitors, it appears that most of these investigational treatments do not carry the same risk of liver and kidney toxicity as some of the more traditional uricosurics. Indeed, a model-based analysis of a systematic review of randomized controlled trials on URAT1 inhibitors concluded that uric acid-lowering efficacy is not an independent factor for the renal safety risk of different URAT1 inhibitors, suggesting instead that structural differences between the drugs could be responsible for the variation [83]. The chemical structures of selected uricosurics are shown in Figure 1. However, since liver and kidney toxicity events are rare, even with the traditional uricosurics of known risk such as benzbromarone, the limited amount of data currently available is not yet sufficient to conclude this firmly.
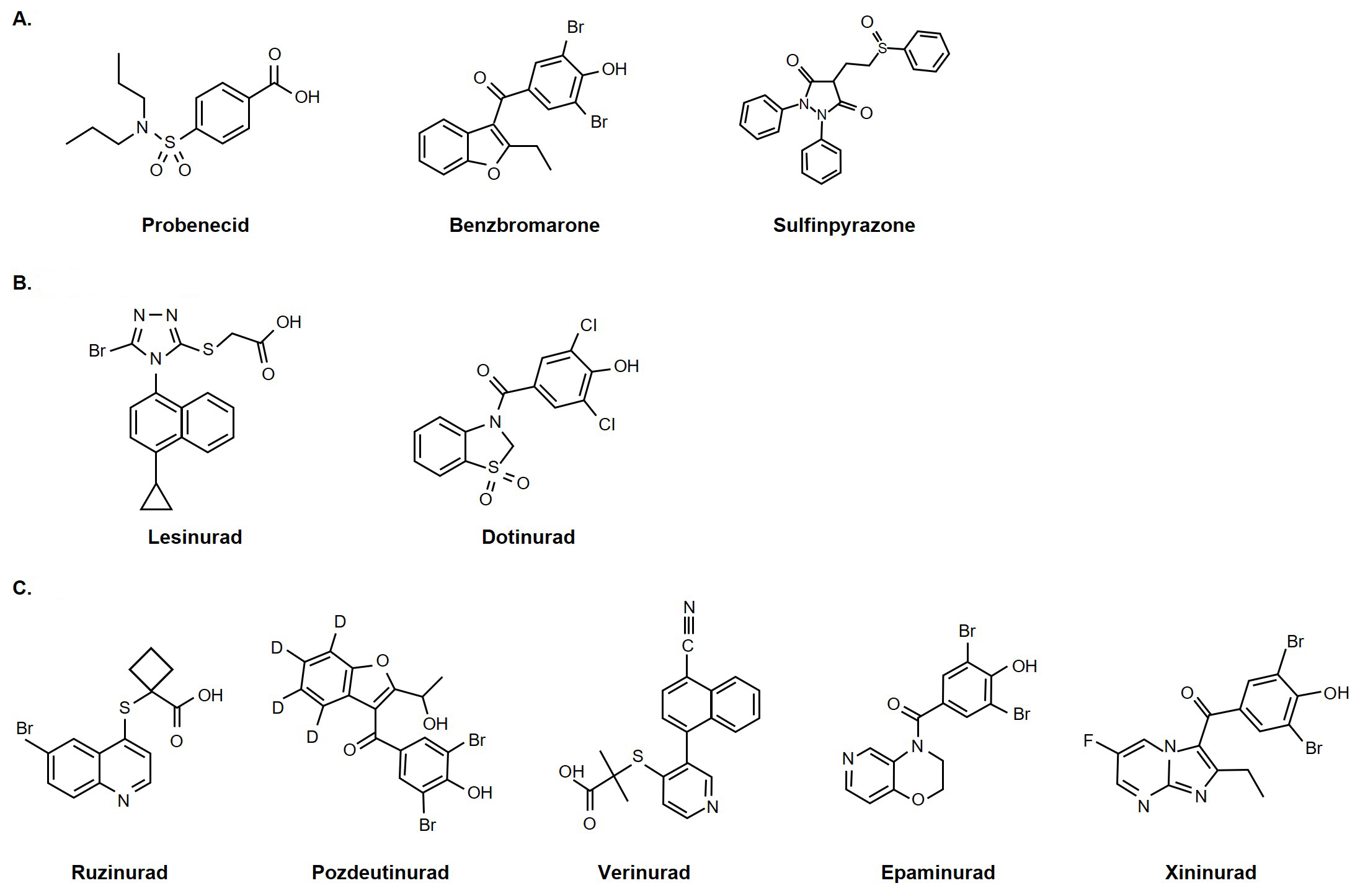
Chemical structures of selected uricosurics. (A) Traditional uricosurics; (B) modern uricosurics; (C) pipeline selective URAT1 inhibitors. Probenecid, benzbromarone, lesinurad, dotinurad, epaminurad were reprinted from [40]. (CC BY 4.0). PubChem source: xininurad, CID 139329673 [118] (https://pubchem.ncbi.nlm.nih.gov/compound/Xininurad); pozdeutinurad, CID 140959988 [119] (https://pubchem.ncbi.nlm.nih.gov/compound/Pozdeutinurad); verinurad, CID 54767229 [120] (https://pubchem.ncbi.nlm.nih.gov/compound/Verinurad); sulfinpyrazone, CID 5342 [121] (https://pubchem.ncbi.nlm.nih.gov/compound/Sulfinpyrazone); ruzinurad, CID 86294127 [122] (https://pubchem.ncbi.nlm.nih.gov/compound/Ruzinurad)
Pharmacokinetic measures that provide useful context for the renal safety profile are the maximum (peak) concentration (Cmax) of the drug after dosing, the measure of steady state (i.e. the rate of drug input is equal to drug elimination) using the area under the concentration curve steady state (AUCss), and the minimum (trough) concentration (Cmin) of a drug after dosing. A comparison of the pharmacokinetic profiles of known URAT1 inhibitors revealed differences between those that have and have not shown a high risk of renal safety issues (Table 4). This suggests that Cmax/AUCss and Cmax/Cmin ratios may provide a helpful tool to recognize the risk: drugs with low Cmax/AUCss and Cmax/Cmin ratios appeared to have a more favorable renal safety profile than those with high Cmax/AUCss and Cmax/Cmin ratios, such as lesinurad or verinurad.
Ratio of Cmax/AUCss or Cmax/Cmin in steady-state pharmacokinetic profile for selected URAT1 inhibitors
| Drug metabolite | URAT1 IC50 (nM) | Cmax/AUCss | Cmax/Cmin | Renal safety profile |
|---|---|---|---|---|
| Lesinurad [123]* | 7,300 | 0.19 | 88 | Serious renal issues, needed to combine with XOIs |
| Verinurad [124]* | < 50 | 0.52 | 33 | Second generation of lesinurad by AstraZeneca; failed development; needed to combine with XOIs |
| Dotinurad [125]* | < 50 | 0.09 | 4.7 | Approved by the Japanese Pharmaceuticals and Medical Devices Agency; no known renal-related safety events |
| Benzbromarone [126]* | 196 | 0.14 | 18 | Used in Asia; no known renal-related safety events |
| 6-OH benzbromarone [126]* | NA | 0.07 | 2.83 | NA |
| Pozdeutinurad (AR882) [81, 127]* | 67 | 0.08 | 2.7 | In development; no renal safety signals identified in phase 2 trials |
* Data have been derived from the cited publications. AUCss: area under the curve steady state; Cmax: maximum concentration; Cmin: minimum concentration; IC50: half-maximal inhibitory concentration; NA: not available; URAT1: urate transporter-1; XOI: xanthine oxidase inhibitor
The current treatment paradigm for chronic gout is suboptimal and there is a need for new treatments with better efficacy and safety. As gout is closely linked to other metabolic diseases, lifestyle modifications regarding diet and exercise can sometimes be beneficial. Bariatric surgery has the potential to be an efficacious intervention that not only manages the weight of patients but can also lead to improvements in chronic gout symptoms over the long term [84, 85]. Allopurinol is the recommended first-line urate-lowering therapy for symptomatic gout [11, 12]; however, a significant proportion of patients (58–79%) treated with allopurinol do not achieve serum urate concentrations of ≤ 6 mg/dL [15, 86]. The recent STOP-GOUT study showed more promising findings (around 80% of patients on both allopurinol and febuxostat achieved this target) but the study had a high rate of participant withdrawal, at around 20% [87]. This is not unusual in studies for gout treatment [88, 89], and more widely, medication adherence among patients with gout is notoriously poor due in part to a lack of efficacy (perceived or real) [90–92].
As well as suboptimal efficacy for some patients, allopurinol and other urate-lowering therapies carry some significant safety risks. For example, allopurinol can cause severe allergic reactions [6], and febuxostat carries a controversial black box warning for increased risk of cardiovascular death, which limits its indication to only those patients with inadequate response or intolerance to allopurinol [89, 93, 94]. Pegloticase, an intravenous recombinant uricase usually reserved for patients who have not responded to XOIs or uricosurics, is frequently associated with infusion reaction, particularly when administered without concomitant immunosuppressant therapy [11, 12, 95, 96].
Current EULAR and ACR guidelines recommend a uricosuric (alone or in combination with a XOI) only after one or two lines of XOIs have failed to adequately control serum urate levels, or were not tolerated [11, 12] (Figure 2). As such, use of uricosurics in practice remains infrequent [11]. Further, the ACR guidelines state that patients with known renal calculi or moderate-to-severe chronic kidney disease (stage > 3) are unsuitable for treatment with existing uricosurics [11]. However, it is important to note that this position could change should new uricosurics with greater efficacy and enhanced safety, particularly in patients with renal disease, become available.
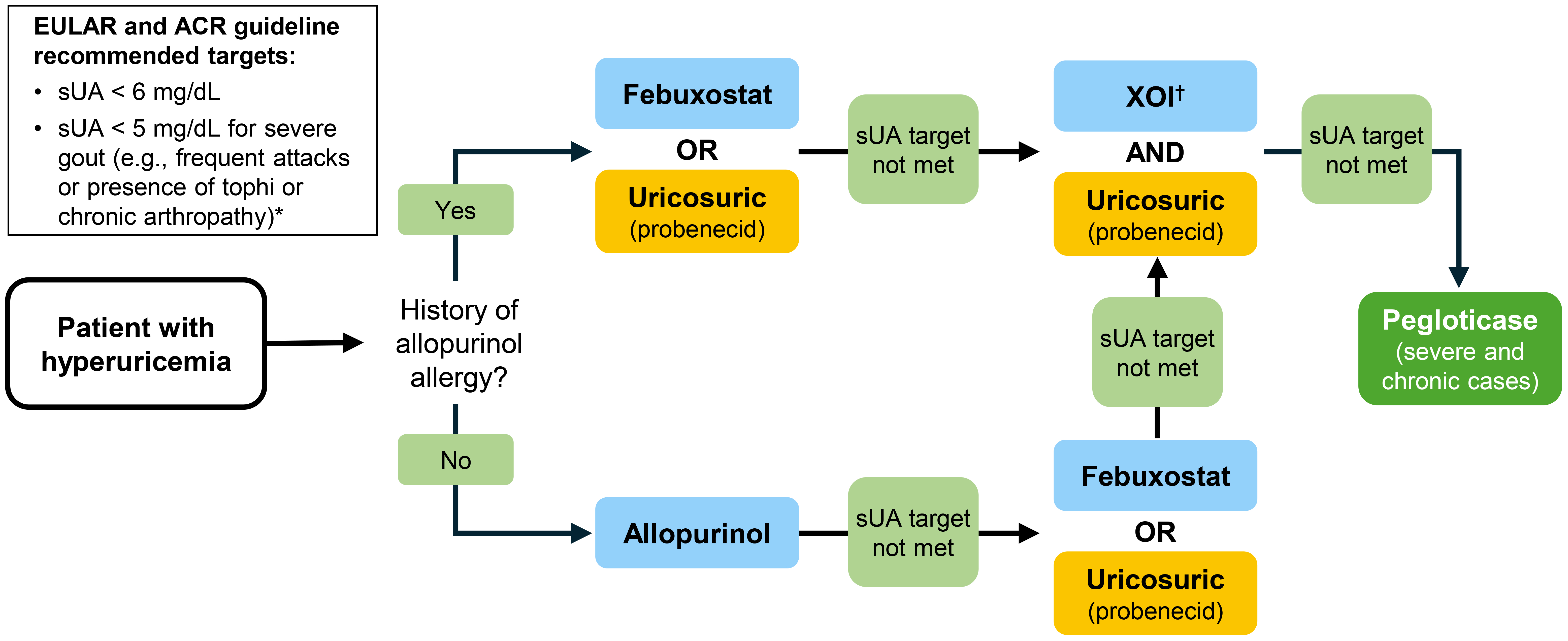
Current position of uricosurics in the treatment of hyperuricemia with urate-lowering therapy in patients with gout in the US and Europe. Adapted with permission from Richette et al. [12]. © 2019 BMJ Publishing Group Ltd & European League Against Rheumatism. * Recommendation appears in EULAR guidelines only. † Febuxostat for patients with history of allopurinol allergy. ACR: American College of Rheumatology; EULAR: European Alliance of Associations for Rheumatology; sUA: serum urate; XOI: xanthine oxidase inhibitor
The ability of the novel, selective URAT1 inhibitors to specifically induce renal uric acid excretion may present an opportunity to improve urate-lowering while maintaining the body in a more homeostatic state in patients with gout, compared with more traditional treatments. Although XOIs remain the current first choice for urate-lowering therapies, they could be superseded by the emerging URAT1 inhibitors if the clinical efficacy and safety seen in phase 2 development can be replicated in large-scale clinical trials. Indeed, early data are promising, but the positioning of future uricosurics may ultimately depend on their potency.
Current evidence suggests a potential benefit of urate-lowering on comorbidities that commonly occur alongside gout, such as renal and cardiovascular disease [14, 17]. Most of this evidence centers on the urate-lowering effects of XOIs [6, 14], but some modern URAT1 inhibitors have also shown evidence of activity beyond simply lowering serum urate. This suggests that some agents may provide additional benefits to patients with gout, or indeed, in indications other than gout.
For example, in patients with T2DM and hyperuricemia, verinurad in combination with febuxostat has demonstrated the ability to reduce both albuminuria and hyperuricemia in patients receiving standard-of-care renoprotective treatment and optimal hypertensive therapy. In a multicenter phase 2 study, the mean percentage change (95% confidence interval) from baseline in urine albumin-to-creatinine ratio after 12 weeks’ treatment was –48.7% (–64.8 to –25.1), compared with –15.3% (–43.2 to 26.4) for placebo, suggesting that verinurad could provide additional renoprotection for patients with T2DM and hyperuricemia [97].
Further, elevated serum urate correlates with poor prognosis in patients with heart failure, potentially due to uric acid crystals causing inflammation in coronary vessel walls [98]. Therefore, verinurad plus allopurinol was investigated in a phase 2b trial (AMETHYST; NCT04327024) including 159 patients with heart failure with preserved ejection fraction [99]. Unfortunately, the study did not meet its efficacy endpoints: verinurad plus allopurinol did not significantly improve exercise capacity or heart failure symptom status after 32 weeks’ treatment, although the treatment was generally well tolerated [100].
As with verinurad, dotinurad has shown potential to be useful for reducing serum urate in patients with diabetes, metabolic syndrome, and cardiovascular disease [101, 102], as well as for reducing inflammation in gouty joints. Dotinurad is known to inhibit the nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing protein-3 (NLRP3) inflammasome in macrophages at clinical concentrations [6, 101]. The NLRP3 inflammasome is a protein complex that mediates the immune response to crystals deposited in the joints of patients with gout [6], by activating interleukin (IL)-1β and IL-18 via the caspase cascade. Its inhibition reduces both caspase-1 and IL-1β production, which may help reduce inflammation, especially important at the beginning of gout treatment when flares are common [101]. Similarly, the NLRP3 inflammasome is implicated in diabetes and metabolic syndrome, and these patients also have elevated serum levels of IL-1β and IL-18 [101].
Pozdeutinurad has demonstrated some early evidence of anti-inflammatory effect in patients with gout. In one phase 2 trial, there was a reduction in gout flares over 3 months in patients treated with pozdeutinurad (with or without allopurinol) compared with allopurinol alone [75].
Traditional uricosurics also have the potential for other therapeutic uses. Probenecid inhibits pannexin1, glycoprotein channels that allow for the flux of small molecules between intra- and extra-cellular spaces, and is an important therapeutic target in inflammation [103]. Indeed, pannexin1 reduces caspase-1 and inflammasome activation in several cell types, thus helping to dampen the inflammatory response. This may be useful in treating the inflammatory sequelae of gout, but also in other inflammatory diseases, such as stroke and central nervous system trauma [104].
Both benzbromarone and sulfinpyrazone exert dual anti-inflammatory and anti-oxidative effects [103, 105, 106]. Both have direct scavenging activity against superoxide radicals, and benzbromarone can reduce the levels of intracellular reactive oxygen species produced by angiotensin II and uric acid in vascular endothelial cells; it has also been shown to reduce oxidative stress in hypertensive rat models [106, 107]. Further, benzbromarone may be protective against propofol-induced inflammation and injury. Propofol can injure human brain microvascular endothelial cells (HBMVECs) and disrupt the blood-brain barrier, leading to severe neurotoxicity. In vitro studies show that benzbromarone can redeem the viability of HBMVECs which propofol has reduced, as well as dramatically suppressing their oxidative stress, and can inhibit the excessive production of pro-inflammatory mediators and adhesion molecules [108].
Uricosurics have a long history in the treatment of chronic gout, but until recently their use has been limited due to suboptimal efficacy, particularly in patients with renal disease and other safety concerns. A host of novel, selective, and potent URAT1 inhibitors show promise as efficacious agents with better efficacy and/or safety profiles than currently available options.
ABCG2: ATP-binding cassette super-family G2
ACR: American College of Rheumatology
AUCss: area under the concentration curve steady state
Cmax: maximum concentration
Cmin: minimum concentration
EULAR: European Alliance of Associations for Rheumatology
FDA: US Food and Drug Administration
GLUT9: glucose transporter 9
HBMVECs: human brain microvascular endothelial cells
IL: interleukin
MRP4: multidrug resistance protein 4
NLRP3: nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing protein-3
NSAID: nonsteroidal anti-inflammatory drug
OAT: organic anion transporter
T2DM: type 2 diabetes mellitus
URAT1: urate transporter-1
XOI: xanthine oxidase inhibitor
The authors thank Nicola Beadle and Emily Atkinson of Alchemy Medical Writing Ltd. for providing medical writing assistance under the direction of the authors (sponsored by Arthrosi Therapeutics Inc.).
RTK: Conceptualization, Writing—original draft, Writing—review & editing. ZS, SY, and LTY: Writing—original draft. MHP: Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
RTK, ZS, SY, and LTY are all employees of Arthrosi Therapeutics Inc. MHP has consulted for Sobi and Amgen. RTK who is the Editorial Board Member of Exploration of Musculoskeletal Diseases had no involvement in the journal review process of this manuscript.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Benjamin Plotz ... Michael H. Pillinger
Hamish Farquhar ... Lisa K. Stamp
Naomi Schlesinger, Dan Kaufmann
Mark D. Russell, James B. Galloway
Robin Christensen ... Lisa K. Stamp
Edward Roddy ... Christian D. Mallen
Philip L. Riches ... Amrey Krause
Emilie Schurenberg ... Kenneth G. Saag
Enrique Calvo-Aranda ... Marta Novella-Navarro
Orsolya I. Gaal ... Tania O. Crișan
