Review
Review

Explor Musculoskeletal Dis. 2025;3:100782 DOI: https://doi.org/10.37349/emd.2025.100782
Received: December 05, 2024 Accepted: January 08, 2025 Published: February 08, 2025
Academic Editor: Valderilio Feijó Azevedo, Federal University of Paraná, Brazil; Fernando Pérez-Ruiz, Cruces University Hospital, Spain
The article belongs to the special issue Biosimilars: State of the Art in the Treatment of Rheumatic Diseases
Routine regulatory requirements for large comparative efficacy trials (CETs) to support marketing approval of monoclonal antibody (mAb) biosimilars have been the focus of extensive debate in the last few years. This review examines the mounting evidence, accumulated over the past decade, focusing on relevant literature and data published in the European Product Assessment Reports (EPARs) for the fifteen anti-TNFα biosimilars approved to date. The potential for residual uncertainties that may require resolution through CETs following comparative physico-chemical, in-vitro potency, and single dose studies in healthy subjects is examined. It is noted that structural and physicochemical differences between biosimilars and reference products are detectable using modern analytical methods at levels well below those that could impact clinical outcomes, and that in vitro potency testing is fully capable of revealing clinically relevant differences. Additionally, comparative pharmacokinetic studies in healthy participants provide a sensitive assessment of potential differences in drug exposure and immunogenicity. The added value of CETs is further questioned in the light of the fact that anti-TNFα’s display a flat dose-response relationship, meaning that unlike cell-based assays, CETs have limited sensitivity to detect potency differences. Initial concerns about extrapolating data from rheumatoid arthritis studies to support marketing approval of other indications such as inflammatory bowel disease for which not all anti-TNFα’s are effective have been alleviated by post-approval studies. Additionally, as of the end of 2024, no cases where clinical efficacy data were necessary to resolve residual quality concerns have arisen following regulatory assessment of 56 mAb biosimilars and fusion proteins. CETs add significant cost and delay to the development of biosimilars and the time is now ripe to re-examine the need for these CET’s and for further evolution in regulatory thinking.
Biological anti-tumor necrosis factor alpha (anti-TNFα) therapies offer a powerful treatment option for inflammatory conditions such as rheumatoid arthritis (RA), juvenile idiopathic arthritis, ankylosing spondylitis (AS), psoriatic arthritis, psoriasis (Ps), hidradenitis suppurativa, Crohn’s disease (CD), ulcerative colitis (UC), and uveitis. Five different anti-TNFα biological entities have been approved for marketing in the United States (US) and European Union (EU); these are the monoclonal antibodies (mAbs) infliximab, adalimumab and golimumab; the pegylated antibody fragment certolizumab pegol and the fusion protein, etanercept. The three mAb anti-TNFα entities are of the IgG1 class. Of these, biosimilars have been approved in major markets for infliximab, adalimumab and etanercept. The introduction of these biosimilars has expanded access to anti-TNFα’s. To date (end of 2024) 15 anti-TNFα biosimilars have been approved — nine adalimumab, three infliximab and three etanercept, offering comparable efficacy and safety to the originators’ products but at significantly lower cost.
This review looks at over a decade of experience in the development and use of anti-TNFα biosimilars and discusses the extent to which clinical data are needed to support future anti-TNFα biosimilar marketing approvals, focusing on published EU experience. Whereas the focus here is on anti-TNFα’s, similar considerations apply to most mAb biosimilars [1]. Regulatory acceptance of a more targeted biosimilar development program, limiting the need for comparative efficacy trials (CETs) would enhance availability and access to life-saving and life-changing therapies, not only for inflammatory diseases but across many therapeutic areas, including cancer.
Compelling evidence has accumulated from the 15 different anti-TNFα biosimilar MA’s (marketing authorization) approved to date [2–17], indicating that CETs provide no additional informational value over comparative analytical, in vitro, and pharmacokinetic (PK) studies; in summary, this evidence includes the following:
Analytical methods detect structural and physicochemical differences between biosimilars and reference products at levels well below those which could impact clinical outcomes.
Therapeutic response in most indications is mediated solely by anti-TNFα neutralization, which can be easily monitored in vitro; only in inflammatory bowel disease (IBD), in which CETs have not been required, might other mechanisms be involved.
Comprehensive in vitro testing programs are capable of detecting all potentially clinically relevant differences. In all cases to date, quality differences have been addressed through cell-based assays without needing clinical data.
Studies in healthy volunteers have provided a sensitive assessment of drug exposure and immunogenicity.
Anti-TNFα’s display a flat dose-response relationship, meaning that, unlike cell-based assays, CETs have limited sensitivity to detect potency differences.
Despite major structural differences, the five approved anti-TNFα’s show similar efficacy in RA, the indication most frequently used in anti-TNFα biosimilar CETs, suggesting that, provided TNFα binding affinity is adequate, even major structural differences have little therapeutic consequence in the single indication most frequently studied to support biosimilarity of anti-TNFα’s.
The concept of biosimilar mAbs, represented by the anti-TNFα’s discussed in this article, initially faced immense skepticism amongst healthcare professionals, regulators and many in industry. A leading immunologist stated in June 2006 that mAb’s “would always need to be considered as unique proteins” [18]. Scientific thinking rapidly evolved and, just two years later, the then-chair of the European Committee for Medicinal Products for Human Use (CHMP) Biosimilar Working Party wrote that “it remains to be seen whether the development of a biosimilar mAb is possible and feasible” [19]. By 2012 the same authors wrote “as of this time, there is no marketing authorization for a biosimilar mAb, but the stage is set!” [20]. Shortly thereafter the marketing application for the first mAb biosimilar (infliximab) was filed in the EU; and was approved by the Commission in September 2013, heralding the dawn of biosimilar mAbs [2].
At the time of this first filing in 2012, knowledge on the structural functional relationships relating to anti-TNFα’s was not fully understood, and CETs were considered by all stakeholders to be essential to support biosimilarity. As such, this first biosimilar anti-TNFα was supported by two clinical trials, the first demonstrating PK equivalence in 250 AS patients and the second showing therapeutic equivalence in 606 patients with RA [2]. That was over a decade ago and the time is now ripe to re-examine the need for these CET’s and for further evolution in regulatory thinking.
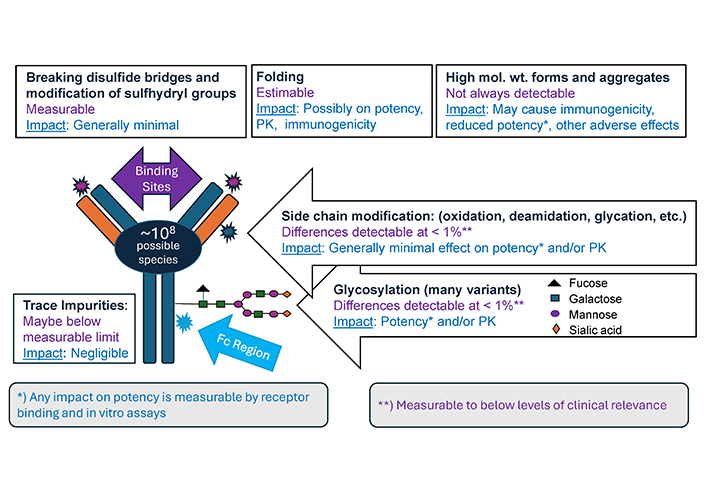
The structure of IgG1 mAbs such as infliximab, adalimumab and golimumab is shown in Figure 1 below. IgG1 comprises two heavy chains and two light chains held together by disulfide bonds and is glycosylated with oligosaccharides of biantennary structure attached to asparagine at position 297 on the heavy chain. mAbs are complex, and some differences from the originator product are inevitable. In fact, differences exist between one batch and the next for any protein-based product and process changes by originators have, on occasion, resulted in significant alterations in key quality attributes, exceeding what might be expected between a biosimilar and its reference product [21, 22].
The first step in developing a biosimilar is to match its structure and quality attributes to the originator product. Typically, over thirty quality attributes are studied to support biosimilarity. These include confirmation of an identical amino acid sequence, comparable folding of the amino acid chains, extent of amino acid side chain modifications, structure of attached sugars (oligosaccharides), disulfide bond formation, changes at the amino- and carboxy-terminals, degree of aggregation and fragmentation, and more. Each batch of a mAb product includes a heterogenous population of many slightly different molecules (see Figure 1). It has been calculated that, approximately 108 potential variants could theoretically exist in a single sample [23]. Understanding of these structural modifications displayed by IgG antibodies has increased tremendously over the past two decades, driven by their increasing therapeutic importance and advancement in analytical techniques [24].
A key attribute is the amino acid sequence, which must exactly match that of the originator product. Correct protein folding may be critical for functionality and numerous methods are available to investigate this.
Glycosylation is influenced by numerous factors relating to the manufacturing process, with variation seen in the oligosaccharides across the population of IgG1 molecules within a product batch. Controlling and optimizing mAb glycosylation is crucial when interactions with immune cells are important in mediating a therapeutic effect but are of limited or no relevance in the case of anti-TNFα’s [24]; this is discussed later. Nevertheless, the glycosylation profile can be well-characterized using methods such as high-pressure liquid chromatography coupled with mass spectrometry. Furthermore, the impact of any glycosylation difference on potency can be accurately measured by binding and cell-based assays [25].
Modifications of amino acid side chains (such as deamidation, oxidation, glycation) can also be measured down to levels well below those that could be clinically significant and are, in any case, mostly of no or very limited clinical impact. Furthermore, most of these modifications form in the body following administration, blurring any difference that may have originally existed [26].
Of all the attributes, aggregation and high molecular weight forms are considered to carry the greatest risk for immunogenicity, although their impact is likely variable depending on their nature [27–29]. Smaller aggregates such as dimers and trimers are not thought to be immunogenic [30]. These variants along with low molecular weight forms or fragments are measurable and are present at very low levels such that differences have had no discernible clinical effect. In CHMP assessments of some anti-TNFα’s, where levels of these variants differed slightly from the originator product, it was concluded that these would not be expected to affect clinical safety or efficacy.
Overall, it is evident that modern analytical methods are fully capable of detecting quality differences at levels well below the threshold of what might impact clinical efficacy or safety. Furthermore, for the 56 mAb and fusion protein biosimilars (excluding replicate MAs), approved in the EU until December 2024 (see Table 1), no case has been observed where therapeutic effect has been impacted by an unidentified quality difference.
Number of mAb and fusion protein biosimilars (excluding replicate MAs), approved until December 2024
| Biosimilars | Number | Biosimilars | Number | Biosimilars | Number |
|---|---|---|---|---|---|
| Adalimumab | 9 | Etanercept | 3 | Rituximab | 5 |
| Aflibercept | 6 | Infliximab | 3 | Tocilizumab | 1 |
| Bevacizumab | 3 | Natalizumab | 1 | Trastuzumab | 7 |
| Denosumab | 3 | Omalizumab | 1 | Ustekinumab | 8 |
| Eculizumab | 2 | Ranibizumab | 5 | ||
Receptor binding and cell-based potency assays provide a second layer of biosimilarity assurance. Where minor differences in quality attributes have occurred and been raised as potential concerns by the CHMP, these have always been satisfactorily addressed using potency assays.
Extremely comprehensive state-of-the-art in-vitro testing programs have been applied in the development of anti-TNFα biosimilars, comparing multiple batches of biosimilar and reference product, and involving a variety of binding and cell-based assays capturing different mechanisms of action under different simulated physiological conditions. As such the biological testing program provides a broad evidence base to support biosimilarity.
To understand the relevance of the biological testing program, it is helpful to have insight into how the application of these bioassays relates to the mechanism by which anti-TNFα’s exert their therapeutic effect. These mechanisms can be divided into two categories. The primary mechanism involves binding to- and neutralization of TNFα, thereby inhibiting subsequent downstream pro-inflammatory effects, such as apoptosis, cell proliferation, and cytokine release. In addition, anti-inflammatory effects may be exerted via reverse signaling causing apoptosis of TNFα expressing cells or activation of regulatory macrophages that dampen the inflammatory response. Assays used to study these effects include soluble and transmembrane TNFα binding affinity assays using methods such as surface plasmon resonance and/or enzyme-linked immunosorbent assay (ELISA) as well as cell-based TNFα binding affinity assays. In addition, sensitive in vitro cell-based assays are used to compare TNFα neutralization activity, TNFα-mediated apoptosis, cytokine release, reverse signaling and regulatory macrophage induction. This basket of assays should have the sensitivity to detect any potentially clinically relevant difference between the biosimilar and originator product.
The second mechanism that may be of relevance involves interaction with cells of the immune system, initiating cytotoxic responses such as antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis and complement-dependent cytotoxicity. While not considered to be of clinical importance in the case of anti-TNFα’s [31], these cytotoxic activities are effectively monitored during anti-TNFα biosimilar development.
Cell-based assays, e.g., ADCC, can be engineered to be highly sensitive, detecting effects that would not occur naturally. For example, under physiological conditions, the density of membrane bound TNFα on the cell surface is likely too low to initiate ADCC. However, assays capable of measuring ADCC induced by TNFα mAbs are routinely part of the biosimilar development program; these cell-based assays that may express supra-physiological levels of membrane bound TNFα are capable of detecting differences in ADCC well below any clinically-relevant threshold [2]. Interestingly, despite major structural differences, both etanercept and certolizumab pegol, which are not mAbs, show similar efficacy to the IgG1 anti-TNFα’s for key indications such as RA, Ps, psoriatic arthritis and AS, but etanercept, in particular, is not effective in IBD [32, 33]. This suggests that response to anti-TNFα treatment in IBD is more sensitive to structural difference compared to the other indications. Possible explanations for this might be that the mechanism by which anti-TNFα’s exert their effect in IBD requires more than just TNFα neutralization and may involve Fc-mediated effects, receptor cross-linking, and/or induction of reverse signaling. Since etanercept is not able to induce reverse signaling, whereas certolizumab can, and since certolizumab displays some efficacy in CD but etanercept does not, reverse signaling is likely a differentiating mechanism of action between IBD and the other indications for which anti-TNFs are approved [34, 35].
The key point is that all these mechanisms can be replicated using in vitro models capable of detecting differences at levels well below the threshold of clinical relevance. This is supported by the observation that in circumstances where anti-TNFα biosimilars have displayed small differences in ADCC, CHMP have always been satisfied that these were of no clinical relevance [2, 3, 5, 6, 16].
The PK profile of a biosimilar can be impacted by structural or binding differences or by differences in immunogenicity and, therefore, comparative PK studies form a key element of the biosimilarity program, providing a sensitive third layer of biosimilarity assurance. For anti-TNFα’s these studies are conducted as a single dose in healthy participants, which afford greater sensitivity than a study in patients, and generally recruit about 50 to 120 participants per arm depending on PK variability.
Differences in active ingredient content have on occasions impacted meeting PK equivalence criteria. Current CHMP guidelines state: “Correction for protein content may be acceptable on a case-by-case basis if pre-specified and adequately justified, with the results from the assay of the test and reference products being included in the protocol” [36].
Experience has shown that immunogenicity is often of no clinical significance, but it could be, and its effect is not easily predictable. The comparative PK studies in healthy participants provide valuable information on immunogenicity, as the sensitivity of immunogenicity assays has vastly improved over the past few decades. Today’s assays are capable of detecting anti-drug antibodies (ADAs) at levels below 10 ng/mL, whereas 100 ng/mL is generally considered to be the threshold for clinical significance [37, 38]. These modern sensitive assays allow for comparison of immunogenicity in a relatively small sample size, often even following a single administration to healthy participants. As an example, in pivotal trials, the repeated administration of the originator adalimumab displayed incidences of ADAs of 0.6% and 12.4% respectively, when administered with and without the immunosuppressant methotrexate [39]. In contrast, comparative PK studies conducted over 10 years later to support approval of adalimumab biosimilars yielded ADA incidences between 70% and over 95% [8–17]. For infliximab this incidence was 30% to 50% for the biosimilars [2–4] versus 13% in the original studies in CD patients, who did not receive immunosuppressive methotrexate [39]. Etanercept showed very low immunogenicity generally [5–7]. In all these cases, development of ADAs had little if any clinical significance, although ADAs have impacted PK parameters.
In head-to-head studies, differences in ADA levels have been seen for biosimilars versus the reference product. The adalimumab biosimilar, ABP 501, represents such an example, displaying lower formation of binding ADAs than the EU-sourced but not the US-sourced originator product [8]. According to the Guideline on similar biological medicinal products containing mAbs non-clinical and clinical issues [36], a lower immunogenicity for the biosimilar would not preclude biosimilarity. Interestingly, the presence of ADAs did impact overall drug exposure, which was approximately 20% to 30% lower for all three treatments in ADA-positive compared to ADA-negative participants. In the case of adalimumab, on average, the plasma half-life was six to seven days in the ADA-positive participants compared to 12 to 15 days in those who were ADA negative [40]. When comparing ABP 501 to EU-sourced originator product, exposure to ABP 501 was slightly higher but still the 90% CIs of the ratios of geometric means were fully contained within 0.80 to 1.25, limits, thereby meeting the requirements for biosimilarity [41]. In the case of another adalimumab biosimilar, GP17-103, the proportion of ADA-positive participants was also lower compared to the originator product, 57.9% vs. 71.1% and this too resulted in similar differences in PK parameters [11].
It is concluded that in the case of anti-TNFα’s, ADAs have not been observed to impact efficacy or safety but have affected PK parameters. In all cases, comparative PK studies in healthy subjects have proved sufficiently sensitive to determine clinically relevant immunogenic differences when comparing reference product and biosimilar.
Dose ranging and proof of concept studies (Phase 2 studies) are not required for the development of biosimilars in that this information can be inferred from the data available on the reference product. However, most international regulations and guidelines suggest that in the case of monoclonal and fusion protein biosimilars, large CETs be conducted in a population representative of the approved therapeutic indications of the original product and be sufficiently sensitive for detecting potential differences between the biosimilar and the original product [36, 42, 43]. In the case of anti-TNFα’s, either RA or Ps have been selected as suitable indications for comparing efficacy of the biosimilar with the originator product. These indications are favored, being common autoimmune disorders affecting over 1.0% of the world’s population [44, 45]. The relatively high magnitude of treatment effect, as well as the abundance of clinical data and literature available on these indications, allows for a deep understanding of the treatment effect.
A key question arising is, are these CETs of value in terms of ensuring efficacy and safety of biosimilars and whether these are generating data of any added value. As already discussed, all points of concern raised by CHMP in the development of anti-TNFα biosimilars have, to date, been addressed by analytical, biological and comparative PK data. Focusing on the CETs supporting the 15 anti-TNFα biosimilar entities approved to date, mean results for the primary endpoints ACR20 in RA and PASI75 in Ps are always within 5% for biosimilar versus reference product (Table 2). The observed slight differences are not statistically significant and likely the result of random intra-participant variability and not reflective of quality differences. ACR 20 (20% improvement in American College of Rheumatology Scale) is universally accepted as a primary outcome measure in trials investigating treatments for RA and similarly PASI75 (75% improvement in Psoriasis Area and Severity Index) is universally accepted as a primary outcome measure in trials investigating treatments for Ps.
Mean primary outcome results (ACR20 for RA and PASI75 for Ps) for the 15 comparative efficacy trials performed on anti-TNFα biosimilars to date. Results are for the Per Protocol Set (PPS) except in the case of ABP 501 and AVT02 where data from Full Analysis Set is provided as PPS data were not available
| Product | Week that 1 endpoint measured | Approvalyear | Biosimilar (B)% | Ref. Product (R)% | Response difference (B – R)% (95% CI) |
|---|---|---|---|---|---|
| Infliximab: Studies in rheumatoid arthritis (N = 478–650; 1:1 randomization) | |||||
| CT-P13 [2] | Week 30 | 2013 | 73.4% | 69.7% | +3.7% (–0.04, 0.12) |
| SB2 [3] | Week 30 | 2016 | 64.1% | 66.0% | –1.88% (–10.26, 6.51) |
| GP-11 [4] | Week 14 | 2018 | 66.7% | 67.2% | –0.58% (–8.42, 7.23) |
| Etanercept: Studies in rheumatoid arthritis (N = 427–481; 1:1 randomization) | |||||
| SB4 [5] | Week 24 | 2015 | 78.1% | 80.3% | –2.2% (–9.41, 4.18) |
| YLB113 [7] | Week 24 | 2020 | 86.0% | 90.6% | –4.6% (–10.1, 0.8) |
| Etanercept: Studies in psoriasis (N = 571; 1:1 randomization) | |||||
| GP15 [6] | Week 16 | 2017 | 73.4% | 75.7% | –2.3% (–9.85, 5.30) |
| Adalimumab: Studies in rheumatoid arthritis (N = 476–648; 1:1 randomization) | |||||
| ABP 501 [8] | Week 24 | 2017 | 74.6% | 72.4% | +2.604% (–4.941, 10.149) |
| SB5 [9] | Week 24 | 2017 | 72.4% | 72.2% | +0.1% (−7.83, 8.13) |
| FKB327 [10] | Week 24 | 2018 | 79.3% | 79.7% | –0.4% (–6.7, 5.9) |
| PF06410293 [13] | Week 12 | 2020 | 70.8% | 75.2% | –4.41% (N/A) |
| CT-P17 [14] | Week 24 | 2020 | 87.0% | 87.0% | +0.06% (–5.60, 5.78) |
| BI 69550 [17] | Week 24 | 2017 | 70.3% | 68.8% | +1.6% (–6.6, 9.8) |
| Adalimumab: Studies in psoriasis (N = 393–441; 1:1 randomization) | |||||
| GP2017 [11] | Week 16 | 2018 | 66.8% | 65.0% | +1.8% (7.46, 11.15) |
| MSB2022 [15] | Week 16 | 2019 | 90% | 92% | –1.86% (–7.82, 4.16) |
| AVT02 [16] | Week 16 | 2022 | 89.2% | 86.9% | +2.3% (–1.34, 5.88) |
The requisite CETs take at least 2 years to complete and to date have, in total, included just under 8,000 patients at an estimated cost of about a billion dollars, yet based on the discussion above, their scientific value is questionable. Moreover, structural differences observed, to date, between biosimilar and reference product, have impacted only Fc functionality in vitro, but not TNFα binding affinity. Yet it is only the latter which is of relevance in the CET test populations of RA or Ps. It is only in the non-tested IBD indications that Fc functionality may have relevance, although even here, the balance of evidence suggests it does not.
Another critical point is that anti-TNFα’s display a relatively flat dose response. While a biological assay can detect differences in potency with high sensitivity, CETs on anti-TNFα’s represent a far blunter approach. In the case of the infliximab originator study, C0168T09, dose response was extremely shallow with a 55% ACR20 response in the low dose (1 mg/kg) group, rising modestly to 77% following a ten-fold dose increase (10 mg/kg); this compares with 12% in the placebo group. In the open-label phase of this study, no difference in response rates was observed between 3, 10 and 20 mg/kg infliximab doses. Similar results were seen with the Phase 3 ATTRACT study, where ACR20 responses at doses between 3 mg/kg every 8 weeks and 10 mg/kg every 4 weeks increased from 42% to 59% compared with 17% in the placebo arm [40]. Since the approved dose of infliximab in RA, which is used in the CET’s, is 3 mg/kg every 8 weeks, it is clear that there will be limited ability to detect differences in potency in the clinical setting. Dose responses are slightly steeper for adalimumab and etanercept, but the same principles apply.
Of further note is the fact that in the case of RA, despite the vastly different structures of the five anti-TNFα’s, ACR20 responses at six months are within a relatively narrow range as shown in Table 3 below.
Mean results for the primary endpoint, ACR20, from the five originator studies conducted in patients with active rheumatoid arthritis who failed previous disease-modifying anti-rheumatic drug (DMARD) therapy comparing anti-TNFα’s in combination with methotrexate versus methotrexate alone
The observations above are in line with those reported by Kirsch-Stefan et al. in 2023 [47] and Bielsky et al. in 2020 [48]. Kirsch-Stefan et al. [47] analysed the marketing authorization applications (MAAs) of all 33 biosimilar mAbs and three fusion proteins that had been evaluated by the European Medicines Agency (EMA) until November 2022. In all the seven cases where differences were observed for the clinical data but not the quality data, these were attributed to factors other than lack of biosimilarity, such as imbalances in the trial arms, immaturity of secondary endpoint results, change in the reference product, or chance findings. Furthermore, in-depth analysis of the marketing authorisation assessments for trastuzumab and rituximab biosimilars, revealed no cases where clinical trial data was necessary to resolve residual uncertainties about the quality part [47].
Bielsky et al. [48] reported in similar vein for 20 biosimilar products approved in the EU up to the end of 2019. These were for the anti-TNFα’s infliximab, adalimumab and etanercept, as well as rituximab, bevacizumab and trastuzumab. They further reported finding no publicly available efficacy trial results that precluded submitting an MAA for a biosimilar candidate [48].
EMA guidance states that if biosimilarity has been demonstrated in one indication, extrapolation to other indications of the reference product could be acceptable with appropriate scientific justification [36]. In the development of the first anti-TNFα biosimilar there was intense debate as to whether for anti-TNFα’s, results from studies in RA could be extrapolated to other indications, particularly to the IBD indications, where the mechanism of action was considered to differ. The CHMP eventually concluded that “extrapolation of the pharmacokinetic, efficacy and safety data generated in the two clinical trials in RA and AS to the other indications of the reference product, including IBD, is considered possible based on the results of the extensive in vitro and ex vivo comparability data on all functionalities of the infliximab molecule, including several experiments especially relevant to IBD” [2].
Nevertheless, there remained considerable skepticism amongst many clinicians as to whether the anti-TNFα biosimilars would provide similar efficacy to the reference product in IBD. This skepticism has considerably diminished over the years as more compelling data generated in clinical trials has shown biosimilar infliximab to provide efficacy against IBD that is comparable to the reference product.
The first clinical evidence for this came from the NorSwitch study conducted in 2014 to 2015 that demonstrated switching patients from the originator infliximab to a biosimilar (CT-P13) had no impact on efficacy or safety. In all 482 patients were enrolled and randomized. Of these 155 (32%) patients in the Full Analysis Set had CD and 93 (19%) had UC. Disease worsening occurred in 53 (26%) patients in the infliximab originator group and 61 (30%) patients in the biosimilar group (per-protocol set; adjusted treatment difference −4.4%, 95% CI −12.7 to 3.9). The frequency of adverse events was also similar between groups [49].
A second study conducted by the sponsor between Aug 2014 and Feb 2017 recruited 220 patients of which 111 were randomized 1:1:1:1 to receive biosimilar or reference product with half being switched to the other therapy at week 6. CDAI-70 response rates at week 6 were non-inferior. Over the total study period, roughly equal numbers of patients in each of the groups experienced at least one treatment-emergent adverse event [50].
Recently a subcutaneous version of a biosimilar infliximab has become available in the US for treating CD and UC. This was made available in the EU in 2019 as an “Extension” post-approval procedure to the biosimilar MA based on a streamlined development program. However, in the US, the only FDA regulatory path for gaining approval for this new route of administration was to license this subcutaneous infliximab as a new biological entity under Section 351(a) of Public Health Service Act. Consequently, two randomized placebo-controlled studies were conducted by the applicant, one in CD and the other in UC. In the case of CD, clinical remission at week 54 was 43.2% in the infliximab group compared to 28.8% in the placebo group (effect size 14.4%) and for UC this this was 63.4% and 26.9% respectively (effect size 36.5%). This compares well with the originator conducted infliximab trials in similar populations under similar conditions where effect sizes were 14.7% for CD and 25.1% for UC, leaving no doubt that also in IBD, an infliximab biosimilar exerts comparable efficacy to that of the reference product [51].
The assessment of safety in clinical trials presents a unique challenge compared to evaluating efficacy. While efficacy endpoints can be clearly defined, safety assessment involves searching for unknown effects amidst a sea of adverse events, many of which are unrelated to the treatment being studied. This complexity necessitates ongoing monitoring of safety even after a drug has received market approval, with post-marketing risk management programs becoming a crucial component of the regulatory process.
The traditional approach to safety assessment for new chemical entities is primarily based on empirically set numerical thresholds. According to the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) E1 guideline [52], a typical safety evaluation requires exposure of over 1,500 participants, with at least 300 of them being treated for more than six months. However, the landscape changes when it comes to biosimilars, which are highly similar to already approved reference products, and as such, the safety profile of biosimilars can be predicted based on the known on-target effects of the reference product. As a result, the focus of clinical studies for biosimilars has shifted away from extensive safety assessments.
Nevertheless, it is important to note that differences in safety profiles have occurred in the past with biosimilars, closely related protein products, and process changes to approved originator biologicals, although not with anti-TNFα’s to my knowledge. These differences are almost invariably associated with immunogenicity. Fortunately, immunogenicity can be monitored effectively in the comparative PK studies as previously discussed, although detection of rare events must rely on post-marketing safety monitoring.
Given this context, the design of clinical studies for biosimilars has not placed significant emphasis on safety as a primary focus. Instead, the approach has been to conduct the necessary efficacy and pharmacological studies, and then scrutinize the resulting clinical data for any indications of safety differences compared to the reference product. This strategy allows for a more efficient development process while still maintaining vigilance for potential safety issues.
In most regions of the world biosimilars are considered interchangeable with the reference product. However, in the US, the Biologics Price Competition and Innovation Act of 2009 [53] introduced interchangeable biosimilars as a separate category to standard biosimilars. Initially, to qualify for interchangeability, FDA required demonstration of equivalent PK at steady state following repeated alternating switching between the biosimilar and reference product. The FDA’s rationale was that repeated switching could enhance the formation of ADAs which could impact PK. However, even if enhanced immunogenicity were to represent a risk, there would need to be an initial difference in immunogenic properties between the biosimilar and the reference product, which should be determinable during the initial biosimilar PK studies.
Herdon et. al. (2023) [54] reviewed results from a total of 5,252 patients who were switched to or from a biosimilar and reference product. Safety data including deaths, serious adverse events, and treatment discontinuation showed no overall risk difference (95% CI ≥ –0.01, ≤ 0.01). Immunogenicity data showed a similar incidence of ADAs compared to patients who were not switched, as was the case for safety data [54].
Initially, switching studies were performed on two adalimumab biosimilars [55, 56] before their approval as interchangeable. Subsequently, in late 2023, FDA approved an ustekinumab biosimilar without clinical switching data, concluding that a switching study was not necessary to support a demonstration of interchangeability [57]. In total 17 of 64 biosimilars have been approved as interchangeable out of which 6 out of 19 were approved as interchangeable in 2024, mostly without clinical switch data [58].
Despite the accumulating evidence indicating that large, costly and time-consuming CETs add little, if any, value to biosimilar development, major regulatory agencies have continued to require such trials when no suitable biomarkers exist, although there are signs this position is changing. In the case of anti-TNFα’s studies of 12 months duration in 450 to 600 patients have been required to gain marketing approval. This adds two to three years to bring a biosimilar anti-TNFα to the market at an additional cost that can approach 100 million dollars, which includes CRO, investigator and other fees plus reference product acquisition costs. FDA has in the past gone further and also required investigating the impact of transitioning patients from the originator to biosimilar, which adds a further six months to the biosimilar development program.
The need for these CETs was debated at the US FDA and the International Pharmaceutical Regulators Program (IPRP) Biosimilars Working Group Conference in September 2023 [59]. FDA advised that they were looking into applying risk-based criteria to assess the need for a CET in every case, but to date nothing on this has been published, although there are signs of progress towards this. Recently EMA have published a Concept Paper for the development of a reflection paper on a tailored clinical approach. This reflection paper is intended to explore how far well-defined analytical and functional data can be predictive for the clinical outcome such that clinical PK/PD trials could prospectively lead to the conclusion of clinical similarity, without the need for large CETs [60]. However, CHMP appear a long way from moving to waiving CETs for any mAbs and fusion proteins [60].
On the other hand, the UK’s Medicines and Healthcare Products Regulatory Agency (MHRA) guidelines note that although each biosimilar development needs to be evaluated on a case-by-case basis, it is considered that, in most cases, a CET may not be necessary if sound scientific rationale supports such an approach. It is expected that in time other agencies will also adopt a similar position, however, at the present rate of progress, this could be five or more years away [61].
Finally, the revised World Health Organization (WHO) guidelines attempt to span these diverging positions, referring to a “tailored clinical data package.” With reference to the paper by Bielsky et al. [48], the WHO guideline goes on to imply that current data suggest that alongside simple proteins such as insulin, teriparatide, filgrastim and somatropin, more-complex proteins such as mAbs could also be sufficiently characterized to eliminate the need for CETs [62].
The rigorous process of establishing biosimilarity through multiple layers of evidence — structural and physicochemical analysis, biological activity testing, and PK studies has proven effective in ensuring the quality, safety, and efficacy of anti-TNFα products. As our understanding of biological products deepens and experience with biosimilars grows, the approach to their development and regulation is expected to evolve towards a more tailored approach with the goal of providing broader access to these important therapies. The discussion and analysis in this article could be equally applied to other mAb biosimilars accelerating development and enhancing access to life changing and lifesaving therapies, so alleviating the costs of treating disease [1].
ACR20: 20% improvement in American College of Rheumatology Scale (20% improvement in tender + swollen joints and 3 of following 5 criteria: patient global assessment, physician global assessment, functional ability measure; visual analogue pain scale, and ESR or CRP)
ADAs: anti-drug antibodies
ADCC: antibody-dependent cellular cytotoxicity
anti-TNFα: anti-tumor necrosis factor alpha
AS: ankylosing spondylitis
CD: Crohn’s disease
CETs: comparative efficacy trials
CHMP: European Committee for Medicinal Products for Human Use
CRP: C reactive protein
EMA: European Medicines Agency
ESR: Erythrocyte Sedimentation Rate
EU: European Union
IBD: inflammatory bowel disease
MA: marketing authorization
MAAs: marketing authorization applications
mAbs: monoclonal antibodies
PASI75: 75% improvement in Psoriasis Area and Severity Index
PK: pharmacokinetic
Ps: psoriasis
RA: rheumatoid arthritis
UC: ulcerative colitis
US: United States
CN: Conceptualization, Investigation, Writing—original draft, Writing—review & editing.
The author declares that he has no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Fernando Perez-Ruiz ... Amaya de Basagoiti-Gorordo
Gilberto Castañeda-Hernández
Fernando Pérez-Ruiz ... Eugenio Chamizo Carmona
Fanny Alcira Reyes Neira ... Andrea Yukie Shimabuco
Leticia A. Shea, Jamshaid S. Ahmed
Lauren N. McGrath ... Steven R. Feldman
Valderilio Feijó Azevedo