 Short Communication
Short Communication

Affiliation:
1Rheumatology Division, Osakidetza, OSI EE-Cruces, Cruces University Hospital, 48903 Barakaldo, Spain
2Biobizkaia Health Research Institute, 48903 Barakaldo, Spain
3Department of Medicine, Medical School, University of the Basque Country, 48903 Barakaldo, Spain
Email: fernando.perezruiz@osakidetza.eus
ORCID: https://orcid.org/0000-0002-5268-1894
Affiliation:
4Medical Affairs, Biogen Spain S.L., 28046 Madrid, Spain
ORCID: https://orcid.org/0009-0000-7614-4949
Affiliation:
5Servicio Extremeño de Salud, Hospital Universitario de Badajoz, 06080 Badajoz, Spain
6Rheumatology Division, Instituto Universitario de Investigación Biosanitaria de Extremadura, Universidad de Extremadura, 06080 Badajoz, Spain
ORCID: https://orcid.org/0000-0002-6322-6846
Explor Musculoskeletal Dis. 2025;3:100784 DOI: https://doi.org/10.37349/emd.2025.100784
Received: October 18, 2024 Accepted: December 21, 2024 Published: February 10, 2025
Academic Editor: Valderilio Feijó Azevedo, Federal University of Paraná, Brazil
The article belongs to the special issue Biosimilars: State of the Art in the Treatment of Rheumatic Diseases
This sub-analysis of the PROPER study aimed to evaluate outcomes following the transition from reference adalimumab (ADL) to SB5 (Imraldi™) in routine clinical practice in Spanish patients with rheumatoid arthritis (RA). Adult Spanish patients (n = 73) with RA who initiated SB5 as part of routine clinical practice following treatment with reference ADL were recruited. Outcome measures included persistence on SB5, clinical characteristics, and disease activity scores at the time of transition to SB5 treatment, clinical management over time, and safety. At Week 48, the Kaplan-Meier [95% confidence interval (CI)] estimate of the probability of persistence on SB5 after switching from reference ADL was 0.84 (0.73–0.90) and 83.6% (46/55) of patients were in remission or had low disease activity. The majority of patients [83.6% (61/73)] experienced no disease flare during the study period and reported that the injection was “simple or very simple” to administer (baseline: 66.7%; Week 48: 69.0%) and were generally “satisfied or very satisfied” with the duration of the injection. In total, 21 patients (21/73, 28.8%) reported at least one drug-related adverse event, which were mild in most cases (17/21, 80.9%). In a Spanish cohort of patients with RA transitioning from reference ADL to SB5, the probability of SB5 persistence was high and treatment effectiveness was maintained for up to 48 weeks. There were no new safety signals and SB5 was well tolerated. These findings suggest that there is no evidence to mitigate against transition from reference ADL to SB5 in patients with RA (Clinicaltrials.gov listing: NCT04089514).
Rheumatoid arthritis (RA) is a systemic, chronic, immune-mediated inflammatory disease (IMID) characterized by chronic joint inflammation, with progressive articular damage if left untreated, as well as the development of extra-articular organ involvement [1–3]. The global RA prevalence is estimated to be 0.21–0.46%, although rates vary considerably among different populations [1, 4]. The EPISER study, conducted in 2016, estimated a prevalence of 0.69% [95% confidence interval (CI): 0.47–0.99] in the adult population in Spain [5]. This aligns with the findings of an observational, retrospective study conducted in six Spanish hospitals (2014 to 2019), which estimated RA prevalence to be 0.49 (95% CI: 0.37–0.60) in the overall population; the RA prevalence was 0.26 (0.19–0.32) in males and 0.71 (0.54–0.87) in females [6].
Through its systemic inflammatory nature, RA is associated with a number of comorbidities that span the cardiovascular, neurologic, respiratory, and metabolic systems [1, 7]. In the Spanish cohort of the COMORA study, 22% of patients had at least one comorbidity at baseline with the most frequent being depression (27.0%) and obesity (26.0%). Myocardial infarction (5.0%), stroke (1.0%), and solid tumors (6.0%) were also observed [8]. In addition to decreased function, patients with RA and comorbidities experience reduced quality of life as well as increased morbidity and mortality [7, 9].
Treatment options for RA have evolved significantly with the introduction of disease-modifying anti-rheumatic drugs (DMARDs). A large number of cytokines, notably tumor necrosis factor (TNF), have been recognized as mediating a wide variety of effector functions relevant to the pathogenesis of RA and are established as therapeutic targets for DMARDs. Given the pivotal role of TNF in RA pathogenesis, anti-TNF therapy is a leading choice of first line biologic treatment in patients with moderate-to-severe RA [2, 10]. Adalimumab (ADL), a subcutaneously administered recombinant human monoclonal antibody, targets and blocks TNF. ADL has become a well-established treatment for adults with RA following approval for use in the European Union (EU) in 2003 [11, 12]. More recently, biosimilars have provided a more cost-effective alternative to off-patent biologic therapies within IMIDs, including RA [13]. The European Medicines Agency has stated that the long-term clinical experience with biosimilars means that they can be used as safely and effectively as their reference molecules [14], and recent EULAR guidelines have advocated for the use of biosimilars where appropriate [15].
SB5 (Imraldi™), an ADL biosimilar, received EU marketing authorization in 2017 [16]. SB5 has been developed as a biosimilar to reference ADL (Humira®), and in phase I and III studies has demonstrated equivalent efficacy and comparable pharmacokinetic, safety, and immunogenicity profiles [17, 18]. Results from clinical trials and real-world evidence suggest transitioning from reference ADL to SB5 does not lead to clinically meaningful differences in terms of pharmacokinetics, efficacy, safety, or immunogenicity [17, 19]. The 48-week, pan-European, real-world PROPER study aimed to evaluate outcomes following transition from reference ADL to SB5 in routine clinical practice in patients with IMID [20]. This sub-analysis focuses on the Spanish RA cohort of the PROPER study.
PROPER was a pan-European, non-interventional, single-cohort, real-world study conducted at specialist clinics across six countries (Germany, Spain, Italy, the UK, Belgium, and Ireland) [20]. The study enrolled 1,033 patients (aged ≥ 18 years) with RA, axial spondyloarthritis, psoriatic arthritis, Crohn’s disease, and ulcerative colitis who initiated SB5 as part of routine clinical practice following a minimum of 16 weeks’ treatment with reference ADL. Clinic visits were anticipated to take place approximately every 3 months (depending on local standards of care; Figure 1). The methods of the PROPER study have been published elsewhere [20].
This sub-analysis of the PROPER study focused on data from 73 patients with RA enrolled in seven centers in Spain. Outcome measures included persistence on SB5, clinical characteristics and disease activity scores at the time of transition to SB5 treatment, clinical management over time, and safety. Persistence on SB5 was estimated using the Kaplan-Meier method, and patient satisfaction with the self-administration device was assessed throughout the study period with the patient satisfaction questionnaire (PSQ).
Disease activity scores were those used routinely for RA at the study sites: Disease Activity Score in 28 joints using C-reactive protein [DAS28-CRP; converted DAS28-CRP (DAS28-CRPconv)] was derived from DAS28 using the erythrocyte sedimentation rate where C-reactive protein was unavailable. Secondary outcome measures included type, dose, regimen, and method of SB5 administration throughout the study; results from the PSQ; use of immunosuppressant and/or use of corticosteroid and/or biologic therapy (other than SB5); incidence of adverse drug reactions (ADRs), defined as non-serious adverse events (AEs) considered to have a causal relationship with SB5; and all serious AEs (SAEs). The study was approved in Spain by the research ethics committee from Hospital La Princesa in Madrid on September 23, 2019.
A total of 73 participants with RA were included across seven sites in Spain. Based on the inclusion criteria, data available for DAS28-CRP change from baseline is available for 62 patients. Mean age at the time of SB5 initiation was 59.6 ± 9.8 years. Patient demographics and other baseline clinical characteristics are presented in Table 1.
Baseline demographic and clinical characteristics
| Variables | Values (n = 73) |
|---|---|
| Age at diagnosis, n, years (SD) | 70*, 43.9 (10.1) |
| Disease duration, n, years (SD) | 59*, 15.0 (7.9) |
| Age at initiation of SB5, n, years (SD) | 73, 59.6 (9.8) |
| Gender, n (%) | |
| 54 (74.0) |
| 19 (26.0) |
| Body mass index, n (%) | |
| 1 (1.4) |
| 59 (80.8) |
| 13 (17.8) |
| Work status at SB5 initiation, n (%) | |
| 26 (35.6) |
| 10 (13.7) |
| 37 (50.7) |
| Tobacco use at SB5 initiation, n (%) | |
| 9 (12.3) |
| 20 (27.4) |
| 44 (60.3) |
| Clinical status based on disease activity score, n (%, n = 41) | |
| 26 (63.4) |
| 5 (12.2) |
| 10 (24.4) |
| 0 |
| Concomitant therapy received by > 5% of the total patient population, n (%) | |
| 41 (56.2) |
| 9 (12.3) |
| 22 (30.1) |
| 7 (9.6) |
| 9 (12.3) |
| 5 (6.8) |
| 6 (8.2) |
| Biologic therapy prior to adalimumab, n (%) | 16 (21.9) |
| Received information on self-administration of SB5, n (%) | 66 (90.4) |
| Aware that SB5 should be removed from the refrigerator 30 min prior to injection, n (%) | 63 (86.3) |
| Aware that SB5 can be stored unrefrigerated < 25ºC for up to 28 days, n (%) | 51 (69.9) |
* n differs from overall n due to incomplete data capture. HMG-CoA: 3-hydroxy-3-methylglutaryl coenzyme A; SD: standard deviation
During follow-up, 68.5% (50/73) of patients received conventional DMARDs as concomitant therapy, methotrexate being the most commonly used (56.2%, 41/73), followed by leflunomide in 12.3% (9/73). The main reasons for the transition from reference ADL to SB5 were cost (43.8%) and physician decision (50.7%) (Table 2). The most common dosing regimen of reference ADL and SB5 at transition was 40 mg every 2 weeks (56.2%). The remaining patients had a dosing regimen of 40 mg every 2 weeks for reference ADL and then 40 mg at a dosing frequency other than every 2 weeks. The majority (98.6%) of patients (n = 72) did not experience a change in dose or frequency of SB5 administration during follow-up.
Reasons for transition
| Cause | Indication | |
|---|---|---|
| RA (n = 73) | ||
| n | % | |
| Mandated by health authority/payer | 2 | 2.7 |
| Cost | 32 | 43.8 |
| Adverse event | 0 | 0.0 |
| Patient decision | 0 | 0.0 |
| Physician decision | 37 | 50.7 |
| Other | 2 | 2.7 |
| Total | 73 | 100.0 |
RA: rheumatoid arthritis
The Kaplan-Meier (95% CI) estimate of the probability of persistence on SB5 at Week 48 after switching from reference ADL was 0.84 (0.73–0.90) (Figure 2). Reasons for discontinuation (16/73) included cost (6/73, 8.2%), AE (5/73, 6.8%), and secondary loss of response (5/73, 6.8%). Sixty-one patients were still receiving SB5 at Week 48.
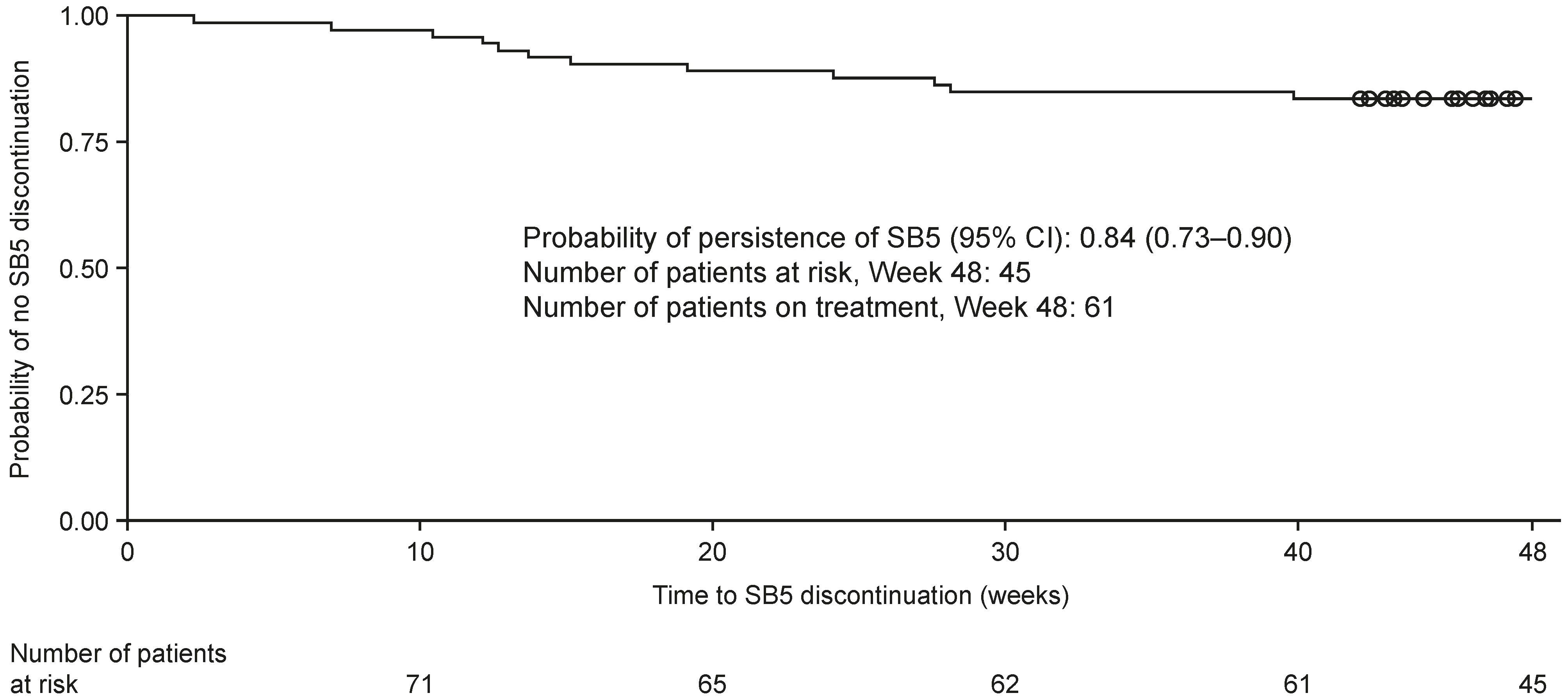
Kaplan-Meier curve for estimating SB5 survival. The X-axis is cut at Week 48. At this time, 45 patients were still at risk. Time to SB5 discontinuation (weeks) = [Date of discontinuation of SB5 or date of censoring – date of SB5 initiation + 1]/7. Patients with ongoing SB5 administration are censored on the date of completion of/withdrawal from the study. Censored events are indicated by circles. CI: confidence intervals
According to the DAS28-CRPconv disease activity index, 80.6% (50/62) of the patients were in remission (score ≤ 2.4) or had low disease activity (2.4 < score ≤ 2.9) at baseline. At Week 48, 83.6% (46/55) of patients with DAS28-CRPconv data available at baseline were in remission or had low disease activity. Mean DAS28-CRPconv ± SD at Weeks 12, 24, and 48 were 2.5 ± 0.7, 2.5 ± 0.8, and 2.3 ± 0.6, respectively. Changes in DAS28-CRPconv from baseline are depicted in Figure 3.
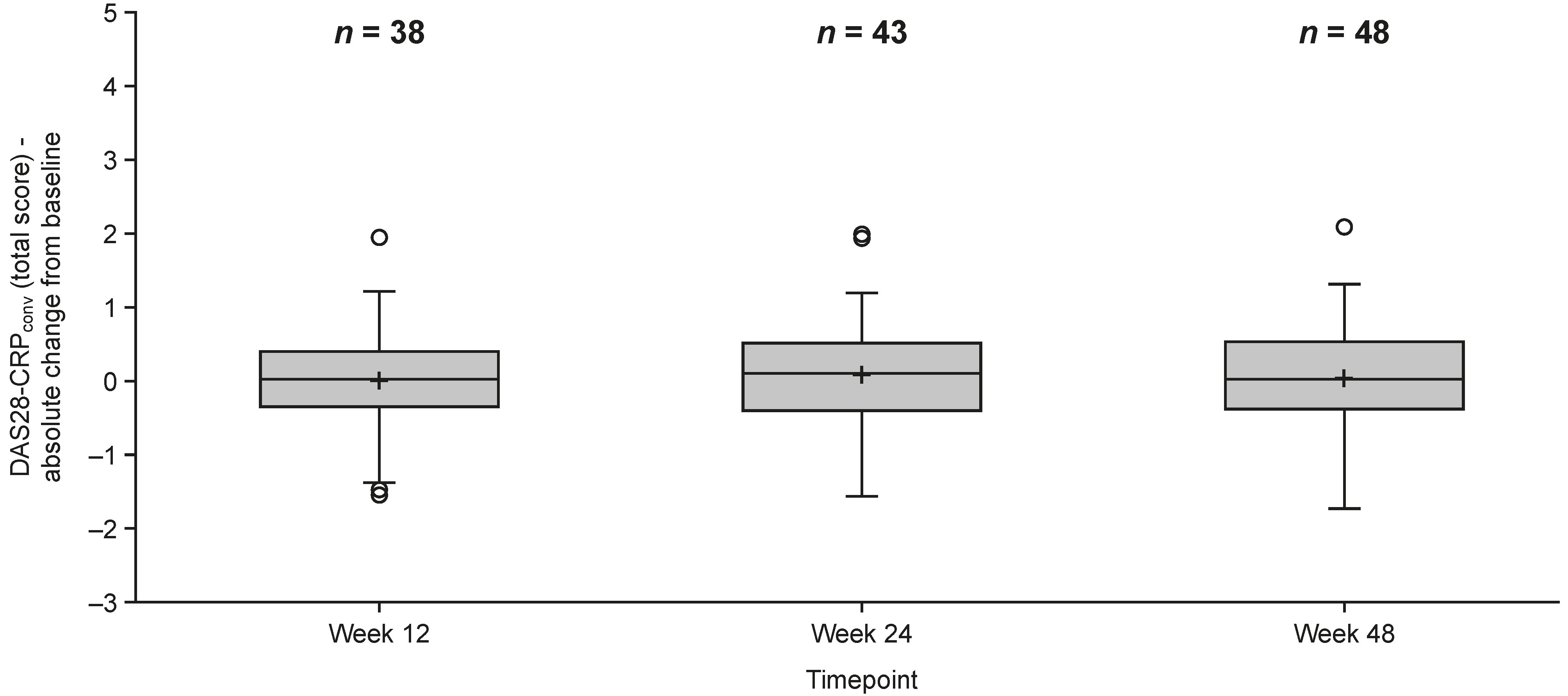
Change in DAS28-CRPconv from baseline. DAS28-CRPconv is calculated by a mathematical formula that includes DAS28-CRP and DAS28-ESR. Box displays 1st and 3rd quartile, median value is indicated by the horizontal line, mean value is displayed using a plus (‘+’), whiskers are drawn from the box to the most extreme point that is less than or equal to 1.5 times the interquartile range. DAS28-CRPconv: converted Disease Activity Score in 28 joints using C-reactive protein; DAS28-ESR: Disease Activity Score in 28 joints-erythrocyte sedimentation rate
83.6% of patients (61/73) experienced no disease flare during the study period, whilst 16.4% of patients (12/73) experienced a single flare episode. Flares were defined according to the investigator’s clinical opinion based on patient-reported symptoms, disease score, or secondary loss of response. The most common manner by which flare was detected was patient-reported symptoms (11/12, 91.7%). Flare-related dose adjustments of biologics and non-biologics were required for 16.7% (2/12) and 58.3% (7/12) of patients experiencing flare, respectively.
Overall, 90.4% of patients had been informed about SB5 administration. SB5 was more commonly administered via pen than by syringe both at baseline and at Week 48 (approximately 3:1 for both time points). There was no major shift in PSQ responses from baseline to Week 48 (Figure 4). The majority of patients reported that the injection was “simple or very simple” to administer (baseline: 66.7%; Week 48: 69.0%) (Figure 4A) and were generally “satisfied or very satisfied” with the duration of the injection (baseline: 55.6%; Week 48: 59.5%) (Figure 4B). The indication on the pen that the injection was complete was generally considered to be “clear or very clear” (baseline: 77.8%; Week 48: 77.5%) (Figure 4C).
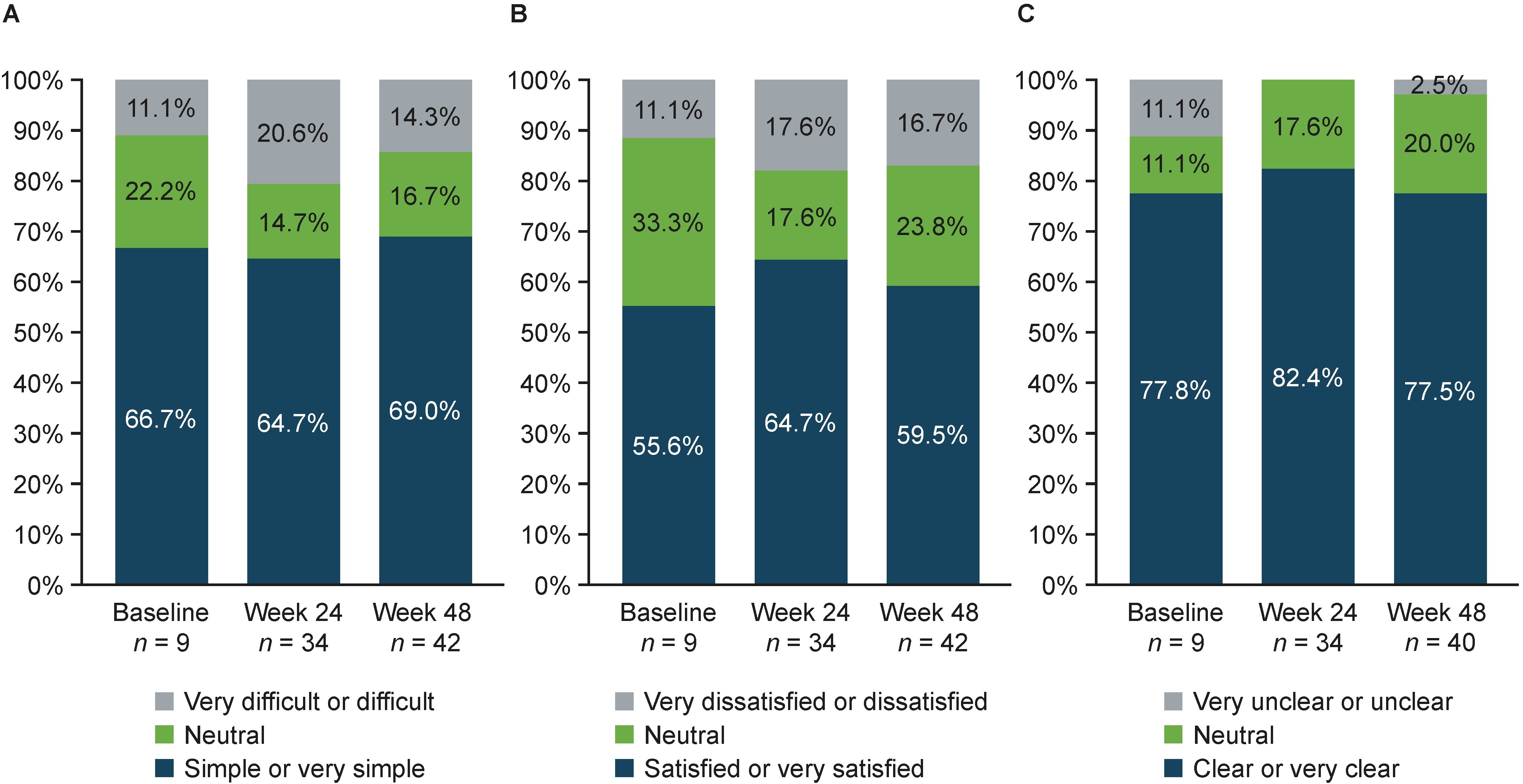
Patient satisfaction with the self-administration device (pen and syringe—from PSQ). (A) Ease of administration of the injection; (B) duration of the injection; (C) indication of completed injection (pen only). PSQ: patient satisfaction questionnaire
In total, 21 patients (21/73, 28.8%) reported at least one drug-related AE, which were mild in most cases (17/21, 80.9%). Amongst the conditions included under the term “general disorders and administration site conditions” (injection site hematoma, hemorrhage, and pain), the most commonly occurring was injection site pain (16/73, 21.9%), which was predominantly mild in severity. Four patients reported SAEs not related to SB5 administration: 1 COVID-19 infection, 1 coronary artery occlusion, 1 diverticulitis, and 1 parvovirus B19 infection. No SAEs that were determined to be related to SB5 were reported.
The findings of this sub-analysis are comparable with those of the main PROPER study, reference ADL real-world evidence studies, and reference ADL-to-SB5 treatment switching studies [20–24]. The findings are also comparable to those for other ADL biosimilars [13]. This analysis represents one of the largest studies of biosimilar ADL switching in RA patients in Spain, with the unique approach of capturing patient experience in terms of persistence and device satisfaction over 48 weeks. Throughout the duration of the study, patients reported high satisfaction with the self-administration device. The probability of persistence with SB5 was high at 0.84 (95% CI: 0.73–0.90), similar to that reported in clinical practice [25]. Of the candidate predictors of persistence evaluated in the pan-European PROPER study, only female sex was associated with increased risk for SB5 discontinuation in RA, with a hazard ratio for discontinuation in females of 3.53 (95% CI: 1.07–11.67) [20], in line with previous findings [21–23].
Long-term effectiveness was maintained up to 48 weeks after switching from reference ADL to SB5, with no meaningful change in doses over time. This is in line with previous findings from an observational treatment-switching study in comparable patient populations and using the same or similar definitions for clinical remission or low disease activity [24].
No new safety signals were observed in this study, and AEs were in line with those reported previously in patients with rheumatic or gastrointestinal IMIDs treated with SB5 [17, 21, 22]. In total, 28.8% (21/73) of patients reported at least one ADR; these were mostly mild in nature with no report of SAEs related to SB5. Injection site reactions were the most common AEs (23.3%, 17/73), in line with findings of other studies; however, patient satisfaction with the device remained high throughout the study [17, 22]. The recent introduction of a high-concentration, low-volume, and citrate-free formulation could potentially enable the reduction of injection site-related reactions and positively affect treatment adherence [26].
The ‘nocebo effect’ occurs when patients have negative expectations of treatment, which can affect their perception of clinical benefits and reduce their willingness to accept treatment. Data collection on patient satisfaction for the adoption of nocebo-reducing strategies is therefore crucial to enhance the acceptance and utilization of biosimilars [27]. Whilst the lower cost of a biosimilar relative to its reference may result in patient misperception of lower effectiveness, and be a risk factor for the nocebo effect, conversely it could enable earlier patient access to effective therapies, thereby reducing disease burden [28].
There were study limitations for this sub-analysis. There were missing baseline data for a number of patients. Flare was reported according to physician discretion; therefore, these results are to be considered with caution. The predominantly female population of the current study could be seen as a limitation. However, as RA is more common in women, these results could more closely reflect the real-world situation. Routine testing for anti-drug antibodies was not done at any study site in Spain. Finally, interpretation of what constituted an ADR was variable between sites highlighting the need for comprehensive safety training and data monitoring to ensure consistency.
In conclusion, the Spanish cohort of patients with RA transitioning from reference ADL to SB5 showed that the probability of SB5 persistence was high and treatment effectiveness was maintained up to 48 weeks. Over 80% of patients were in remission or had low disease activity throughout the study, and the majority of patients did not require any dose modification. Furthermore, episodes of flare were infrequent/low, and no new safety signals were observed, highlighting that SB5 was well tolerated. These findings suggest that there is no evidence to mitigate against transition from reference ADL to SB5 in patients with RA.
ADL: adalimumab
ADR: adverse drug reaction
AE: adverse event
CI: confidence interval
DAS28-CRP: Disease Activity Score in 28 joints using C-reactive protein
DAS28-CRPconv: converted Disease Activity Score in 28 joints using C-reactive protein
DMARD: disease-modifying anti-rheumatic drug
EU: European Union
IMID: immune-mediated inflammatory disease
PSQ: patient satisfaction questionnaire
RA: rheumatoid arthritis
SAE: serious adverse event
TNF: tumor necrosis factor
The authors would like to thank Janet Addsion (Biogen IDEC, Maidenhead, UK) for her contribution to the content of the manuscript and critical review, and Maximilian Utzinger (Biogen GmbH, Munich, Germany) for the critical review of the manuscript.
BOP: Data curation, Writing—original draft, Conceptualization, Methodology, Supervision, Writing—review & editing, Validation. FPR: Writing—original draft, Conceptualization, Supervision, Writing—review & editing, Validation. ECC: Writing—original draft, Conceptualization, Supervision, Writing—review & editing, Validation. All authors read and approved the submitted version.
FPR: speaker and advisor for Biogen, advisor for Fresenius-Kabi; member of the Corporative Pharmacy Commission, Basque Health Service-Osakidetza; member of the OSI-EEC Pharmacy Commission; President, OSI EEC/BS Commission for Adequate Clinical Practice and Assistance Implementation; Editor-in-Chief of Exploration of Musculoskeletal Diseases, but had no involvement in the journal review process of this manuscript. BOP is an employee of and may hold stock in Biogen. ECC declares that there are no conflicts of interest.
The study was approved by the ethics committee from Hospital La Princesa and then registered in the Spanish Agency for Medicines and Health Products (AEMPS. Agencia Española de Medicamentos y Productos Sanitarios) EPA-SP BIO-IMR-2019-01.
Written and oral information about the study was provided to patients and written, informed consent was obtained from each patient prior to participation in the study.
Not applicable.
Due to internal regulations from the sponsor, the data pertaining to this research will not be shared. Further details on the sponsor’s clinical trial transparency and data-sharing policy can be found at https://clinicalresearch.biogen.com/. In addition, the authors are willing to answer reasonable questions.
This study was funded by Biogen International GmbH (Baar, Switzerland). Medical writing assistance was provided by Chloe Schon and Iain Bartlett of Springer Healthcare Ltd, UK, and was funded by Biogen International GmbH in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3). The funder developed the methodology of the PROPER study and conducted the formal investigation and data analysis.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Fernando Perez-Ruiz ... Amaya de Basagoiti-Gorordo
Gilberto Castañeda-Hernández
Fanny Alcira Reyes Neira ... Andrea Yukie Shimabuco
Leticia A. Shea, Jamshaid S. Ahmed
Lauren N. McGrath ... Steven R. Feldman
Valderilio Feijó Azevedo

