
Abstract
A short overview of the main features of progressive myoclonus epilepsies (PMEs), such as Lafora disease (LD), neuronal ceroid lipofuscinoses (NCLs), and myoclonus epilepsy with ragged-red fibers (MERRF) is given. The stress of this review paper is put on one of the PME’s, the Unverricht-Lundborg disease (ULD)—EPM1, which is caused by mutations in the human cystatin B gene (stefin B is an alternative protein’s name). However, different other genes/proteins were found mutated in patients presenting with EPM1-like symptoms. By understanding their function and pathophysiological roles, further insights into the underlying processes of EPM1 can be obtained. On a broader scale, common pathophysiological mechanisms exist between ULD, LD and NCLs, such as, reactive glia, synaptic remodeling, neuronal hyperexcitability, impairements in the lysosomal/endocytosis system, cytoskeletal functions, and mitochondria. Oxidative stress is also in common. By understanding the underlying molecular and cellular processes, early interventions, better therapies and eventually, by using modern stem cell, gene editing or replacement methods, a cure can be expected.
Keywords
Progressive myoclonus epilepsy, neurodegeneration, ubiquitin proteasome system, mitochondrial impairment, autophagy, monogene diseases, oxidative stress, cystatin BIntroduction
Proteinopathies comprise a large number of different diseases where proteins misfold and aggregate or condense [1]. Proteostasis (i.e., balance between protein synthesis, folding, and degradation when proteins aggregate) goes awry in many entry points [2]. Impairments of mitochondrial, ubiquitin proteasomal and lysosomal systems can occur. Together with gut-brain axis unbalance the brain can get affected with neuroinflamamtion, oxidative stress (OS), cytokine release, and glia cells activation. This latter causes excitotoxicity and hyper-excitability of neurons. A subclass of proteinopathies are amyloidoses, systemic and local, among them neurodegenerative diseases (NDs), such as Alzheimer’s, Huntington’s, and Parkinson’s disease. Apart from NDs, protein aggregation and subsequent accompanying consequences have recently been reported for major mental health diseases, such as schizophrenia and depression. Not to dismiss, traumatic brain injury also causes protein aggregation (taupathy), neurodegeneration and epilepsy [3]. Progressive myoclonus epilepsies also have common pathology resembling proteinopathies, with autophagy and mitochondrial systems impaired [4]. We assume that protein aggregation and various inclusions observed in these type of epileptic syndromes contribute to the common pathology of “gain in toxic function” as also observed in other neurodegenerative conditions. These commonly observed processes are: OS, mitochondria, and autophagy impairements. As for the “loss of function” of the genes affected, either by their absence or mutation/aggregation, even though they have different functions, they also are part of some common molecular and cellular processes over progressive myoclonus epilepsies (PMEs).
What can we learn from familial forms of PMEs
There are several types of PMEs. They are all characterized by myoclonic and tonic-clonic seizures. Later symptoms comprise muscle rigidity, problems with balance and, in some PMEs mental decline and even behavioral, and psychiatric problems. Patients can become wheelchair-bound. The most known among PMEs are Lafora disease (LD) and Unverricht-Lundborg disease (ULD), the latter also called progressive myoclonic epilepsy type 1 (EPM1). Other, less common forms of PMEs include mitochondrial encephalopathies, neuronal ceroid lipofuscinoses (NCLs), sialidoses, myoclonus epilepsy with ragged-red fibers (MERRF), neuronopathic Gaucher disease, Farber disease and, Dentatorubral-pallidoluysian atrophy. The causal genes are known for ULD, LD, NCLs, MERRF, Farber disease and sialidoses [5]. EPM1 will be described separately.
LD (also marked as EPM2) is caused by mutations in EPM2A and NHLRC1 genes, which encode laforin and malin, respectively [6, 7]. Laforin is a dual specificity phosphatase and malin is an E3 ubiquitin ligase. Both proteins are involved in glycogen processing, and due to their mutations and consequent loss of function, insoluble bodies of glycogen (Lafora bodies) accumulate in the brain and some other tissues, such as the liver and skin. Lohi et al. [8] 2005, proposed that laforin might direct two negative feedback loops: polyglucosan-laforin-GSK3β-GS to inhibit glycogen synthase (GS) activity and polyglucosan-laforin-malin-GS to remove GS through proteasomal degradation. In addition, autophagy is impaired in LD [9]. The ER stress and unfolded proteins response are activated, due to protein aggregation (Figure 1) Oxidative stress and autophagy impairment follow increased protein aggregation, due to less efficient ubiquitin proteasome clearance [10]. The disease onset is in late childhood or adolescence. It is characterized by progressive neurologic deterioration and mental health symptoms, leading to death within 10 years. Fortunately, by using modern gene managing techniques there are several options, how to treat LD [11].
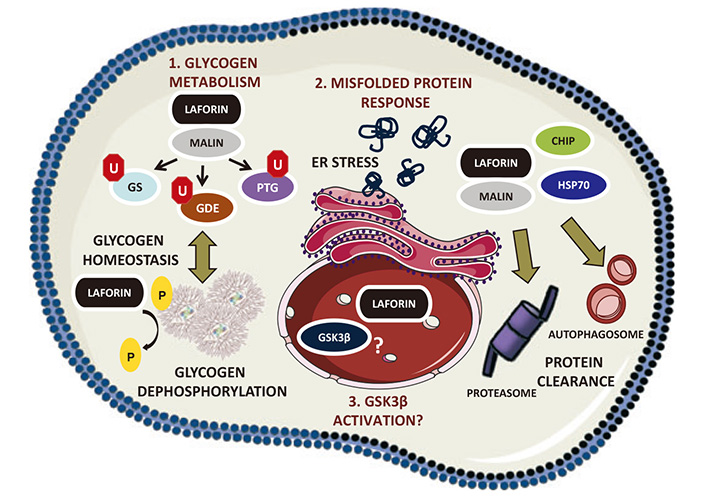
Laforin functions in cell physiology [12]. Schematic view of the different functions of laforin. GS: glycogen synthase; GDE: glycogen debranching enzyme; HSP70: heat shock protein 70; U: ubiquitin; P: phosphate
Note. Reprinted with permission from “Laforin, a protein with many faces: glucan phosphatase, adapter protein, et alii” by Gentry MS, Romá-Mateo C, Sanz P. FEBS J. 2013;280:525–37 (https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2012.08549.x). © 2012 FEBS.
Among the fatal myoclonus epilepsies fall different NCLs, a group of monogenic, autosomal recessive neurodegenerative disorders [13]. The majority of the 14 NCL genes [14, 15] discovered thus far encode proteins, which are part of endo/lysosomal pathways. CLN1/PPT1, CLN2/TPP1, CLN5, CLN10/CTSD, CLN13/CTSF, and CLN11/GRN are lysosomal proteins, more specifically: lysosomal enzymes (CLN1, CLN2, CLN10, and CLN13), a soluble protein (CLN5), and a protein in the secretory pathway (CLN11), whereas CLN3, CLN7/MFSD8, CLN12/ATP13A2 are membrane proteins [14, 15]. Two NCL genes encode endoplasmic reticulum membrane proteins (CLN6, CLN8) [14, 15]. The phenotype of lipofuscinoses depends somewhat on the mutated gene and the mutation. Apart from the myoclonus, they present with psychomotor delay, progressive loss of vision and dementia. Patients die early. The name comes from lipofuscin, an indigestible mixture of fluorescent cross-linked proteins and lipids. The debris affects neuronal cytoskeleton and cellular trafficking. It triggers lysosomal membrane permeabilization and subsequent atypical necroptosis.
MERRF is caused by mutations in the mitochondrial DNA (mtDNA), with A8344G mutation in the tRNA(Lys) gene being the most common [16, 17]. mtDNA is transmitted vertically from the mother to the offspring. The disease onset is in childhood and early adolescence. MERRF is a rare disease, multisystem mitochondrial syndrome characterized by progressive myoclonus and generalized epileptic seizures. Cerebellar ataxia, weakness of muscles-myopathy, cardiac arrhythmia, hearing loss, optic atrophy, and dementia occur concomitantly. Furthermore, lactic acidosis and stroke-like episodes (MELAS) are established phenotypes of mitochondrial encephalopathies [17], which are also caused by mtDNA and tRNA(Leu) mutation.
A myoclonus epilepsy EPM7, albeit usually more severe than EPM1, results from mutations in the KCNC1 gene [18] encoding the potassium ion channel subunit of the Kv3 subfamily of voltage-gated tetrameric channels.
Yet another rare myoclonus epilepsy is the Dravet syndrome, previously termed severe myoclonic epilepsy of infancy. It is caused by a mutation of the sodium voltage-gated channel alpha subunit (SCN1A) gene [19].
EPM1 on the molecular and cellular level: what are consequences of the loss of cystatin B function to neural physiology
ULD or EPM1 is caused by mutations in CSTB gene, coding for cystatin B [20, 21]—equaly assigned as cysB, stefin B, stB, a cysteine protease inhibitor. The EPM1 disease in most cases starts in childhood or early adolescence. Usually, an earlier onset of symptoms predicts a more severe course. In addition to myoclonus and tonic-clonic seizures, gait unstability, muscle rigidity, symptoms comprise dysarthria (trouble speaking clearly), dysphagia (difficulty swallowing), and tremor. The phenotype can be mild or more severe, whereas patients become wheelchair-bound. They do not have major cognitive decline but depression is common.
At least 14 genetic mutations CSTB gene [20, 21] have been found. The group of Lehesjoki has determined many other mutations apart from dodecamer repeats, among them are missense, nonsense, frameshift, and deletion mutants [22]. Up-date of the mutations and splice-variants in CSTB gene has been gathered recently by Singh and Hämäläinen [23]. The patients heterozygous for the dodecamer repeat and a point mutation, either frameshift, stop or missense with loss of function, are differently affected [24]. Patients homozygous for two null alleles manifest the most severe neonatal-onset progressive encephalopathy [25].
We have shown that some of the mutations change the protein’s stability, folding/unfolding and propensity to aggregate. They may attain cytotoxic function similar to other amyloid-forming proteins—as we have proposed already in 2005 [26]. Our later studies have shown different inclusions upon expression of selected EPM1 mutants in cell culture, which proved more or less toxic, depending on the unfolding intermediate and type of oligomers formed [27, 28]. We also have studied stefin B knock-out (KO) mice primary astrocytes, and have observed more protein aggregates by other proteins and an impaired autophagic flux [29]. As possible confirmation of gain in toxic function, patients bearing one allele of the usual dodecamer repeat expansion and one allele of R68X in the cystatin B gene, show more severe pathology and clinical symptoms [30]. This mutation leads to an unfolded protein, which aggregates extensively [31], therefore, it is reasonable that cells degrade it fast. Patients who are heterozygous for the dodecamer repeat and Q71P or G50E mutation, which forms aggresomes in cell culture [28], may be expected to show more severe symptoms, due to gain in toxicity. However, most of the mutations also affect the protein’s function. Thus, G4R, Q71P and G50E mutations showed loss of lysosomal localization [22, 32], which may be reflected in a normal course of the EPM1 disease, which is due to decreased activity of this protein.
Function-wise, human stefin B [33] is a cysteine protease inhibitor [34, 35] but there is more and more evidence that this protein may exert alternative functions [36, 37]. We have proposed it may act as an amateur chaperone, helping other proteins to fold or prevent their unfolding [38, 39]. It readily forms domain-swapped dimers, similar to stefin A [40, 41], and tetramers composed from two such entities [42]. We have shown that stefin B tetramers interact with amyloid-beta peptide and reduce its amyloid fibril formation [43]. Importantly, the group of Melli has confirmed the oligomeric nature of this protein in cells [44] and that the oligomers depend on the redox state of the cell [44, 45].
Furthermore, the Italian groups have reported that cystatin B has a role in vesicular transport [46] as well as in neural proliferation and interneuron migration [47], in synapse physiology [46, 48] and in neurogenesis [47]. That cystatin B has important roles in the brain [23], contributing to synaptic plasticity has been confirmed more recently in studies of human cerebral organoids [49]. Already in 2002, by Di Giaimo et al. [50], it has been shown that cystatin B in the rat cerebellum interacts with proteins, which play a role in cellular growth, proliferation and differentiation. By yeast two-hybrid system cystatin B was shown to interact with brain β-spectrin, a cytoskeletal protein, with NF-L (neurofilament light chain), which is important for cytoskeleton dynamics and with RACK-1 (receptor for activated C kinase 1), which mediates the interaction of the activated C kinase embedded in the cell membrane, with the cytoskeleton. The three proteins—RACK-1, β-spectrin, and NF-L co-localize in Purkinje cells and in Bergmann glia, where in complexes with cystatin B they contribute to cell development and differentiation [23].
In stefin B (cystatin B) KO cells [51], animal models and patients [52], OS was found to be increased. Therefore, anti-oxidant treatment with N-acetyl cysteine (NAC) has been tried to treat EPM1 patients with variable outcomes [53]. Maher et al. [54], showed that murine stefin B deficient macrophages were significantly more sensitive to the lethal lipopolysaccharide (LPS)-induced sepsis due to increased caspase-11 expression and NLRP3 inflammasome activation [54]. They also showed that stefin B deficiency resulted in the destabilization of the mitochondrial membrane and elevated mitochondrial reactive oxygen species (ROS) generation [51, 54], leading to cell death. Recently, the same group has shown that stefin B inhibits NLRP3 inflammasome activation via AMPK/mTOR signalling [55].
In conclusion, stefin B (cystatin B) function has been associated with many biological processes such as apoptosis [56], the response to OS [52], cell cycle regulation [57, 58], inflammation [54, 55]. However, the molecular mechanisms by which stefin B mediates these processes remain largely unknown. Which of the stefin B functions are expressed first and upstream of OS is not clear as yet. In addition, nuclear localization and function(s) [57, 58] of this protein are most important in neurons and astrocytes.
Importantly, CSTB gene is part of chromosome 21 (Ch21q22.3) together with some other important neural system genes, such as amyloid-beta precursor protein (APP), Copper/zinc superoxide dismutase 1 (SOD-1), glutamatergic receptor GluR5, and prion. An additional copy of chromosome 21 is present in Down syndrome (DS) patients. Due to the triplicated gene, SOD-1 increased function causes OS in people with DS, leading to early Alzheimer’s disease (AD). Cystatin B increased amount and function does not seem to improve symptoms of concominant AD [59]. Whereas EPM1 symptoms would be milder or more severe in people with DS has not been reported as yet but the so-called late-onset myoclonic epilepsy (LOMEDS) is frequent after the age of 40 in people with DS and leads to worse cognitive outcomes and early mortality [59]. It would be of interest to check for an increased protein aggregation in DS patients.
Other genes mutations causing EPM1-like symptoms
Another mutation producing similar symptoms as EPM1 was identified in Prickle1 protein [60, 61]. Apart from progressive myoclonus syndrome, Prickle paralogs have been associated with several neurological and NDs, including autism spectrum disorder and AD, and with autoimmune diseases, such as rheumatoid arthritis. Prickle family of proteins take part in planar cell polarity (PCP) signalling [62]. Prickle binds to the transmembrane protein Vangl and the Vangl-Prickle complexes accumulate at the plasma membrane, where they regulate the actin cytoskeleton [63, 64]. In addition, Prickle inhibits Dishevelled (Dsh) [62] in the cytoplasm and the transmembrane protein Frizzled (Fz) (see Figure 2).
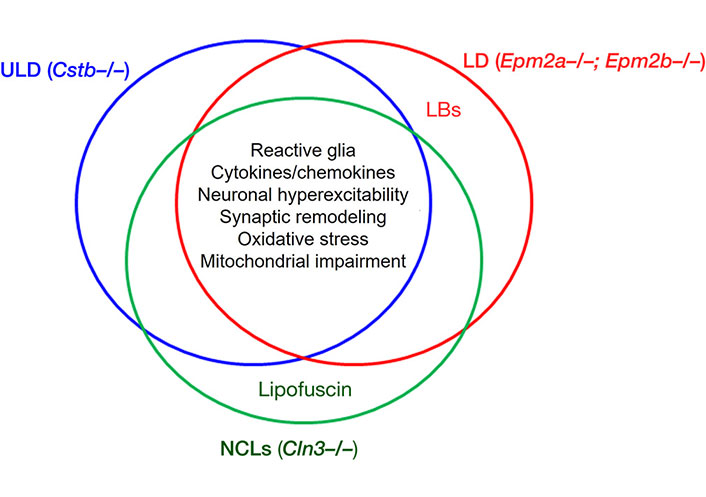
Schematic view of the common pathophysiological mechanisms of Unverricht-Lundborg disease (ULD), Lafora disease (LD) and neuronal ceroid lipofuscinoses (NCLs) [4]. LB: Lafora bodies. In the intersection, are given the common pathological processes affected in the three disorders, according to the data obtained with animal models of the diseases. These are: reactive glia, release of cytokines/chemokines, synaptic remodeling, oxidative stress, neuronal hyperexcitability—and, as added, the mitochondrial impairment
Note. Adapted from “Neuroinflammation and progressive myoclonus epilepsies: from basic science to therapeutic opportunities” by Sanz P, Serratosa JM. Expert Rev Mol Med. 2020;22:e4 (https://www.cambridge.org/core/journals/expert-reviews-in-molecular-medicine/article/neuroinflammation-and-progressive-myoclonus-epilepsies-from-basic-science-to-therapeutic-opportunities/FFDB1F62EBB390CC1CEF30A5AB5B35CE). CC BY.
Another mutation producing similar symptoms was found in the SCARB2 gene [65]. Function of the SCARB2 gene coding for lysosomal integral membrane protein—LIMP2, is only partially resolved. A better-known function is membrane transport at the endosomal/lysosomal compartment [66]. It has been discovered that LIMP2, apart from lysosomal membranes, resides at intercalated discs of the heart, where it interacts with N-cadherin. Knockdown of LIMP2 with RNA interference (RNAi), decreased the binding of N-cadherin to the phosphorylated form of β-catenin, and LIMP2 overexpression had the reverse effect [67].
Signaling pathways in common between PMEs and NDs
In EPM1, EPM2, and MERRF increased OS and mitochondrial impairment is observed. For example, mitochondrial impairment with increased OS happens in stefin B KO mice upon LPS challenge [51]. Neuroinflammation happens in EPM1 and also some other PMEs, such as NCLs and LD [4]. In EPM1, represented by model stefin B KO mice, neuroinflammation is a key hallmark [68]. As well, innate immunity response and inflammasome activation were observed [69, 70]. Activated glia with accompanying excitotoxicity are also in common. Together, these pathological phenomena: mitochondrial impairment, increased OS, neuroinflammation and activated (reactive) glia, likely end-points of other upstream events, are in common with several PMEs (Figure 2) but also with many other neurodegenerative conditions.
A common signaling pathway impaired in PMEs, NDs with AD as prototype, as well as in neuropsychiatric diseases (NPDs) is the Wnt signaling, as observed and suggested by Polajnar and Žerovnik [71, 72]. Wnt signaling is an autocrine paracrine signal transduction pathway. It plays a role in cytoskeleton and midbrain development, among others. Ablation of Wnt-1 results in severe defects of the midbrain, the cerebellum and the spinal cord while ablation of Wnt-3a results in a total loss of the hippocampus. One can distinguish canonical and non-canonical Wnt signaling pathways. The canonical (classical) pathway is described in the legend to Figure 3. In short: the Wnt-protein ligand binds to a Fz family receptor, which passes the signal to the protein Dsh. Dsh negatively regulates GS kinase-3 beta (GSK3β), which inhibits β-catenin. At least two non-canonical Wnt pathways exist: the PCP pathway, in which Fz acts through monomeric GTPase and Jun N-terminal kinase (JNK) to regulate the cytoskeleton [73] and the Wnt/Ca2+ signaling pathway, in which Fz activation leads to increased intracellular Ca2+ [74].
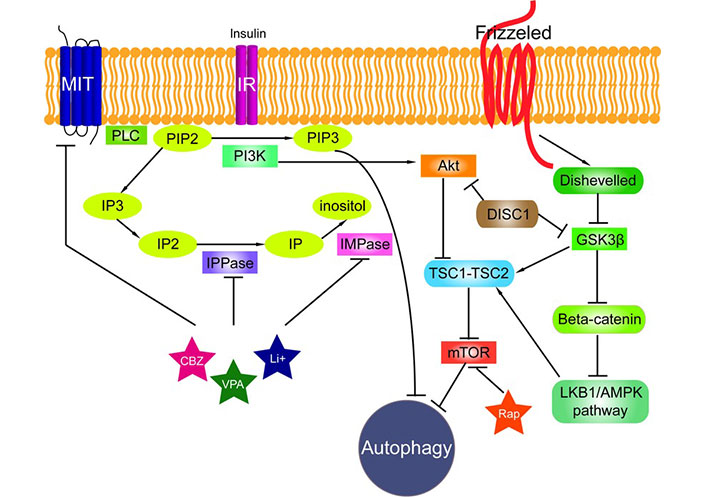
The signalling pathways connected to autophagy [71]. Phosphatidylinositol signalling pathway is regulated by Class I phosphoinositide 3-kinases (PI3Ks), which are activated by kinase receptors like insulin receptors (IRs) and responsible for the production of phosphatidylinositol (3,4,5)-triphosphate (PIP3) from phosphatidylinositol 4,5-bisphosphate (PIP2). Phospholipase C (PLC) cleaves the PIP2 into diacyl glycerol and inositol 1,4,5-trisphosphate (IP3). Inositol polyphosphate 1-phosphatase (IPPase) catalyses inositol bisphosphate (IP2) to inositol monophosphate (IP), which is further dephosphorylated by inositol monophosphatase (IMPase) to inositol. High-affinity inositol transport is additionally catalysed by the active myo-inositol/H+ transporter (MIT). MIT, IPPase and IMPase are all inhibited by carbamazepine (CBZ), valproic acid (VPA) and lithium (Li+). PI3Ks have been linked to an extraordinarily diverse group of cellular functions through regulation of the Akt/TSC1-TSC2/mTOR pathway. Disrupted in Schizophrenia 1 (DISC1) in its wild-type form also negatively regulates both GSK3β and Akt (also known as protein kinase B, PKB). Tuberous sclerosis protein 1 and 2 (TSC1/2) form a complex that like rapamycin (Rap) inhibits mammalian target of rapamycin (mTOR) kinase. Wnt signalling activation is mediated through binding of a Wnt-protein ligand to a Frizzled family receptor, which passes the biological signal to the protein Dishevelled (Dsh). Dsh negatively regulates glycogen synthase kinase-3 beta (GSK3β), which alternatively inhibits β-catenin, one of the central proteins of the Wnt signalling pathway. β-Catenin, however, negatively regulates LKB1/AMPK pathway (liver kinase B1/5’ adenosine monophosphate-activated protein kinase) that indirectly regulates autophagy
Note. Reprinted from “Impaired autophagy: a link between neurodegenerative and neuropsychiatric diseases” by Polajnar M, Zerovnik E. J Cell Mol Med. 2014;18:1705–11 (https://onlinelibrary.wiley.com/doi/10.1111/jcmm.12349). CC BY.
mTOR signaling is indeed affected in LD and in EPM1, where in addition Prickle gene is part of PCP signaling [62] and stefin B is also involved in PCP signaling [37] and in autophagy [29]. A recent paper reports that stefin B over-expression downregulated mitochondrial ROS and lowered inflammasome activation. This occurred concomitantly with AMPK phosphorylation and downregulation of mTOR activity, leading to the induction of autophagy [55].
To be noted, in AD and in schizophrenia the disrupted Wnt signaling pathway leads to poor working memory. Namely, inhibition of Wnt signaling impairs long-term potentiation (LTP) while its activation facilitates LTP, as a study on a mouse model showed. Huperzine A and lithium, both inhibit (GSK3β) and thus stabilise levels of β-catenin [74].
What are the prospects for a cure or early treatment of various monogenic PMEs?
Treatments for PMEs thus far have been supportive and palliative. With recent advances in enzyme replacement methods, short RNA-based therapies, stem cell and gene therapies, prospects are brighter [5]. In more detail, enzyme replacement therapy by providing active enzymes into the affected cells seems a likely way forward for ULD, NCLs, Farber disease and sialidoses. To replace the impaired gene either by viral delivery or by mRNA embedded in liposomes might soon become possible for ULD [75] and Batten disease [76] and, if side effects are manageable, this would represent a cure. Gene replacement has been successfully applied also in LD mouse model [77].
As autophagy is impaired in LD, we suggested this might be helped by autophagy-enhancing molecules, such as rapamycin analogs [72]. Recently, indeed such a trial for LD model mice was performed by using trehalose [78]. This treatment resulted in lower susceptibility to PTZ-induced seizures but did not reduce the burden of Lafora bodies [5].
In LD and ULD proteostasis is also impaired (see Figures 1 and 3), therefore, heat shock proteins would seem beneficial. Such a trial was performed on LD model mice, where heat shock factor-1 (HSF1) was restored by using dexamethasone treatment [79].
Neuroinflammation, which is a hallmark of most of PMEs can be decreased by using known anti-inflammatory drugs but also by other modulators of inflammation [80] as was shown in a study of LD model mice.
Another common impairment of PMEs is OS. Anti-oxidants, such as NAC [81], and different vitamins have been applied to improve EPM1, EPM2, and MERRF, however, often with no clear benefit. The reason might be that oxidative damage has already occurred. An early treatment would likely bring more benefit, however, it depends on the time of diagnosis. Based on case studies NAC proved beneficial for ULD [82, 83].
Of note, some anti-oxidant molecules can also decrease the number of protein aggregates, which may increase along with the decrease of autophagic flux and increase in OS (this makes a vicious circle, negative feedback). We have performed a study where polyphenols and some vitamins affect on aggregation of stefin B [84].
Conclusion
PMEs entered into an exciting era when the disease cure may be expected. By more understanding of the underlying molecular and cellular processes, which will lead to early interventions, better supportive therapies and eventually, by using stem cells, enzyme replenishment (delivery of liposome enwrapped proteins or by mRNA vaccination) and lowering gene expression (by RNAi) or totally new gene incorporation, i.e., replacement of the mutated gene, familial PMEs might get cured at the core cause. Similar development is seen with some of the so called orphan diseases [85].
Abbreviations
| AD: | Alzheimer’s disease |
| CSTB gene: | cystatin B gene |
| DS: | Down syndrome |
| Dsh: | Dishevelled |
| EPM1: | Progressive myoclonic epilepsy type 1 |
| Fz: | Frizzled |
| GS: | glycogen synthase |
| GSK3β: | glycogen synthase kinase-3 beta |
| KO: | knock-out |
| LD: | Lafora disease |
| MERRF: | myoclonus epilepsy with ragged-red fibers |
| mtDNA: | mitochondrial DNA |
| NAC: | N-acetyl cysteine |
| NCLs: | neuronal ceroid lipofuscinoses |
| NDs: | neurodegenerative diseases |
| NPDs: | neuropsychiatric diseases |
| OS: | oxidative stress |
| PCP: | planar cell polarity |
| PMEs: | progressive myoclonus epilepsies |
| ULD: | Unverricht-Lundborg disease |
Declarations
Acknowledgments
I thank all the collegues and PhD students, now successful scientists, who contributed to molecular and cellular studies of stefin B. Especially are to be acknowledged: Dr. S. Čeru, Ass. Prof. N. Kopitar Jerala, Dr. M. Polajnar, Ass. Prof. A. Verčič-Taler. For the in vitro work on recombinant stefin B, I acknowledge contributions of Dr. S. Rabzelj, Dr. S. Jenko Kokalj, Prof. D. Turk, all from the Dept. Biochemistry and Molecular and Structural Biology, Jožef Stefan Institute, 1000 Ljubljana, Slovenia. From foreign collaborators I would like to expose Prof. A. Smajlovic Kriještorac, Dr. S. Hasanbašić, Dr. A. Jahić Mujkić and Prof. S. Berbić from the Uni. Tuzla BiH and dr. R.A. Staniforth and prof. J. P Waltho from the Uni. Sheffield, UK.
Author contributions
EŽ: Conceptualization, Investigation, Writing—original draft, Writing—review & editing.
Conflicts of interest
The author declares that there is no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Financial support was provided by the ARIS funded research program: Proteolysis and its regulation in health and disease [P1-0140] (led by Prof. V. Turk by 2009 and after that by Prof. B. Turk). Part of the work on stefin B was also covered by the ARIS research project: Oligomers of amyloidogenic proteins from a to z: biophysical properties, structure, function and mutual interactions [J7-4050] (led by E. Žerovnik). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2024.