
Abstract
Purinergic signaling, mediated by ATP and adenosine receptors, plays a crucial role in cellular communication and homeostasis within the central nervous system (CNS), particularly by regulating synaptic activity, glial cell functions, and neuroplasticity. Glial cells, including astrocytes and microglia, contribute to both short-term processes, such as neurotransmission and neuroinflammation, and long-term functions, including synaptic remodeling, tissue repair, and behavioral adaptation. Dysregulation of purinergic signaling in these cells has been implicated in the pathogenesis of various neurodegenerative and neuropsychiatric disorders. This article explores the evolving concept of the synapse, highlighting the active role of glial cells in synaptic modulation and emphasizing the significance of purinergic signaling in synaptic function and responses to conditions such as injury and neurotoxicity. Specifically, it examines the roles of ATP and adenosine receptors—such as P2X4, P2X7, P2Y1, and P2Y12—in mediating key astrocytic and microglial functions, including neuroinflammation, phagocytosis, synaptic plasticity, and neuronal damage. Furthermore, the article discusses the involvement of purinergic receptors in neurological disorders such as epilepsy, Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, ischemic stroke, Rett syndrome, and autism spectrum disorder, as well as potential therapeutic strategies targeting these receptors to mitigate inflammation, promote tissue repair, and improve clinical outcomes.
Keywords
Astrocytic and microglial purinergic signaling, immune responses, neuroinflammatory processes, quadripartite synapse model, Alzheimer’s disease, stroke and injury, epilepsy, multiple sclerosis (MS)Introduction
Purinergic signaling, mediated by ATP and adenosine through their respective receptors, plays a central role in regulating diverse physiological processes across various cell types [1, 2]. Initially recognized for its involvement in energy metabolism, ATP has emerged as a crucial signaling molecule in the central nervous system (CNS), influencing a wide range of functions, from neurotransmission to inflammation [3, 4]. The discovery of purinergic receptors has significantly advanced our understanding of how nucleotides and nucleosides facilitate cellular communication. These receptors modulate essential processes such as neurotransmitter release, synaptic plasticity, and cell proliferation [5, 6].
In CNS, purinergic signaling is vital for communication between neurons and glial cells. ATP, acting through P2 receptors, regulates excitatory neurotransmission and modulates glial cell functions, including the responses of astrocytes and microglia to injury or disease [5, 7, 8]. Conversely, adenosine, acting through P1 receptors, exerts neuromodulatory effects by influencing neurotransmitter release and promoting neuroprotective responses under stress. These dual roles underscore the complexity of purinergic signaling, where disruptions in neuronal-glial communication and inflammatory responses contribute to the pathogenesis of neurodegenerative disorders [9, 10].
The concept of synaptic communication has evolved from a simplistic neuron-to-neuron model to a more intricate framework that includes non-neuronal cells, such as astrocytes and microglia [11]. This expanded model emphasizes the pivotal roles of glial cells in synaptic function, maintenance, and plasticity, with purinergic signaling acting as a key mediator. It offers new insights into neuroimmune interactions that underlie neurological disorders, such as epilepsy and neurodegeneration, through the modulation of purinergic pathways and glial cell responses [12].
Moreover, purinergic signaling plays a critical role in regulating immune responses during inflammatory processes by modulating immune functions in both the peripheral and CNS. ATP, released in response to cellular injury, activates purinergic receptors on immune cells, including microglial cells, thereby initiating inflammatory cascades [13, 14]. In contrast, adenosine exerts immunosuppressive effects, balancing pro-inflammatory and anti-inflammatory responses. Disruptions in purinergic signaling are associated with neuroinflammatory disorders such as epilepsy and multiple sclerosis (MS) [10, 15]. Targeting purinergic signaling pathways offers promising therapeutic strategies for managing CNS-related diseases.
This article provides a comprehensive analysis of glial purinergic signaling, emphasizing the roles of ATP and adenosine in cellular communication and their broader implications for CNS function. It explores the evolving concept of synaptic communication, highlighting the critical involvement of astrocytes and microglia in regulating neuroinflammatory responses and synaptic plasticity. The review details the functional roles of glial purinergic receptors, including P2X, P2Y, and P1 subtypes, in mediating key physiological processes such as phagocytosis, calcium signaling, and cytokine release. Additionally, it examines how dysregulated glial purinergic signaling disrupts CNS homeostasis and contributes to the pathophysiology of neurological disorders, including neurodegenerative diseases, stroke, and traumatic brain injury (TBI). By emphasizing the therapeutic potential of selectively targeting purinergic receptors, the article provides insights into strategies for promoting CNS repair in various neurological conditions.
Functional diversity of purinergic signaling and its implications for cellular communication and glial cell mediation
Purinergic receptors in the central nervous system: mechanisms of synaptic modulation, neuroplasticity, and neuroinflammation
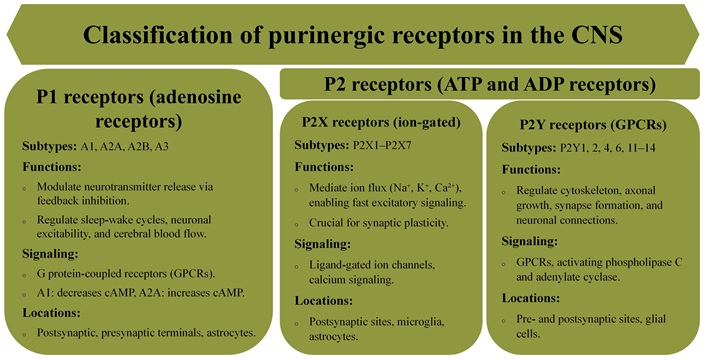
The discovery of purinergic receptors has significantly advanced our understanding of how nucleotides and nucleosides regulate a variety of physiological processes, particularly in the CNS. These receptors are classified into two major families: P1 receptors, which are selective for adenosine, and P2 receptors, which primarily respond to ATP and ADP (Figure 1). While ATP is well-known for its role in cellular energy metabolism, it also acts as a crucial extracellular signaling molecule in the CNS. Astrocytes and neurons are the primary sources of ATP release into the surrounding environment [16, 17]. Early studies faced challenges due to ATP’s rapid degradation into adenosine, which masked the distinct roles of ATP and adenosine in signaling. Later research clarified that P1 receptors, inhibited by methylxanthines, mediate adenosine’s effects, while ATP activates P2 receptors, establishing two distinct signaling pathways [1, 18, 19].
Purinergic signaling plays a fundamental role in cellular communication across various species, underscoring its evolutionary conservation. Homologs of P2X receptors have been identified in organisms like Dictyostelium and Schistosoma, and ATP signaling is even present in plants, highlighting its importance in maintaining cellular functions and enabling adaptive responses across biological systems [20, 21]. In the healthy brain, purinergic signaling coordinates processes between both neural and non-neural cells, promoting homeostasis and facilitating adaptive responses to physiological and environmental changes. Glial cells are central to orchestrating these processes, interacting with neurons and regulating synaptic activity, metabolism, and immune responses [7, 8, 22].
Purinergic receptors exhibit remarkable diversity, with specific subtypes performing distinct cellular functions (Figure 1). The P1 receptor family includes four subtypes (A1, A2A, A2B, and A3), each with varying affinities for adenosine, influencing pathways such as modulation of adenylate cyclase activity [1, 23, 24]. The P2 receptor family consists of P2X receptors, which are ligand-gated ion channels, and P2Y receptors, which are G protein-coupled receptors (GPCRs) [25]. The P2X receptor family consists of seven subtypes (P2X1–7), which can form both homo- and heteromeric channels [26, 27]. Notably, in addition to heteromeric channels, the P2X7 receptor forms homotrimeric channels that mediate cellular responses such as proliferation, apoptosis, and inflammation. P2X receptors facilitate ion flux (Na+, K+, Ca2+), resulting in increased intracellular calcium levels and membrane depolarization [28, 29]. In contrast, the eight subtypes of P2Y receptors (P2Y1, 2, 4, 6, and 11–14) initiate downstream signaling via GPCR pathways, regulating targets like phospholipase C (PLC) and adenylate cyclase [30, 31].
The activity of purinergic receptors is precisely regulated by ecto-nucleotidases, such as nucleoside triphosphate diphosphohydrolase-1 (CD39) and ecto-5’-nucleotidase (CD73), which hydrolyze ATP and ADP into adenosine, thus activating P1 receptors and maintaining the balance between ATP and adenosine signaling [32–34]. Adenosine’s effects are further fine-tuned by enzymatic degradation and uptake through equilibrative nucleoside transporters (ENT1–4) [35, 36], ensuring that purinergic signaling maintains cellular homeostasis and facilitates appropriate cellular responses.
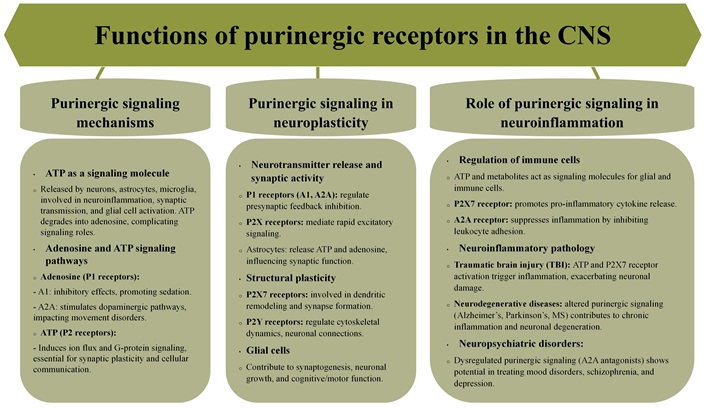
Purinergic receptors are crucial for neuroplasticity, influencing neurotransmission, structural plasticity, and behavioral adaptation (Figure 2) [5, 24]. Glial cells play key roles in these processes due to their expression of purinergic receptors, extending the influence of purinergic signaling beyond neurons [37]. P2 receptors, particularly P2X receptors localized to postsynaptic sites, mediate rapid excitatory signaling in response to ATP release [38–40]. P1 receptors, such as A1 and A2A, modulate neurotransmitter release through feedback inhibition at presynaptic sites, fine-tuning synaptic activity, especially during prolonged neuronal firing, and contributing to synaptic plasticity [41, 42]. Astrocytes actively participate by releasing ATP and adenosine in response to neuronal activity, influencing synaptic function and maintaining extracellular homeostasis [43, 44].
Functions of purinergic receptors in CNS. CNS: central nervous system; MS: multiple sclerosis
Purinergic signaling also regulates structural changes in neurons. P2X7 receptors are involved in dendritic remodeling and synapse formation, processes critical for learning and memory [45, 46]. P2Y receptors regulate the cytoskeleton and promote axonal growth, facilitating synapse formation and neuronal connections [47, 48]. Glial cells support structural plasticity by releasing ATP in response to neuronal activity, promoting synaptogenesis and neuronal growth [49, 50]. Microglia utilizes purinergic signaling to mediate neuroinflammatory responses that can impact synaptic plasticity [51, 52]. These glial-mediated effects emphasize the crucial role of glial cells in shaping neuronal networks during development, learning, and injury repair [50, 53].
Purinergic signaling is also implicated in regulating behavioral processes such as attention and memory [54]. Glial cells, particularly astrocytes, contribute to behavioral plasticity by modulating synaptic transmission through purine release, directly affecting neuronal excitability and cognitive functions [55].
Purines produced in the CNS regulate the infiltration, trafficking, and activation of peripheral immune cells under both physiological and pathological conditions. ATP and its metabolites, including ADP and adenosine, act as key extracellular signaling molecules through P2X and P2Y purinergic receptors expressed on glial and immune cells [1, 56]. Under normal conditions, purinergic signaling maintains CNS homeostasis by modulating microglial surveillance and synaptic activity [7]. However, in neuroinflammatory states such as TBI and neurodegenerative diseases, excessive ATP release from damaged cells acts as a damage-associated molecular pattern (DAMP), activating microglia and infiltrating immune cells via P2X7 receptors [57]. Purines also influence leukocyte adhesion and transmigration across the blood-brain barrier (BBB) by modulating endothelial activation. Adenosine, through A2A receptors, can suppress inflammation by inhibiting leukocyte adhesion and promoting the anti-inflammatory M2 microglial phenotype [58]. Conversely, ATP-P2X7 receptor signaling amplifies inflammation by promoting pro-inflammatory cytokine release, such as interleukin-1β (IL-1β) and tumor necrosis factor-alpha (TNF-α), further enhancing leukocyte recruitment [57].
Experimental models have provided critical insights into the multifaceted roles of purinergic signaling in CNS pathology. In experimental autoimmune encephalomyelitis (EAE), a widely used model for MS, the blockade of P2X7 receptors has been shown to attenuate neuroinflammation by reducing T cell activation and subsequent CNS infiltration [59, 60]. T cell activation in EAE is a highly coordinated process initiated by antigen-presenting cells (APCs), including dendritic cells, microglia, and macrophages, which process and present myelin-derived peptides via major histocompatibility complex (MHC) molecules to naïve CD4+ T cells [61]. This interaction, in conjunction with co-stimulatory signaling (e.g., CD28/B7) and cytokine-mediated polarization, drives the differentiation of naïve T cells into pathogenic effector subsets, particularly T helper 1 (Th1) and Th17 cells [62, 63]. ATP, acting through P2X7 receptors on both APCs and T cells, plays a pivotal role in modulating this activation process. P2X7 receptor activation promotes the assembly of the NOD-like receptor protein 3 (NLRP3) inflammasome, leading to the release of pro-inflammatory cytokines such as IL-1β and interleukin-18 (IL-18), which further amplify T cell expansion and effector function [64]. Moreover, P2X7 receptor signaling enhances the metabolic reprogramming of T cells, favoring glycolysis-driven proliferation, a key requirement for sustaining the inflammatory response. The blockade of P2X7 receptors disrupts this inflammatory cascade by impairing inflammasome activation, attenuating IL-1β release, and limiting the expansion of pathogenic Th1 and Th17 cells [65]. Consequently, the reduction in pro-inflammatory T cell infiltration across the BBB alleviates CNS inflammation and tissue damage, highlighting P2X7 as a potential therapeutic target for autoimmune neuroinflammatory disorders [66].
Similarly, in spinal cord injury (SCI), ATP release drives microglial activation and secondary tissue damage [67, 68]. In TBI, purinergic receptor activity exhibits both protective and detrimental effects depending on receptor subtype and activation timing, underscoring the complex and context-dependent nature of purinergic signaling in neuroinflammation [69].
Short- and long-term roles of purinergic signaling in the CNS mediated by astrocytes and microglia
Purinergic signaling plays a central role in regulating both short-term and long-term cellular functions in the CNS, supporting a diverse range of physiological and pathological processes. Short-term effects, ranging from milliseconds to seconds, involve rapid synaptic transmission and acute cellular responses, while long-term effects, spanning minutes to hours or even days, include sustained cellular changes such as gene expression modulation, neuroinflammation, and prolonged adaptations in synaptic plasticity and cellular proliferation [1, 5, 70]. Initially, purinergic signaling was primarily studied in the context of short-term events such as neurotransmission, neuromodulation, secretion, chemoattraction, and acute inflammation. These processes are primarily mediated by ATP, which engages P2X ionotropic and P2Y metabotropic receptors to trigger rapid cellular responses necessary for maintaining homeostasis under acute conditions [24, 25, 71]. However, emerging evidence highlights that purinergic signaling also has long-term (trophic) roles, such as cell proliferation, differentiation, migration, and apoptosis [72, 73]. These trophic functions are critical for developmental processes, tissue repair, and the progression of pathological states like cancer and atherosclerosis (Table 1).
Short-term vs. long-term functions of purinergic signaling in the CNS
| Function | Short-term roles | Long-term roles | Cellular involvement | References |
|---|---|---|---|---|
| Neuromodulation | - Modulation of synaptic transmission- Enhanced synaptic plasticity during neuronal firing | - Maintenance of synaptic homeostasis over time- Regulation of long-term potentiation | Neurons, astrocytes, microglia | [7, 38, 74] |
| Synaptic plasticity | - Modulation of neurotransmitter release at synapses- Regulation of ion flux via P2X receptors | - Structural changes like dendritic remodeling- Synapse formation and stabilization | Neurons, astrocytes, oligodendrocytes | [6, 40, 70] |
| Inflammatory responses | - Activation of microglia for injury response- Chemotactic signaling through P2Y12 receptors | - Persistent neuroinflammatory responses in neurodegenerative disorders, such as Alzheimer’s and Parkinson’s disease | Microglia, astrocytes, neurons | [24, 75, 76] |
| Cognitive and behavioral adaptation | - Modulation of neuronal excitability- Regulation of neurotransmitter release during short-term stress or stimulus | - Long-term effects on learning and memory- Potential involvement in psychiatric disorders (e.g., schizophrenia, anxiety) | Astrocytes, neurons, microglia | [24, 54, 70, 77] |
| Tissue repair and regeneration | - Acute ATP release to mediate damage response and recruit glial cells | - Long-term neurogenesis, axonal growth, and synaptic remodeling | Astrocytes, microglia, oligodendrocytes | [78, 79] |
Astrocytes and microglia are key mediators of purinergic signaling in both short-term and long-term contexts [80]. Astrocytes release ATP in response to neuronal activity and various physiological stimuli. In short-term contexts, ATP activates P2 receptors on neighboring neurons and glial cells, modulating synaptic transmission and enhancing synaptic plasticity. These processes are integral to neuromodulation, a crucial aspect of dynamic neuronal communication [75, 78]. Astrocytic purinergic signaling is also essential for long-term functions such as regulating cell migration, angiogenesis, and tissue repair. ATP released by astrocytes is metabolized to adenosine, acting on trophic receptors to influence cell proliferation and neurogenesis, supporting developmental and reparative processes [78, 81, 82].
Microglia are similarly regulated by purinergic signaling. In short-term responses, ATP acts as a “find-me” signal, guiding microglial migration to injury or infection sites through activation of P2Y12 receptors [75, 83]. This chemotactic response initiates inflammatory processes and facilitates the clearance of cellular debris, essential for resolving acute damage. Beyond these immediate actions, microglia are involved in long-term functions, including synaptic remodeling and pruning, crucial for maintaining synaptic integrity and plasticity [22, 51, 84]. These processes are regulated by ATP and adenosine signaling, with dysregulation contributing to neurodegenerative diseases. Chronic activation of P2X7 receptors on microglia, for example, has been associated with sustained inflammatory responses and neurotoxicity, highlighting the dual nature of purinergic signaling in microglial physiology [52, 76, 85, 86].
By integrating both short-term and long-term functions, purinergic signaling orchestrates a complex network of cellular processes in the CNS, with astrocytes and microglia playing critical roles. Astrocytes primarily contribute to neuromodulation and structural support, while microglia specialize in immune surveillance and synaptic remodeling [74, 80, 87]. Together, these glial cells modulate the dynamic and adaptive responses of the CNS to physiological demands and pathological challenges.
The quadripartite synapse and the role of purinergic receptors in neuro-immune communication
The concept of the synapse has evolved considerably, transitioning from an understanding rooted solely in neuronal interactions to a more complex model that incorporates non-neuronal cell types [11, 37]. Early studies emphasized synaptic communication as a direct interaction between pre- and post-synaptic neurons, focusing on neurotransmitter release, receptor activation, and subsequent downstream effects. However, over time, research began to reveal astrocytes as active participants in synaptic function, leading to the emergence of the tripartite synapse model [11, 88]. In this model, astrocytes are recognized not only as structural supports but as active modulators of synaptic transmission through the release of gliotransmitters. Their processes extend into synaptic spaces, where they detect and respond to changes in neurotransmitter levels, influencing synaptic activity and plasticity (Table 2) [55, 89–91].
Evolution of synaptic models: from neuronal communication to the role of glial cells in synaptic function and neurological disorders
| Era/Model | Key concepts | Key players | Main mechanisms | Notable findings | Implications for neurological disorders | References |
|---|---|---|---|---|---|---|
| Early synaptic model | Synapse as a site of communication between pre- and post-synaptic neurons | Neurons | Neurotransmitter release, receptor activation | Focus on direct synaptic communication via neurotransmitter release and downstream effects | Limited scope, failed to account for contributions from glial cells in synaptic function | [92, 93] |
| Tripartite synapse model (astrocytes and neurons) (2000s) | Inclusion of astrocytes as active participants in synaptic communication | Neurons, astrocytes | Gliotransmitter release, astrocyte involvement in synaptic plasticity | Astrocytes play an active role in synaptic activity by detecting and responding to neurotransmitter levels, modulating plasticity | Introduced the idea of glial cells’ involvement in synaptic modulation | [94–96] |
| Modified tripartite synapse model (neurons and microglia) (2000s–2010s) | Recognition of microglia as regulators of synaptic function and plasticity | Neurons, microglia | Microglial surveillance, synaptic pruning, cytokine-mediated signaling | Microglia monitor and eliminate weak synapses, playing a role in synaptic remodeling | Dysregulated microglial activity contributes to neurodevelopmental disorders and neurodegenerative diseases | [97, 98] |
| Quadripartite synapse model (2010s) | Extension to include microglia in synaptic function | Neurons, astrocytes, microglia | ATP-driven purinergic signaling, synaptic maintenance and pruning | Microglia actively participate in synaptic signaling and plasticity, contributing to both health and pathology | Provides a more holistic view of synaptic communication, involving immune and neuroinflammatory responses | [22, 99] |
| Purinergic signaling in quadripartite synapse | ATP as a key signaling molecule among neurons, astrocytes, and microglia | Neurons, astrocytes, microglia | ATP release during neuronal activity, microglial recruitment via P2Y12 receptors | ATP release directs microglial processes toward active synapses, influencing plasticity and hyperactivity in disorders like epilepsy | Disruption of ATP signaling and glial synchronization can lead to disorders like epilepsy and neurodegeneration | [75, 80, 100] |
| Astrocyte-microglial crosstalk | Dynamic interaction between astrocytes and microglia via ATP | Astrocytes, microglia | Modulation of microglial responses to neurotransmission (glutamatergic and GABAergic) | Astrocytes can enhance or suppress microglial activity, maintaining synaptic balance | Disruptions in crosstalk contribute to pathologies like epilepsy, and neurodegenerative diseases | [80, 101] |
| Neuroimmune modulation | Role of glial cells in neuroprotection and response to injury | Astrocytes, microglia | Release of protective molecules (IL-6) and ATP in response to inflammation or injury | Glial cells coordinate protective responses to neurotoxic stimuli and maintain neuronal health | Imbalances in glial responses contribute to inflammatory diseases and worsen neuronal damage in conditions like neurodegeneration and injury | [80, 102, 103] |
| Disruptions in synaptic homeostasis | Alterations in glial cell function contribute to disease | Neurons, astrocytes, microglia | Disruption of synaptic balance via glial dysfunction | Loss of synchrony between astrocytes and microglia can lead to hyperexcitable synapses, as in epilepsy | Implicates glial dysfunction in diseases such as epilepsy, neurodegeneration, and neuroinflammatory conditions | [74, 104, 105] |
IL-6: interleukin-6
Despite the progress introduced by the tripartite model, it quickly became evident that even this framework did not capture the full complexity of synaptic interactions. Microglia have garnered attention for their roles in synaptic pruning, neuro-surveillance, and modulation of neuronal circuits [22, 106]. Their involvement in synaptic health has led to the development of the quadripartite synapse hypothesis, which includes neurons, astrocytes, and microglia in a cooperative network [99, 107]. This model proposes that microglia, alongside astrocytes, participate in synaptic signaling, maintenance, and plasticity through intricate interactions with neurons [74]. The quadripartite model provides a more holistic view of synaptic communication, recognizing the importance of glial cells and highlighting their contributions to both synaptic homeostasis and pathological conditions (Table 2) [108].
Microglial activity is crucial in synaptic remodeling, particularly in synapse elimination, but recent evidence suggests that synaptic pruning is a complex and multifaceted process that may not always depend on microglial engulfment. For instance, in the rodent cortex, synapse elimination continues even in the absence of microglia, with dendritic spines turning over at a rate of 5–10% within a few days [109, 110]. Moreover, Synapse elimination is largely unaffected in mice lacking the complement receptor C3, a key mediator in the proposed microglial synapse engulfment pathway [111]. This observation suggests that synaptic pruning may involve alternative mechanisms, such as tunneling nanotubes, which facilitate bidirectional transfer of intracellular molecules between microglia and neurons, thereby enabling synaptic component exchange [112, 113]. While microglia are known to actively engulf synaptic elements through processes mediated by the complement system, particularly C1q and C3, the precise mechanisms by which complement-tagged synapses are targeted and eliminated in the developing visual system remain an area of active investigation [114]. Foundational studies in this field have opened avenues for exploring molecular cues that regulate microglia-synapse interactions, including purinergic signaling. Specifically, purinergic receptors, especially P2Y12, are implicated in microglial motility and their capacity to detect ATP/ADP gradients released by active neurons, guiding their processes toward synaptic sites [115]. This signaling cascade is essential for both physiological synaptic pruning and pathological conditions, where dysregulated purinergic signaling and complement activation contribute to synaptic loss in neurodegenerative diseases [116]. Consequently, the intersection of complement-mediated pruning and purinergic receptor signaling represents a critical regulatory axis in neurological disease, providing a potential framework for therapeutic interventions aimed at mitigating aberrant synapse elimination. Moreover, microglia may engage in presynaptic trogocytosis or directly phagocytose neurons and dendrites, indicating that synaptic pruning extends beyond traditional microglial engulfment processes. Overall, these observations underscore the complexity of synaptic pruning, revealing diverse mechanisms that may or may not rely on microglial activity [117].
Purinergic signaling has emerged as a critical regulator of microglial functions during synaptic pruning. Both P2X and P2Y receptors are essential in modulating microglial responses to neuronal activity. Activation of P2X receptors induces a rapid influx of calcium ions, significantly influencing microglial motility and their interactions with synaptic sites. This calcium signaling is crucial in determining whether synapse elimination or stabilization occurs, depending on the local environment. In contrast, P2Y receptors regulate microglial chemotactic migration, guiding them to areas where synaptic pruning is taking place. These receptors also modulate the microglial response to ATP released during neuronal activity, thereby influencing the dynamics of synaptic remodeling. Furthermore, microglia contribute to learning-dependent synapse formation via microglial brain-derived neurotrophic factor (BDNF) [110] and dendritic spine remodeling through TNF-α produced by CX3CR1+ monocytes [109]. Therefore, although microglia play a crucial role in synaptic pruning, their functions extend beyond mere synapse elimination. Purinergic receptors act as key regulatory mechanisms that modulate microglial activity in synaptic remodeling and developmental plasticity.
Purinergic signaling is central to the interactions within the quadripartite synapse, particularly the role of ATP as a signaling molecule among neurons, astrocytes, and microglia [99]. During heightened neuronal activity—such as when NMDA receptors are activated—neurons release ATP into the extracellular environment [17, 118]. This ATP release acts as a beacon, signaling microglia and astrocytes to mobilize toward active synapses [119, 120]. Specifically, ATP binds to P2Y12 receptors on microglia, directing their processes toward areas of increased neuronal activity [83]. This recruitment process is crucial in conditions of synaptic hyperactivity, as seen in diseases like epilepsy, where microglial interactions with hyperactive synapses may exacerbate neuronal hyperexcitability [118, 121, 122].
ATP-driven purinergic signaling is not limited to neuron-microglia interactions but extends to astrocyte-microglial crosstalk, influencing overall synaptic modulation. Astrocytes, in response to ATP signaling, release molecules that can enhance or suppress microglial activity, depending on the needs of the surrounding neurons [75, 80, 123]. Experimental studies using agents like probenecid and suramin have further clarified ATP’s role in coordinating microglial responses to both excitatory (glutamatergic) and inhibitory (GABAergic) neurotransmission [124–126]. Through these purinergic pathways, astrocytes and microglia work synergistically to maintain synaptic balance, responding dynamically to shifts in neuronal signaling and ensuring synaptic stability [51, 89].
Astrocytes and microglia engage in complex, ATP-mediated communication that plays a significant role in maintaining synaptic integrity and modulating the brain’s response to injury and neurotoxicity [37, 127]. In inflammatory conditions, such as those induced by lipopolysaccharide (LPS), activated microglia release ATP, signaling astrocytes to engage in protective responses that help shield neurons from potential damage [128, 129]. For instance, astrocytes release interleukin-6 (IL-6) in response to certain neurotoxic stimuli, such as exposure to methylmercury, a process regulated through ATP released by microglia [130, 131]. This crosstalk not only sustains neuronal health but also provides an adaptive response to neurotoxic environments, underscoring the neuroprotective roles of glial cells in the brain’s immune response.
Disruptions in this delicate neuro-immune balance can result in neurological disorders. In epilepsy, for example, the loss of astrocyte-microglial synchrony contributes to the hyperexcitable neuronal environment characteristic of the disorder [132, 133]. Factors such as TNF-α—often released by microglia—can increase glutamate release, exacerbating network hyperactivity [134]. Additionally, P2Y1 receptor signaling has been linked to abnormal synaptic function, implicating purinergic receptor dysfunction in epileptogenesis [10, 135]. Beyond epilepsy, the quadripartite synapse model offers insights into other neurodegenerative and neuroinflammatory conditions, highlighting how glial cell contributions to synaptic function can both support and disrupt neuronal networks, depending on the cellular environment [105, 136, 137].
Purinergic signaling in neuron-glia interactions: mechanisms regulating synaptic function, neuroinflammation, and brain homeostasis
Purinergic signaling, involving ATP and its breakdown product adenosine, is crucial for regulating neuronal and glial functions within the brain. This signaling system interacts with P2 and P1 receptors, which respond to extracellular ATP and adenosine, respectively, and plays a key role in synaptic transmission, plasticity, and neuroinflammatory processes (Table 3).
Mechanisms of purinergic signaling in neuron-astrocyte-microglia interactions
| Mechanism | Cell types involved | Key receptors | Function/Role | References |
|---|---|---|---|---|
| Purinergic signaling in brain homeostasis | Astrocytes, microglia, neurons | P2 receptors (P2Y1, P2Y12), A1Rs, A2ARs | Purinergic signaling regulates synaptic pruning, neuroprotection, maintaining brain development, and homeostasis | [75, 138, 139] |
| Neuron-astrocyte communication for homeostasis | Neurons and astrocytes | P2X, P2Y, A1Rs, A2ARs | Bidirectional communication: neurons release neurotransmitters causing Ca2+ changes in astrocytes, which release ATP to modulate synaptic activity and maintain brain homeostasis | [44, 140, 141] |
| ATP in synaptic transmission | Neurons and astrocytes | P2XRs, NMDA receptors, A1Rs, A2ARs | ATP mediates excitatory neurotransmission; P2XRs regulate synaptic plasticity, with roles in both LTP and synaptic depression | [44, 142, 143] |
| Astrocytic ATP release and synaptic regulation | Astrocytes and neurons | P2X, P2Y, A1Rs, A2ARs | Astrocytes release ATP, influencing synaptic excitability, ion balance, and synaptic plasticity through interaction with adenosine receptors (A1Rs, A2ARs) | [44, 144, 145] |
| Adenosine receptor interactions | Astrocytes and neurons | A1Rs, A2ARs | A2ARs enhance NMDA receptor activity and glutamate release, while A1Rs inhibit synaptic transmission, offering neuroprotective effects | [42, 146, 147] |
| ATP and calcium (Ca2+)-mediated communication | Astrocytes | P2Y, P2X | ATP activates purinergic receptors (P2X, P2Y) to induce Ca2+ increases in astrocytes, propagating Ca2+ waves that influence synaptic modulation | [148–150] |
| Neurovascular coupling and redox homeostasis | Astrocytes and blood vessels | A1Rs, A2ARs | Astrocytes regulate blood flow and redox balance in response to neuronal activity, utilizing ATP and adenosine signaling | [151, 152] |
| Reactive astrocytes in injury | Astrocytes | A1Rs, A2ARs | During injury, ATP and adenosine modulate astrocytic responses: A1Rs suppress excessive excitation; A2ARs promote synaptic facilitation and inflammation | [153, 154] |
| Microglial activation and neuroinflammatory responses | Microglia | P2X4, P2Y12, A2AR | ATP activates microglia through P2X and P2Y receptors, initiating immune responses; P2X4Rs mediate pro-inflammatory responses, while P2Y12Rs guide microglial migration | [155–157] |
| Adenosine modulation of microglial inflammation | Microglia | A2AR | Adenosine via A2ARs counterbalances ATP-induced activation of microglia, reducing inflammation and promoting neuroprotection | [157–160] |
| Astrocyte-microglia crosstalk during injury | Astrocytes and microglia | P2Y1, P2Y12 | ATP release from damaged cells activates purinergic receptors on both astrocytes and microglia, amplifying the inflammatory response and modulating synaptic activity | [37, 75, 80] |
LTP: long-term potentiation
Astrocytic ATP release and purinergic signaling: key mechanisms in synaptic plasticity and intercellular communication
ATP serves as a critical mediator of synaptic function, particularly in excitatory neurotransmission. Ionotropic P2XRs mediate rapid synaptic responses, with hippocampal slice studies demonstrating their involvement in regulating synaptic plasticity. However, their role in long-term potentiation (LTP) and synaptic depression is debated, with some evidence suggesting P2XRs facilitate LTP, while others suggest they induce synaptic depression [38, 39, 142, 161–163].
Astrocytes contribute to synaptic regulation by releasing ATP, which can occur via exocytosis, modulating neuronal excitability and synaptic activity [44, 55, 145, 164]. The interaction between ATP-mediated P2 receptor activation and adenosine receptors (A1Rs and A2ARs) creates a dynamic balance that modulates synaptic transmission [165, 166]. A2ARs typically enhance NMDA receptor activity and glutamate release, promoting synaptic plasticity, while A1Rs inhibit synaptic transmission, offering neuroprotective effects [167–169]. This interplay is vital for maintaining functional neuronal networks and may influence neurodegenerative processes.
In addition to synaptic regulation, astrocytes use purinergic signaling to coordinate calcium (Ca2+)-mediated intercellular communication [170]. ATP acts as a critical extracellular messenger activating purinergic receptors like P2Y and P2X, inducing intracellular Ca2+ increases in astrocytes [38, 145]. These Ca2+ waves propagate through astrocyte networks, influencing synaptic modulation and metabolic support for neurons [171].
ATP signaling also plays a key role in neurovascular coupling, where astrocytes regulate blood flow in response to neuronal activity, and in maintaining redox homeostasis [152]. During injury, astrocytes undergo reactive changes influenced by ATP and adenosine signaling [172]. Adenosine, produced from ATP breakdown by ecto-nucleotidases on astrocyte surfaces, acts on A1 and A2A receptors, modulating astrocytic responses. A1Rs suppress excessive excitation, while A2ARs promote synaptic facilitation and inflammation [166, 169, 173].
ATP and adenosine mediated crosstalk between neurons, astrocytes, and microglia
Neuron-astrocyte communication is essential for brain homeostasis, with both neurons and astrocytes capable of releasing ATP and responding to its extracellular concentrations [4]. Synaptically released neurotransmitters trigger Ca2+ changes in astrocytes, leading to the release of gliotransmitters like ATP, which modulate synaptic activity [174]. This bidirectional communication enables astrocytes to regulate synaptic environments by clearing neurotransmitters, maintaining ion balance, and modulating synaptic strength [175]. ATP conversion to adenosine further refines synaptic transmission through P1 receptors (A1Rs and A2ARs) [144, 176].
The fine-tuning of synaptic activity by this purinergic system is vital for regulating excitatory and inhibitory synapses, which is essential for maintaining neural network activity [177, 178]. Dysregulation of purinergic signaling has been implicated in various pathological conditions, including neurodegenerative diseases [6].
Purinergic signaling is also essential for microglial function, particularly in neuroinflammatory processes. ATP acts as danger signal activating microglia via P2X and P2Y receptors, initiating immune responses [75, 179]. P2X4 receptors mediate pro-inflammatory responses, contributing to conditions like neuropathic pain, epilepsy, and stroke, while P2Y12 receptors guide microglial migration to injury sites, modulating their interaction with neurons and promoting neuroprotective processes [75, 180, 181].
Adenosine, through A2ARs, counterbalances ATP-mediated microglial activation, reducing inflammation by inhibiting cytokine release and regulating potassium currents [157, 182]. This balance ensures that microglia respond appropriately to injury without exacerbating inflammation, highlighting the purinergic system’s role in neuroprotection.
Astrocytes and microglia interact closely during both physiological and pathological conditions, with ATP playing a crucial role in this crosstalk [37, 80]. In response to brain injury, damaged cells release ATP, activating purinergic receptors on both astrocytes and microglia. Microglial ATP release can activate P2Y1 receptors on astrocytes, triggering additional ATP release and amplifying the response [75, 183, 184]. This interaction influences synaptic activity, with astrocyte-derived ATP modulating microglial motility and activation, often via P2Y12Rs.
The bidirectional communication between astrocytes and microglia, mediated by purinergic signaling, is critical for brain development, homeostasis, and injury responses [7]. The balance between ATP and adenosine signaling through P2 and P1 receptors shapes glial activity, impacting processes such as synaptic pruning, and neuroprotection [6].
Purinergic signaling in neuroinflammation: glial regulation and immune dynamics
Purinergic signaling orchestrates an extensive range of immune and inflammatory responses. In the CNS, glial cells serve as pivotal regulators of these processes, shaping the neuroinflammatory landscape (Figure 3). Dysregulation of glial-mediated purinergic signaling plays a central role in the progression of neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and MS [3, 6, 76]. Deciphering the intricate mechanisms underlying this signaling network is essential for advancing therapeutic strategies targeting neuroinflammatory and immune-related disorders.
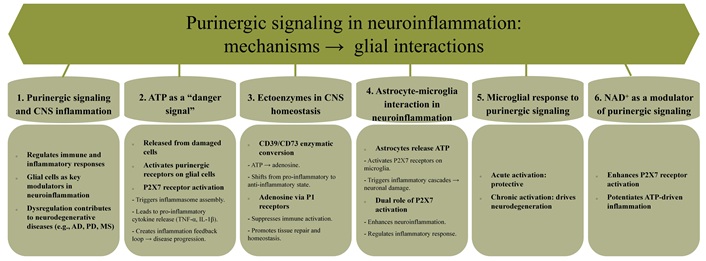
Purinergic signaling in neuroinflammation: mechanisms and glial cell interactions. CNS: central nervous system; NAD+: nicotinamide adenine dinucleotide; CD39: nucleoside triphosphate diphosphohydrolase-1; CD73: ecto-5’-nucleotidase; AD: Alzheimer’s disease; PD: Parkinson’s disease; MS: multiple sclerosis; TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1β
In the CNS, ATP released from damaged or necrotic cells acts as a potent “danger signal”, triggering immune responses via purinergic receptor activation on glial cells (Figure 3) [185, 186]. Among these receptors, P2X7 stands out for its pivotal role in assembling inflammasomes—multiprotein complexes that mediate the production and release of pro-inflammatory cytokines, including TNF-α and IL-1β [2, 179, 187]. These cytokines amplify immune responses, creating a feed-forward loop of inflammation that exacerbates neurodegenerative disease pathology [9, 188, 189].
The extracellular environment of the CNS is meticulously regulated by ectoenzymes such as CD39 and CD73, which sequentially hydrolyze ATP into adenosine (Figure 3). This enzymatic conversion shifts the signaling milieu from a pro-inflammatory state dominated by ATP to an anti-inflammatory state mediated by adenosine [33, 190–192]. Acting through P1 receptors expressed on glial cells, adenosine suppresses immune activation, fosters tissue repair, and preserves CNS homeostasis [160, 193, 194]. This dynamic interplay highlights the dual and context-dependent roles of ATP and adenosine in neuroinflammation.
Astrocytes play dual roles during neuroinflammatory events, acting as both supportive and detrimental players in the pathology of neurodegenerative diseases. In conditions such as amyotrophic lateral sclerosis (ALS), astrocytes release ATP, which activates P2X7 receptors on neighboring microglia [195, 196]. This activation initiates a cascade of inflammatory responses that can exacerbate neuronal damage. While P2X7 receptor activation may promote neuroinflammation, it also serves a regulatory role in modulating the inflammatory response, underscoring its complex involvement in the neuroinflammatory process [26]. In vitro studies using astrocyte cell cultures have demonstrated that P2X7 receptor activation leads to the release of pro-inflammatory cytokines, further highlighting the importance of these receptors in the neuroinflammatory milieu.
This interaction amplifies the release of inflammatory cytokines, creating a positive feedback loop that intensifies neuroinflammation [37, 85]. Concurrently, microglia respond to purinergic signals by mounting immune responses. While this activation can be protective during acute phases, prolonged microglial activation contributes to chronic inflammation and tissue damage, which are hallmarks of neurodegenerative diseases [3, 197, 198].
Nicotinamide adenine dinucleotide (NAD+) emerges as a critical modulator of purinergic signaling, primarily through its enhancement of P2X7 receptor activation. Elevated NAD+ levels potentiate ATP-driven inflammatory signaling, positioning NAD+ metabolism as a promising therapeutic target for modulating immune responses and neuroinflammation in the CNS [199–201].
Targeting purinergic signaling offers a compelling approach to managing neuroinflammation and related neurodegenerative disorders. Key strategies include inhibiting ATP release, blocking P2 receptor activation, and modulating ectoenzymes like CD39 and CD73 to regulate glial activation, mitigate inflammation, and slow disease progression. A comprehensive understanding of its role in glial immune regulation is essential for advancing effective therapeutic interventions.
Purinergic signaling in astrocytes: mechanisms, neuroinflammatory modulation, and implications for CNS disorders
Purinergic signaling in astrocytes is a pivotal mechanism for maintaining brain homeostasis and modulating neuroinflammatory responses [15, 78, 195]. Astrocytes, as the most abundant glial cells in the CNS, engage in a variety of processes through purinergic receptors (Figure 4). Ionotropic P2X receptors, particularly P2X4 and P2X7, mediate rapid responses to extracellular ATP, a vital intercellular signaling molecule. P2X4 facilitates calcium influx, synaptic plasticity, and astrocytic migration, functions essential for neuronal health and tissue repair [38, 66, 202, 203]. Furthermore, P2X4 activation triggers the release of BDNF, which supports neuronal survival and plasticity. The upregulation of P2X4 in neuroinflammatory conditions like SCI and MS positions it as a potential therapeutic target for modulating astrocytic function and mitigating inflammatory damage [204–207]. Conversely, P2X7 plays a central role in gliotransmitter release and cellular stress responses but is often dysregulated in neuroinflammatory diseases such as Alzheimer’s and epilepsy [66, 208]. Persistent activation of P2X7 by elevated ATP levels leads to NLRP3 inflammasome activation and the release of pro-inflammatory cytokines [23, 209, 210], exacerbating disease progression and making it another critical target for intervention.
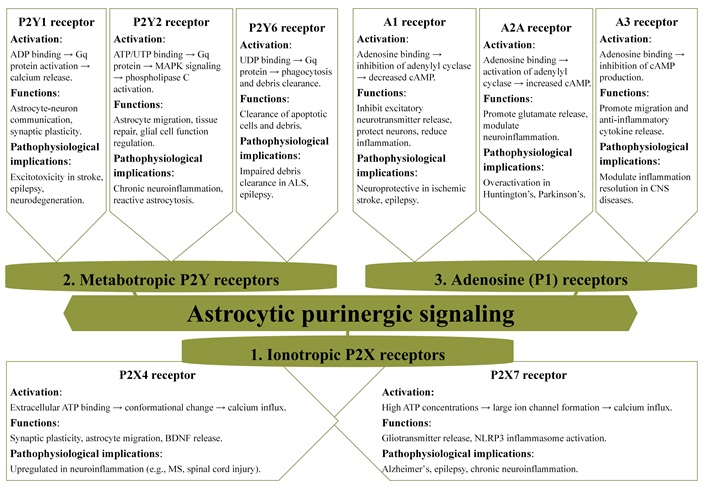
Purinergic receptor expression in astrocytes: mechanisms of activation and their roles in CNS function and pathophysiology. MAPK: mitogen-activated protein kinase; ALS: amyotrophic lateral sclerosis; CNS: central nervous system; BDNF: brain-derived neurotrophic factor; MS: multiple sclerosis; NLRP3: NOD-like receptor protein 3
Metabotropic P2Y receptors facilitate slower but sustained responses in astrocytes [150]. P2Y1, a receptor involved in ADP-induced calcium signaling, is integral to synaptic plasticity and memory formation. However, its dysregulation is linked to excitotoxicity, contributing to neuronal damage in conditions such as stroke and epilepsy [54, 211]. P2Y2 is crucial for astrocyte migration and tissue repair, mediated through the activation of mitogen-activated protein kinase (MAPK) and PLC signaling pathways. While P2Y2 promotes healing, its prolonged activation can result in reactive astrocytosis, a pathological hallmark of chronic neuroinflammation [47, 212, 213]. Although less studied, P2Y4 is believed to play a role in regulating nutrient uptake and energy metabolism, suggesting its importance in cellular energetics [214, 215].
The P1 adenosine receptors add another layer of complexity to purinergic signaling in astrocytes [7, 78]. Among these, the A1 receptor exerts neuroprotective effects by inhibiting excitatory signaling and calcium wave propagation, which helps prevent excitotoxicity. This receptor emerges as a promising target for neuroprotection in ischemic stroke and epilepsy, where excessive excitatory signaling can lead to neuronal damage [169, 216, 217]. Conversely, the A2A receptor exacerbates neuroinflammation by enhancing glutamate release, contributing to excitotoxicity and disease progression in conditions like Huntington’s disease [218, 219]. The A3 receptor, on the other hand, promotes astrocyte migration and the release of anti-inflammatory cytokines, offering neuroprotection in neuropathic pain conditions [220, 221]. Together, these receptors regulate a delicate balance between excitotoxic and anti-inflammatory pathways in astrocytes, influencing their role in CNS physiology and pathology.
The interplay between P2 and P1 receptors orchestrates critical astrocytic processes, including calcium dynamics, energy metabolism, and immune responses. Dysregulation of these signaling pathways is implicated in a variety of CNS disorders. In neurodegenerative diseases such as Alzheimer’s and Huntington’s, aberrant P2X7 and A2A receptor activity drives chronic neuroinflammation, a key factor in disease progression [222, 223]. Similarly, in ischemic stroke, the neuroprotective effects of the A1 receptor stand in contrast to the tissue repair-promoting functions of P2Y2 [224, 225]. In chronic pain and epilepsy, alterations in P2X4 and P2Y1 receptor signaling contribute significantly to disease pathology [184, 226]. The diverse roles of these purinergic receptors underscore their therapeutic potential, offering opportunities to target neurodegeneration, inflammation, and injury repair effectively (Figure 4).
The pivotal roles of purinergic signaling in microglial function: mechanisms, receptor dynamics, and neuroinflammatory implications
Microglia relies heavily on purinergic signaling to regulate their diverse physiological and pathological roles [75, 227]. Purinergic receptors play a central role in modulating microglial functions and processes, including cell dynamics, phagocytosis, chemotaxis, and the release of inflammatory mediators [75, 228]. Different P2Y and P2X receptor subtypes are critical for these activities, with the involvement of each receptor varying according to the activation state of the microglia in response to ATP or ADP signaling (Figure 5) [229, 230]. These dynamics enable both homeostatic regulation and responses to pathological stimuli within the brain. Each receptor subtype contributes uniquely to microglial functions, including cellular motility and interactions with neurons [25].
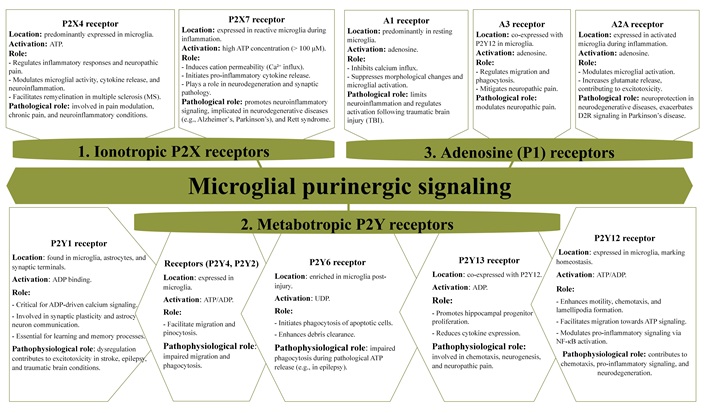
Purinergic receptor subtypes in microglia: expression, activation, and their functional and pathophysiological roles. D2R: D2 receptor; NF-κB: nuclear factor kappa B
P2X receptors in microglia
P2X receptors, particularly P2X4 and P2X7, are expressed in microglia and play crucial roles in injury, inflammation, and pain responses [180, 231, 232].
P2X4 receptors play a pivotal role in regulating inflammatory responses and are closely associated with neuropathic pain. Activation by ATP induces calcium influx through the receptor, which triggers the p38 MAPK pathway and leads to the release of BDNF [181, 207, 233–235]. BDNF enhances dorsal horn neuron excitability, contributing to pain sensitivity, especially following nerve or SCI. Additionally, P2X4 receptors modulate microglial activity, amplifying pro-inflammatory signaling and cytokine release, which exacerbates neuroinflammation and promotes further neuronal damage. This dual role in pain modulation and glial activation underscores the importance of P2X4 in chronic pain and neuroinflammatory conditions [236, 237].
P2X7 receptors are minimally expressed in homeostatic microglia, with quantitative analyses indicating low surface expression and transcript levels under normal physiological conditions [238, 239]. However, these receptors are highly expressed in meningeal macrophages, highlighting cell-type-specific expression patterns within the CNS [239]. In the context of inflammation and neurodegeneration, microglial P2X7R expression may become upregulated, suggesting a role in pathological states [238]. Notably, P2X7R has been implicated in synaptic pathology and behavioral deficits. In a mouse model of Rett syndrome, pharmacological inhibition of P2X7R ameliorated dendritic spine abnormalities and improved social behavior, further supporting its involvement in neurodevelopmental disorders [238].
P2X7R plays a crucial role in neuroinflammatory signaling. Activated by high extracellular ATP concentrations (> 100 μM), P2X7R induces cation permeability, particularly for Ca2+, and activates diacylglycerol lipase (DAGL)-mediated inflammatory pathways [240–243]. These receptors are essential for the release of pro-inflammatory cytokines, emphasizing their significance in neuroinflammation.
Electrophysiological studies have provided no evidence supporting the formation of a large pore by P2X7R. The receptor’s single-channel conductance (15–20 pS) remains consistent even during prolonged ATP exposure, indicating that pore dilation is not an intrinsic property of P2X7R. Instead, the observed increase in membrane permeability to large molecules during sustained P2X7R activation may involve alternative mechanisms, such as the recruitment of pannexin-1 channels, the involvement of other ion channels and transporters, or specific intrinsic properties of the P2X7R channel [244–246]. High-resolution single-channel recordings further distinguish the unitary conductance of P2X7R (approximately 15 pS) from other surface channels in whole-cell recordings of rodent astrocytes.
P2Y receptors in microglia
The P2Y12 receptor is a crucial marker of microglial homeostasis. Under physiological conditions, its activation by ATP or ADP enhances microglial motility and structural responsiveness, promoting lamellipodia formation, which is essential for chemotaxis. This enables microglia to migrate toward ATP, signaling cellular distress or injury [115, 228, 247–249]. Impaired migration in P2Y12-deficient microglia underscores its critical role in chemotaxis [250]. P2Y12 signaling engages pathways such as phosphatidylinositol 3-kinase (PI3K)-Akt and PLC, which are integral to ATP/ADP-driven chemotaxis [251, 252]. Additionally, P2Y12 activation supports cellular protrusions by promoting integrin-β1 accumulation at microglial terminals [253]. While pharmacological inhibition or genetic deletion of P2Y12 disrupts injury-induced chemotaxis, basal activity remains unaffected [190, 191]. Interestingly, state-dependent dynamics reveal reduced microglial motility during wakefulness, mediated by β2-adrenergic receptors rather than P2Y12 [254].
Beyond chemotaxis, P2Y12 contributes to pro-inflammatory signaling, including nuclear factor kappa B (NF-κB) activation via potassium efflux and THIK-1 channels, which affects inflammasome activation [255, 256]. It also influences synaptic plasticity in the visual cortex by promoting microglial process arborization and maintaining neuron-microglia structural connections. Notably, ischemic events reduce microglial-neuron interactions upon P2Y12 inhibition [115, 257, 258]. Both P2Y12 and P2Y13 receptors mediate microglial process motility. In their surveying state, microglia extend and retract processes to monitor the CNS environment. Upon neuronal injury, ATP or ADP release activates P2Y12, driving chemotactic responses through PI3K and PLC signaling cascades, calcium influx, and cytoskeletal rearrangements [259–262]. These mechanisms are crucial in neurodegenerative conditions like MS, where microglia migrate to engulf apoptotic neurons and modulate inflammation [121, 263]. The absence of P2Y13 impairs microglial morphology and ramification, highlighting its chemotactic role [259, 264]. P2Y13 also influences hippocampal neurogenesis and reduces pro-inflammatory cytokine expression in neuropathic pain models [265, 266]. Despite these diverse roles, basal microglial surveillance persists in the absence of P2Y12 and P2Y13, suggesting that normal ATP/ADP levels may not fully activate these receptors [259, 267].
P2Y6 receptors are also vital in regulating microglial functions. P2Y6R initiates phagocytosis of apoptotic cells and neuronal debris. This process involves PLC activation, InsP3 production, and calcium release, enhancing debris clearance [268–270]. Excessive ATP release during pathological conditions, such as epilepsy, may desensitize microglia to ATP signals, impairing phagocytosis [268–270]. Furthermore, P2Y6R shifts microglial behavior from migration to phagocytosis based on environmental cues, demonstrating a complementary role to P2Y12 in microglial dynamics [179, 271–273]. Other P2Y receptors, such as P2Y1 and P2Y4, also contribute to microglial activities. P2Y1 activation by ADP supports migration, while P2Y4 facilitates pinocytosis and migration [274, 275].
A1 and A3 adenosine receptors in microglia
A1R is highly expressed in microglia and suppresses ATP-induced morphological changes without regulating cytoskeletal alterations like A2ARs. A1R activation inhibits calcium influx, reducing ATP-induced cell rounding and membrane ruffling [159, 166, 276, 277]. In TBI and EAE models, A1R activation limits microglial activation and neuroinflammation, as evidenced by comparisons between wild-type and A1R knockout mice [278–280].
A3R interacts with P2Y12R to mediate microglial process extension and migration in response to ATP and adenosine. A3R activation also mitigates neuropathic pain by suppressing microglial activation in the spinal cord dorsal horn [75, 281]. ATP released by microglia is degraded into adenosine via ectoenzymes like CD39 and CD73, establishing a feedback loop that recruits microglia to injury sites [282, 283].
In CD39-deficient mice, microglial migration toward ATP is impaired, while phagocytic activity increases independently of ATP stimulation [283, 284]. Combined CD39 and CD73 deficiency reduces microglial network complexity, which can be restored by elevated adenosine levels [283, 285].
In PD models, CD73-derived A2AR signaling modulates microglial morphology and activation. CD73 deficiency attenuates pro-inflammatory responses to LPS while promoting microglial process extension in injury and PD models [75, 286–288]. Understanding A1R and A3R roles offers promising therapeutic opportunities for managing neuroinflammatory processes in neurological disorders.
A2A receptors in microglia and neuroinflammation
Microglia are central to maintaining CNS homeostasis, dynamically monitoring their microenvironment through process extension and retraction [289, 290]. These movements are driven by chemoattractants like ATP and ADP, which activate P2Y12R on microglia [75, 83]. In response to inflammatory stimuli, microglia transition from a ramified, resting state to an amoeboid, activated form. This transition is characterized by process retraction, increased phagocytosis, and the release of bioactive molecules, which play a key role in neuroinflammatory processes [291, 292]. A2AR regulates this transition. Inflammatory conditions upregulate A2AR expression while reducing P2Y12R levels, promoting reactive microglial states [51, 291, 293]. Adenosine, derived from ATP, activates A2AR pathways, modulating microglial behavior and increasing their sensitivity to inflammatory signals [287, 294].
A2ARs have diverse roles in CNS, including enhancing glutamate release, which exacerbates excitotoxicity, and promoting microglial activation [295, 296]. A2AR antagonists have shown neuroprotective effects in preclinical models of neurodegenerative diseases [297, 298]. In PD, they are particularly effective when combined with dopamine D2 receptor (D2R) agonists, enhancing weakened D2R signaling in the striatum caused by dopaminergic neuron loss and reducing neuroinflammation in the extrapyramidal system [288, 299–303].
The activation of A2ARs exacerbates neuroinflammation in various contexts. In perinatal brain injury models, A2AR activation increases pro-inflammatory markers such as IL-1β, IL-6, and TNF-α, effects mitigated by A2AR antagonists [75, 159]. These antagonists also reverse glucocorticoid-induced microglial hyper-ramification and associated anxiety-like behaviors, while reducing cytokine levels following LPS exposure [75].
The role of glial purinergic receptors in neurological disorders: mechanistic insights and therapeutic implications
Purinergic receptors play a central role in the pathophysiology of numerous neurological disorders by regulating processes such as neuronal excitability, neuroinflammation, and glial cell function [24, 54, 75]. These receptors are key mediators in amplifying inflammatory cascades and exacerbating neuronal dysfunction, which contribute to the progression of conditions such as epilepsy, MS, AD, PD, ALS, and traumatic brain and spinal cord injuries (TBI/SCI) [3]. Their critical involvement in these pathologies highlights their therapeutic potential, with recent advances in targeting purinergic signaling offering innovative strategies for improving clinical outcomes (Figure 6).
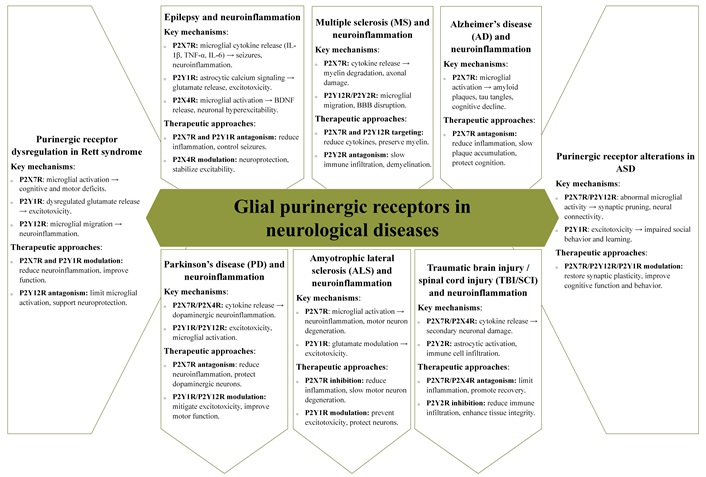
The role of glial purinergic receptors in disease progression and their potential as therapeutic targets in neurological disorders. IL-1β: interleukin-1β; TNF-α: tumor necrosis factor-alpha; IL-6: interleukin-6; BDNF: brain-derived neurotrophic factor; BBB: blood-brain barrier; ASD: autism spectrum disorder
The role of glial purinergic receptors in epilepsy: mechanisms of neuroinflammation and therapeutic opportunities
In epilepsy, glial purinergic receptors play pivotal roles in modulating neuronal excitability and neuroinflammation—two interconnected processes critical to seizure initiation and propagation [226]. The P2X7R, predominantly expressed on microglia, serves as a key driver of neuroinflammatory responses. Its activation induces the release of pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-6 via inflammasome activation, exacerbating neuronal excitability and contributing to increased seizure frequency and severity [304–306]. Furthermore, P2X7R activation facilitates ATP release from glial cells, amplifying purinergic signaling and sustaining a pro-inflammatory microenvironment that worsens seizure pathology [240, 307]. Targeted antagonism of P2X7R has shown significant promise in preclinical models by reducing neuroinflammation, dampening seizure severity, and protecting against neuronal loss.
The P2Y1R also plays a critical role in epilepsy pathophysiology. P2Y1R activation regulates astrocytic calcium signaling, which influences glutamate release into the synaptic cleft. Excessive glutamate release, driven by P2Y1R, can lead to excitotoxicity, a process that damages or kills neurons and contributes to epilepsy progression [15, 211, 308]. Therapeutic strategies targeting P2Y1R have demonstrated potential in mitigating excitotoxic damage by modulating glutamate dynamics [211, 309], thereby providing neuroprotection and enhancing seizure control.
Similarly, the P2X4R, primarily localized to microglia, is implicated in neuroinflammatory processes associated with epilepsy. Activation of P2X4R triggers the release of BDNF [235, 310], which exacerbates hyperexcitability in neuronal circuits. Targeting P2X4R may attenuate neuroinflammation and reduce seizure susceptibility [10, 311], offering a dual approach to improving seizure control and limiting neuronal damage.
These findings underscore the critical roles of glial purinergic receptors in epilepsy. Their involvement in maintaining the delicate balance between neuroinflammation and excitability not only drives disease progression but also highlights their potential as therapeutic targets. Advances in purinergic receptor modulation offer promising avenues for developing novel, glial-focused interventions aimed at reducing seizure burden and preserving neuronal integrity in epilepsy.
Glial purinergic receptors in MS: mediators of neuroinflammation and therapeutic targets
In MS, glial purinergic receptors, including P2X7R, P2Y1R, P2Y12R, and P2Y2R, play crucial roles in modulating neuroinflammatory responses and driving disease progression [85, 179, 312]. These receptors regulate the activation of glial cells, particularly microglia and astrocytes, which are central to the inflammatory processes underlying demyelination and neurodegeneration [129, 227].
The P2X7R, predominantly expressed on microglia, serves as a key mediator of pro-inflammatory signaling in MS [59, 85]. Activation of P2X7R triggers the release of cytokines such as IL-1β, TNF-α, and IL-6 via inflammasome pathways, exacerbating the inflammatory cascade. This heightened inflammatory response contributes to myelin sheath degradation and axonal damage, hallmark features of MS pathology [85, 313, 314]. Moreover, sustained P2X7R activation promotes ATP release, creating a feedback loop that perpetuates glial activation and inflammation. Preclinical studies suggest that P2X7R inhibition can significantly reduce pro-inflammatory cytokine production, attenuate microglial activation, and mitigate neuroinflammation, thereby offering neuroprotection and reducing myelin loss [59, 202].
MS is primarily driven by the infiltration and activation of autoreactive lymphocytes, particularly CD4+ T cells of the Th1 and Th17 subsets, which secrete pro-inflammatory cytokines such as interferon-gamma (IFN-γ) and IL-17, contributing to CNS tissue damage and immune tolerance breakdown [315, 316]. Purinergic signaling plays a pivotal role in regulating T cell activity during MS pathogenesis, where ATP released from damaged and activated immune cells acts as a DAMP by binding to P2X and P2Y receptors on CD4+ T cells, modulating their activation, differentiation, and cytokine secretion [76]. Activation of P2X7R on Th1 and Th17 cells further enhances IL-1β and IL-18 release, amplifying neuroinflammation and lesion progression [57]. Beyond direct T cell modulation, purinergic signaling influences immune cell infiltration and BBB integrity in MS, with adenosine signaling through A2A receptors inhibiting T cell adhesion and reducing cytokine production, while ATP-P2X7 signaling promotes BBB disruption and T cell trafficking into the CNS [58]. EAE, a widely used MS model, has demonstrated that pharmacological blockade of P2X7R reduces both CD4+ T cell infiltration and disease severity, highlighting the therapeutic potential of targeting purinergic pathways in MS [32].
The P2Y12R, predominantly expressed on resting microglia, plays a dual role in MS pathophysiology. Under physiological conditions, P2Y12R facilitates microglial surveillance [317]; however, during MS, its activation promotes microglial migration and aggregation at injury sites. This response amplifies inflammatory signaling and contributes to tissue damage [318].
Similarly, the P2Y2R is implicated in astrocytic activation and immune cell recruitment, further aggravating the inflammatory environment in MS. P2Y2R activation induces the release of chemokines and adhesion molecules, facilitating leukocyte infiltration into the CNS and exacerbating demyelination [318–320]. Targeting P2Y2R may help reduce astrocytic-driven inflammation and immune cell infiltration, thereby slowing disease progression.
The P2Y1R is another critical player in MS pathology, primarily through its role in modulating astrocytic calcium signaling. P2Y1R activation influences the release of pro-inflammatory mediators, exacerbating neuronal excitotoxicity [15, 184, 211]. Inhibiting P2Y1R activity has the potential to attenuate astrocyte-mediated neuroinflammation and protect neurons from further damage.
These findings underscore the complex roles of glial purinergic receptors in MS. Their involvement in regulating glial activation, neuroinflammation, and tissue damage highlights their potential as therapeutic targets. Advances in receptor-specific modulation offer promising strategies to mitigate inflammation, slow disease progression, and preserve neurological function in MS patients.
Role of P2X7 receptor-mediated neuroinflammation in AD pathogenesis and therapeutic targeting
In AD, purinergic signaling, particularly through the P2X7R, plays a critical role in the neuroinflammatory processes that contribute significantly to neuronal death and cognitive decline. P2X7R, predominantly expressed on microglia, is activated by extracellular ATP, which is released during cellular stress or injury [66, 202, 321, 322]. Upon activation, P2X7R triggers a cascade of pro-inflammatory cytokine release, primarily through inflammasome-mediated pathways. This cascade exacerbates neuroinflammation, a key pathological feature of AD, and accelerates disease progression by amplifying the inflammatory microenvironment surrounding neurons [189, 240].
Chronic activation of P2X7R contributes to a self-perpetuating cycle of neuroinflammation, characterized by persistent microglial activation. This, in turn, leads to further cytokine production, oxidative stress, and the recruitment of peripheral immune cells into the brain [3, 52, 197, 209]. These processes drive synaptic dysfunction, a critical early event in AD pathology, and neuronal loss, ultimately resulting in the cognitive deficits characteristic of the disease. The neuroinflammatory response triggered by P2X7R activation also contributes to the formation of amyloid plaques and tau tangles, the hallmark proteinopathies of AD. The inflammation surrounding these plaques exacerbates their toxic effects on neurons [3, 52], further accelerating disease progression.
Recent studies have highlighted the potential of targeting P2X7R as a therapeutic strategy to mitigate neuroinflammation and slow the progression of AD. Inhibition of P2X7R has shown promise in preclinical models by reducing the release of pro-inflammatory cytokines, dampening microglial activation, and protecting synaptic integrity [307, 323, 324]. Modulating this receptor may help prevent the amplification of neuroinflammatory pathways that contribute to neuronal damage. Additionally, P2X7R antagonism could help preserve cognitive function by reducing synaptic loss and protecting neurons from excitotoxicity induced by excessive glutamate release.
Purinergic receptors in PD: mechanisms of neuroinflammation and therapeutic implications
In PD, purinergic receptors such as P2X7R, P2X4R, P2Y1R, and P2Y12R play essential roles in the neuroinflammatory processes that drive dopaminergic neuron degeneration, a hallmark of the disease [24, 179, 266]. Activation of P2X7R and P2X4R, both predominantly expressed on microglia, triggers the release of pro-inflammatory cytokines, including IL-1β, TNF-α, and IL-6. These cytokines perpetuate a chronic neuroinflammatory environment that exacerbates neuronal damage, especially in dopaminergic pathways [325]. This microglial activation is a critical contributor to the progressive loss of dopaminergic neurons, leading to motor dysfunction and other PD-related symptoms [52, 326]. Additionally, P2Y1R activation is implicated in the exacerbation of glutamate-induced excitotoxicity, a process that significantly damages neurons and contributes to the ongoing neurodegeneration in PD [309, 327].
P2Y12R, another key receptor expressed on microglia, modulates glial cell responses and amplifies the inflammatory cascade, thereby promoting sustained neuroinflammation and neuronal loss [83, 227]. This receptor’s role in regulating microglial activation and migration further highlights its contribution to the chronic inflammatory environment that characterizes PD pathophysiology.
Given the central roles of these purinergic receptors in promoting neuroinflammation and neuronal damage, targeting them presents a multifaceted approach for mitigating disease progression. Antagonism of P2X7R has shown promise in preclinical models by reducing microglial activation and pro-inflammatory cytokine release, potentially preserving dopaminergic neurons and improving motor function. Similarly, modulating P2Y1R and P2Y12R activity could help limit excitotoxicity and attenuate neurodegeneration, further contributing to the protection of dopaminergic neurons [224, 328, 329]. These therapeutic strategies hold significant potential for slowing the progression of PD and improving patient outcomes by addressing the underlying neuroinflammatory mechanisms that drive disease pathogenesis.
Purinergic receptors in ALS: modulation of P2X7R and P2Y1R as therapeutic strategies to mitigate neuroinflammation and excitotoxicity
In ALS, purinergic signaling, particularly through the P2X7R, plays a pivotal role in driving microglial activation and neuroinflammation, both of which are key contributors to the progressive degeneration of motor neurons [66, 240, 330]. The activation of P2X7R on microglia leads to the release of a variety of pro-inflammatory cytokines, which further amplify the inflammatory milieu in the CNS. This enhanced neuroinflammatory environment exacerbates motor neuron damage, accelerating disease progression [331]. Additionally, P2Y1R contributes to excitotoxicity by modulating glutamate signaling, thereby intensifying neuronal injury [332].
Recent preclinical studies have underscored the potential therapeutic benefits of targeting P2X7R, with antagonists showing promise in inhibiting microglial activation, reducing neuroinflammation [333, 334], and slowing the rate of motor neuron degeneration [335]. Inhibition of P2Y1R, on the other hand, could help reduce glutamate-mediated excitotoxicity, offering further protection against neuronal loss [184, 309]. Together, these strategies provide a multifaceted approach to managing ALS, highlighting the potential of purinergic receptor modulation as a means to delay disease onset, slow progression, and ultimately preserve motor neuron function. These findings suggest that targeting glial purinergic receptors could become a valuable therapeutic strategy in the fight against ALS.
Purinergic receptors in TBI/SCI: pathophysiological roles and therapeutic potential
Following TBI/SCI, purinergic signaling plays a crucial role in the initiation and propagation of neuroinflammatory cascades that exacerbate secondary neuronal damage and tissue degeneration [52, 336–338]. In these contexts, the activation of purinergic receptors, particularly P2X7R, P2X4R, and P2Y2R, is central to the neuroinflammatory response that not only contributes to the immediate tissue injury but also impedes the long-term recovery process [56, 339, 340].
Activation of the P2X7R triggers the release of a wide array of pro-inflammatory cytokines from microglia and astrocytes. This cytokine release amplifies the neuroinflammatory milieu, further exacerbating neuronal injury and impairing the ability of the CNS to effectively repair itself [179, 208]. The inflammatory environment that emerges from P2X7R activation also plays a significant role in the disruption of the BBB and the recruitment of peripheral immune cells [59, 240, 341], thereby prolonging inflammation and increasing the extent of secondary damage.
P2X4R and P2Y2R also contribute to glial cell activation and modulate neuroinflammatory responses. P2X4R is involved in the regulation of microglial activity, where its activation leads to the release of additional pro-inflammatory mediators, such as reactive oxygen species (ROS) and cytokines [75, 232, 342]. P2Y2R, on the other hand, has been implicated in the regulation of astrocytic response to injury and modulating the release of glutamate [150, 343], further intensifying excitotoxicity and exacerbating neuronal death. These purinergic receptors collectively hinder the resolution of inflammation and complicate tissue repair processes, contributing to the progression of both brain and spinal cord injuries.
Targeting these glial purinergic receptors offers a promising strategy to mitigate the harmful effects of neuroinflammation and excitotoxicity following TBI/SCI. In particular, antagonism of the P2X7 receptor has shown potential in preclinical models by reducing microglial activation, cytokine production, and neuronal damage, thereby limiting the extent of secondary injury [240, 313, 344]. Additionally, modulating P2X4R and P2Y2R activity could help in fine-tuning the inflammatory response [14, 342, 345], facilitating glial cell reactivity and promoting tissue repair mechanisms.
Therapeutic approaches that focus on modulating purinergic signaling pathways hold the potential to enhance functional recovery by reducing neuroinflammation, promoting neuroprotection, and supporting tissue repair. By targeting purinergic receptors, it may be possible to mitigate the deleterious effects of inflammation while simultaneously enhancing the endogenous repair mechanisms that are critical for long-term recovery and functional outcomes following traumatic injury to the brain or spinal cord. These strategies, still under investigation, may offer novel therapeutic avenues for improving both the short- and long-term prognosis of TBI/SCI patients.
P2X receptor dysregulation in Rett syndrome and autism spectrum disorder
Emerging evidence increasingly implicates P2X receptors, particularly P2X7 and P2X4, in the regulation of neuroinflammatory processes associated with Rett syndrome and autism spectrum disorder (ASD) [346–349]. P2X receptors mediating Ca2+ and Na⁺ influx to regulate neuroimmune signaling, including NLRP3 inflammasome activation, cytokine release, and cell survival pathways [350, 351].
In Rett syndrome, mutations in the methyl-CpG-binding protein 2 (MECP2) gene impair synaptic plasticity and drive chronic neuroinflammation, a key pathological feature of the disorder [352]. P2X7 receptor activation in astrocytes and microglia has been implicated in excessive pro-inflammatory cytokine release, contributing to synaptic dysfunction and neuronal injury. Astrocytic P2X7 activation disrupts synaptic homeostasis by promoting excessive gliotransmitter release (e.g., glutamate and ATP), heightening excitotoxicity and neuronal stress [66, 85]. Concurrently, microglial P2X7 activation amplifies ATP-driven calcium influx, oxidative stress, and a self-sustaining inflammatory feedback loop. This chronic microglial reactivity, characterized by elevated pro-inflammatory cytokines and ROS, exacerbates neurodegeneration, synaptic instability, and phenotypic severity in Rett syndrome [179, 238]. Moreover, P2X7-mediated calcium dysregulation can disrupt neurodevelopmental processes, including dendritic growth and synapse formation, further compounding the disorder’s neurological deficits [238, 353].
Similarly, ASD has been increasingly linked to altered purinergic signaling through the dysregulation of P2X7 and P2X4 receptors [347, 354]. Elevated extracellular ATP levels and overactivation of microglial P2X7 receptors have been observed in ASD models, correlating with increased expression of pro-inflammatory cytokines such as IL-6 and IL-1β. This neuroinflammatory state can impair synaptic pruning and neurogenesis, critical processes during early brain development [355]. Microglial cells exhibit exaggerated activation due to P2X7 stimulation, contributing to aberrant microglial engulfment of synaptic elements and a failure to appropriately regulate neuronal connectivity [86, 304]. Astrocytes, similarly, become reactive in ASD models, leading to excessive ATP release and disruption of normal neuronal-glial signaling pathways [356].
P2X4 receptor involvement in microglial motility and synaptic plasticity has also been implicated in ASD, with aberrant activity contributing to altered neuronal circuit formation and behavioral abnormalities, such as deficits in social interaction and sensory processing [310]. Notably, microglial P2X4 activation has been associated with impaired phagocytic clearance of apoptotic cells and debris, further compromising the neurodevelopmental environment [357].
Therapeutically, targeting the P2X7 receptor has emerged as a promising strategy for mitigating neuroinflammation and its pathological consequences in both Rett syndrome and ASD. Pharmacological inhibitors of P2X7, such as Brilliant Blue G and selective small-molecule antagonists, have shown potential in reducing microglial activation and pro-inflammatory cytokine release in preclinical models [358]. Additionally, genetic approaches, such as conditional knockout models, have demonstrated reduced neuroinflammation and improved synaptic function [222], highlighting the receptor’s role as a viable therapeutic target. Targeting astrocyte activation through P2X7 inhibition may also restore synaptic homeostasis and limit glutamate excitotoxicity in both disorders [359].
Exploring purinergic signaling in glial cells as a therapeutic target for CNS disorders
Glial cells exhibit significant involvement in pathological states [360, 361]. Purinergic signaling emerges as a key regulator of neuroinflammation, neurodegeneration, and synaptic dysfunction, especially under conditions of stress, injury, or inflammation [3, 6]. This positions purinergic receptors as promising therapeutic targets for CNS disorders (Table 4).
Glial purinergic receptors: roles in CNS function, and therapeutic potential in neurological disorders
| Receptor type | Role in CNS | Therapeutic potential | References |
|---|---|---|---|
| P2X1 | Regulates cerebral blood flow, neurovascular coupling, and blood-brain barrier function | Modulation could improve ischemic stroke outcomes by enhancing blood flow and neurovascular responses | [362–364] |
| P2X4 | Involved in neuropathic pain signaling, synaptic plasticity, microglial activation, and cytokine release | Antagonists are being studied for neuropathic pain and chronic inflammatory conditions | [180, 181, 365] |
| P2X5 | Modulates glial cell proliferation and inflammatory responses | Targeting P2X5 receptors could regulate gliosis, neuroinflammation, and neurodegeneration | [366–368] |
| P2X7 | Drives inflammasome activation, cytokine release, and neuroinflammation. Involved in Alzheimer’s, Parkinson’s, and multiple sclerosis pathophysiology | Antagonists (e.g., BBG) reduce neuroinflammation and neurodegeneration in various CNS diseases | [66, 240, 313, 326, 369, 370] |
| P2Y1 | Modulates microglial activation, neuronal excitability, and blood-brain barrier integrity | Targeting P2Y1 receptors could aid in conditions like multiple sclerosis and traumatic brain injury | [104, 119, 371] |
| P2Y6 | Facilitates phagocytosis, aiding in the clearance of cellular debris and neurotoxic aggregates | Enhancing P2Y6 activity could help clear amyloid plaques in Alzheimer’s and alpha-synuclein in Parkinson’s | [272, 372–374] |
| P2Y12 | Essential for microglial chemotaxis and inflammatory response | Targeting P2Y12 receptors may improve recovery and reduce damage in stroke, Alzheimer’s, and other CNS injuries | [115, 375–377] |
| P2Y13 | Modulates microglial migration, activation, synaptic plasticity, and neuroinflammation | Antagonists may regulate glial activation in neurodegenerative diseases and acute CNS injuries | [264, 267, 378] |
| A1 adenosine | Reduces excitotoxicity, regulates neurotransmitter release, and provides neuroprotection, particularly during ischemic events | A1 receptor agonists are potential treatments for ischemia, epilepsy, and neurodegenerative diseases | [379–382] |
| A2A adenosine | Modulates neuroinflammation, synaptic plasticity, and myelination. Suppresses pro-inflammatory cytokine release | A2A antagonists could reduce neuroinflammation and improve dopaminergic signaling in Parkinson’s disease | [194, 302, 382, 383] |
BBG: Brilliant Blue G; CNS: central nervous system
Astrocytes play a protective role by releasing ATP, which is converted to adenosine through ectoenzymes such as CD39 and CD73. Adenosine, acting via A1 adenosine receptors, reduces excitatory neurotransmission, protecting neurons from excitotoxicity [173, 384, 385]. Enhancing ectoenzyme activity represents a potential strategy for conditions like epilepsy, characterized by neuronal hyperexcitability [386].
ATP interacts with microglial receptors, such as P2X7 and P2Y12, to drive neuroinflammation by promoting the release of pro-inflammatory cytokines. This pathological cascade exacerbates neuronal damage in conditions like AD, MS, and stroke [52, 179, 202]. Modulating P2Y12 receptors has been proposed as a strategy to prevent secondary neuronal damage following stroke [387, 388]. Additionally, enhancing astrocytic adenosine production can reduce neuronal excitability and suppress seizures [389, 390], while inhibiting P2X7 receptor activity may alleviate inflammation and protect against demyelination in MS [59, 391]. Therapeutic strategies targeting ATP release, including inhibition of Pannexin-1 channels or modulation of microglial purinergic receptors, have shown promise in reducing immune activation and preserving neuronal integrity [392]. Furthermore, purinergic signaling presents promising therapeutic opportunities for neuropathic pain, with targeting P2X4 receptors on microglia demonstrating potential in alleviating pain hypersensitivity in neuropathic conditions [180, 181, 393].
In chronic neurodegenerative disorders, sustained P2X7 receptor activation drives inflammasome assembly and perpetuates inflammation [370, 394]. Selective P2X7 antagonists have demonstrated efficacy in reducing neuroinflammation and neuronal damage in preclinical models [326, 395]. Similarly, modulating A2A receptors may attenuate inflammation in Alzheimer’s and enhance dopaminergic signaling in PD [298, 396].
Future therapeutic strategies should prioritize the development of receptor-specific agonists and antagonists to precisely modulate purinergic signaling pathways. This approach could allow for targeted interventions that either enhance or inhibit purinergic signaling in a controlled manner, improving clinical outcomes in a variety of neurological conditions. Furthermore, enhancing ectoenzyme activity to regulate adenosine levels offers another promising avenue, as it could fine-tune the balance between purine nucleotides and their receptors, ultimately influencing neuroinflammatory processes, synaptic plasticity, and neuronal excitability. Exploring receptor crosstalk within the quadripartite synapse, which involves interactions between neurons, astrocytes, and microglia, could uncover novel mechanisms of cellular communication and identify new targets for intervention.
Additionally, targeting cell-type and region-specific purinergic responses may further optimize therapeutic precision, minimizing unintended effects on non-target cells and enhancing the efficacy of treatment. Advances in single-cell genomics, optogenetics, and spatial transcriptomics could be particularly valuable in dissecting the heterogeneity of purinergic receptor expression across different brain regions and cell types. However, significant challenges remain in achieving tissue specificity and minimizing off-target effects, which continue to pose obstacles to the development of safe and effective therapies. To address these challenges, extensive preclinical validation using animal models, as well as robust translational studies, will be essential for ensuring the safety and efficacy of novel purinergic-based therapies in treating CNS disorders. This multifaceted approach holds the potential to not only advance our understanding of purinergic signaling but also to lead to breakthroughs in the treatment of neurological diseases.
Conclusions
Purinergic receptor signaling plays a pivotal role in regulating cellular functions across physiological and pathological states, particularly within the CNS. Its involvement in neuroinflammation, neurodegeneration, and synaptic communication highlights its therapeutic potential for addressing a range of neurological and cardiovascular diseases. The dynamic interplay between ATP and adenosine, mediated by P1 and P2 receptors, is crucial for maintaining neural network functionality and homeostasis through the modulation of neuronal and glial interactions.
Emerging insights into purinergic pathways emphasize the delicate balance between pro-inflammatory and anti-inflammatory signaling. Distinct receptor subtypes, including P2Y, P2X, and adenosine receptors, orchestrate complex microglial and astrocytic responses that influence disease progression. Targeting these pathways offers promising avenues for therapeutic intervention, such as modulating key receptors like P2X7 or regulating ectoenzymes CD39 and CD73 to balance ATP and adenosine levels. These strategies could enable tailored approaches for managing chronic neuroinflammatory conditions, including epilepsy, MS, and neurodegenerative diseases.
Despite these advances, significant research limitations persist. Many studies rely heavily on in vitro models or rodent systems, which may not fully capture the intricacies of human purinergic signaling. The heterogeneity of purinergic receptor subtypes and their context-dependent roles complicate the identification of universally effective therapeutic targets. Additionally, the off-target effects and systemic consequences of modulating purinergic signaling remain poorly understood, limiting the translational potential of current findings.
Future research must focus on addressing these challenges by employing more sophisticated models, such as human organoids or advanced in vivo systems, to better replicate the complexity of CNS environments. Longitudinal studies exploring receptor dynamics in chronic disease states are crucial for understanding the temporal and spatial specificity of purinergic signaling. Furthermore, the development of highly selective modulators with minimal side effects will be critical to advancing therapeutic applications.
Critically, while literature has illuminated key aspects of purinergic signaling, gaps remain in understanding the interplay between purinergic receptors and other signaling pathways. A more integrative approach, combining molecular, genetic, and systems-level studies, is essential for uncovering novel regulatory mechanisms. By addressing these limitations and pursuing innovative directions, the field holds the potential to transform our understanding of CNS pathophysiology and develop effective treatments that enhance neuroprotection, restore synaptic integrity, and improve clinical outcomes in a wide array of neurological disorders.
Abbreviations
| AD: | Alzheimer’s disease |
| ALS: | amyotrophic lateral sclerosis |
| APCs: | antigen-presenting cells |
| ASD: | autism spectrum disorder |
| BBB: | blood-brain barrier |
| BDNF: | brain-derived neurotrophic factor |
| CD39: | nucleoside triphosphate diphosphohydrolase-1 |
| CD73: | ecto-5’-nucleotidase |
| CNS: | central nervous system |
| D2R: | dopamine D2 receptor |
| DAMP: | damage-associated molecular pattern |
| EAE: | experimental autoimmune encephalomyelitis |
| GPCRs: | G protein-coupled receptors |
| IL-18: | interleukin-18 |
| IL-1β: | interleukin-1β |
| IL-6: | interleukin-6 |
| LPS: | lipopolysaccharide |
| LTP: | long-term potentiation |
| MAPK: | mitogen-activated protein kinase |
| MS: | multiple sclerosis |
| NAD+: | nicotinamide adenine dinucleotide |
| NLRP3: | NOD-like receptor protein 3 |
| PD: | Parkinson’s disease |
| PI3K: | phosphatidylinositol 3-kinase |
| PLC: | phospholipase C |
| ROS: | reactive oxygen species |
| SCI: | spinal cord injury |
| TBI/SCI: | traumatic brain and spinal cord injuries |
| TBI: | traumatic brain injury |
| Th1: | T helper 1 |
| TNF-α: | tumor necrosis factor-alpha |
Declarations
Author contributions
MMN: Conceptualization, Writing—original draft, Writing—review & editing.
Conflicts of interest
The author declares that there are no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2025.
Publisher’s note
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.