
Abstract
The SAR-CoV-2 virus has evolved to co-exist with human hosts, albeit at a substantial energetic cost resulting in post-infection neurological manifestations [Neuro-post-acute sequelae of SARS-CoV-2 infection (PASC)] that significantly impact public health and economic productivity on a global scale. One of the main molecular mechanisms responsible for the development of Neuro-PASC, in individuals of all ages, is the formation and inadequate proteolysis/clearance of phase-separated amyloid crystalline aggregates—a hallmark feature of aging-related neurodegenerative disorders. Amyloidogenesis during viral infection and persistence is a natural, inevitable, protective defense response that is exacerbated by SARS-CoV-2. Acting as chemical catalyst, SARS-CoV-2 accelerates hydrophobic collapse and the heterogeneous nucleation of amorphous amyloids into stable β-sheet aggregates. The clearance of amyloid aggregates is most effective during slow wave sleep, when high levels of adenosine triphosphate (ATP)—a biphasic modulator of biomolecular condensates—and melatonin are available to solubilize amyloid aggregates for removal. The dysregulation of mitochondrial dynamics by SARS-CoV-2, in particular fusion and fission homeostasis, impairs the proper formation of distinct mitochondrial subpopulations that can remedy challenges created by the diversion of substrates away from oxidative phosphorylation towards glycolysis to support viral replication and maintenance. The subsequent reduction of ATP and inhibition of melatonin synthesis during slow wave sleep results in incomplete brain clearance of amyloid aggregates, leading to the development of neurological manifestations commonly associated with age-related neurodegenerative disorders. Exogenous melatonin not only prevents mitochondrial dysfunction but also elevates ATP production, effectively augmenting the solubilizing effect of the adenosine moiety to ensure the timely, optimal disaggregation and clearance of pathogenic amyloid aggregates in the prevention and attenuation of Neuro-PASC.
Keywords
Adenosine moiety effect, adenosine triphosphate (ATP), mitochondrial dynamics, glymphatic system, neurodegenerative disorders, protein hydration, slow wave sleep, viral persistenceIntroduction
Neuro-PASC: symptoms, challenges, and solutions for viral persistence in the brain
The classification of Neuro-post-acute sequelae of SARS-CoV-2 infection (PASC) describes a wide range of neurological manifestations observed in individuals with PASC, or “long COVID” [1]. Cognitive impairment including brain fog and frontal network dysfunctions involving deficits in attention, memory, and executive function, can persist beyond one year in recovered individuals regardless of infection severity or hospitalization status [2–11]. Optical clearing and imaging studies observed the persistent presence and accumulation of the SARS-CoV-2 spike (S) and nucleocapsid (N) proteins at the skull-meninges-brain axis of postmortem skulls from patients who died from COVID-19. Whereas the S protein from SARS-CoV-2 was detected in 10 out of 34 postmortem skull samples obtained from patients who did not die of COVID-19 causes. The fact that skull marrow cells, meningeal cells, and brain tissue cells all tested positive for the S protein at rates of 2.3%, 2.8%, and 1%, respectively, implies that viral persistence is maintained in many individuals long after the clearance and resolution of infection. Similarly, healthy mice aerosol-inoculated with SARS-CoV-2 (Omicron B, XBB sub-lineage) virus exhibited clear persistence of the S protein in the head at 28 days post-infection after full recovery from acute symptoms and viral clearance from the skull marrow and brain cortex [12].
A prospective longitudinal cohort study of 475 adults (mean age 58.26 years) hospitalized with a clinical diagnosis of COVID-19 in the UK reported that participants experienced not only the emergence of new psychiatric and cognitive symptoms over the first 2–3 years but also the continued deterioration of existing symptoms present at 6 months post-hospitalization [13]. In 2023, an ambispective cohort study evaluated post-hospitalization Neuro-PASC (PNP, n = 50) and non-hospitalized Neuro-PASC (NNP, n = 50) patients in Medellin, Columbia diagnosed with COVID-19 between 2020 and 2023. The analysis of the data collected revealed that 64% of PNP, compared to 42% of NNP presented abnormal neurological symptoms that included brain fog (60%) and myalgia (42%). While numbness/tingling and abnormal body sensations were more prevalent in PNP (58% and 42%, respectively) than in NNP (24% and 12%, respectively). Conversely, brain fog was more common in NNP (64%) than PNP (56%). Both groups presented identical levels of fatigue (74%) and sleep dysregulation (46%) during examinations [14]. In November 2024, a large cross-sectional study involving 200 PNP and 1100 NNP patients reported that younger (18–44 years) and middle-aged (45–64 years) subjects, regardless of infection severity, were disproportionately affected by neurological symptoms compared to older (65+ years) subjects [15].
The potential mechanisms responsible for neurological and even structural alterations of the brain by the SARS-CoV-2 virus [16–21], remain to be fully elucidated. Nonetheless, an exhaustive list of reviews, hypotheses, and experimental work examining mechanisms including, but not limited to, neuroinflammation [22–25], mitochondrial quality control [26, 27], mitochondrial DNA content [28, 29], and endoplasmic reticulum stress [30] have been presented as causes responsible for the activation of Neuro-PASC [31–37]. Overwhelming evidence supports the existence of a strong, positive correlation between SARS-CoV-2 infection and neurological dysfunction [38–43]. A systematic analysis of resting-state electroencephalogram (rsEEG) in Long COVID patients revealed striking abnormalities resembling those exhibited in early developmental stages of neurodegenerative diseases (NDDs) including Alzheimer’s disease (AD) and related dementia (ADRD). The shift to lower frequencies in the rsEEG (reduced alpha rhythm, increased slow waves) and epileptiform-like EEG signals imply potential overlapping pathophysiology in both Neuro-PASC and ADRD [44]. Although a systematic review comprising 18 studies involving 412957 COVID-19 patients aged ≥ 65 years reported the identification of new-onset cognitive impairment in 64% of the patients [45], viral infection-induced cognitive impairment may not be unique to the SARS-CoV-2 virus, as recent advances link viral amyloidogenesis to neurodegeneration [46].
A dominant association exists between viral infections and NDDs
Many viral infections are associated with elevated risks in the development of NDD. A thorough data analysis of cohorts (> 800000) in the FinnGen and the UK Biobank was conducted to identify longitudinal and cross-sectional associations between viral exposures and common NDDs including AD, amyotrophic lateral sclerosis (ALS), dementia, Parkinson’s disease (PD), and multiple sclerosis (MS). The study reported 45 viruses, including viral encephalitis, viral hepatitis, influenza, pneumonia, herpes simplex, and varicella-zoster virus are associated with an elevated risk of NDD up to 15 years post-infection [47]. An analysis of transcriptomic and interactomic profiles of brain samples from the Gene Expression Omnibus repository of AD patients who died of COVID-19, AD without COVID-19, COVID-19 without AD, and control individuals found 21 interactome key nodes are dysregulated in AD patients who died of COVID-19, with the implication that SARS-CoV-2 infection exacerbated the AD condition due to the presence of higher levels of amyloid-beta (Aβ), inflammation, and oxidative stress [48]. Whether the SARS-CoV-2 virus features characteristics distinct from other viruses and exacerbates NDD is unclear and requires elucidation.
The development of Neuro-PASC is not specific to age or infection severity
Although age-related hippocampal atrophy and neuroinflammation can augment cognitive impairment [49], the development of Neuro-PASC is observed across all ages and health groups. A systematic review and meta-analysis of 11 studies involving > 6.8 million controls and 0.94 million infected subjects reported a higher risk in the development of new-onset dementia in adults aged > 60 years at different intervals post COVID-19 diagnosis [50]. Whereas, a longitudinal multimodal MRI study in Lombardy Italy, investigating the neural activities in mildly-infected, healthy young adults (mean age 24 years) 12 months post-infection, discovered persistent structural and functional changes in specific brain areas resulting in deficits in olfaction, visual/verbal domain of short-term memory, and reduction in spatial working memory compared to uninfected controls [51]. Imaging evidence from ultra-high field 7 T MRI of COVID-19 survivors (median time of 6.5 months post-hospitalization), showed magnetic resonance susceptibility abnormalities at multiple regions of the brainstem, including the medulla oblongata, pons, and midbrain [52].
A registered clinical trial (NCT04865237), conducted in the United Kingdom between March 2021 and July 2022, examined the changes in cognitive functions in 34 healthy, seronegative young adults aged 18–30 years, inoculated with Wildtype SARS-CoV-2. The study reported that subjects with mild infections exhibited statistically lower cognitive scores that persist for one year or longer, compared to uninfected test subjects [53]. SARS-CoV-2 intranasally inoculated ferrets demonstrated subclinical to mild respiratory disease. Even though there is no evidence of brain infection detected via in situ hybridization or immunohistochemistry, the presence of viral RNA in the brain, 7 days post infection, induced neurovirulence associated with microglial activation, astrocytic deactivation, and Alzheimer type II astrocytes [54]. Additionally, the discovery of substantial abnormal neuroimaging findings in a significant number of young children with neurological symptoms post COVID-19 infection [55] all point to the potential involvement of multifaceted, universal molecular mechanisms associated with phase separation and amyloidogenesis in the development of Neuro-PASC that may affect infected/exposed subjects of all ages and health conditions.
Melatonin as a cost-effective, time-tested solution for the potential treatment of Neuro-PASC
Melatonin (N-acetyl-5-methoxytryptamine) is an evolutionarily conserved molecule [56] that can effectively counter the development of Neuro-PASC by rebalancing the complex energetic scales of amyloidogenesis promoted by the SARS-CoV-2 virus. The impressive, extensive array of functions and features associated with melatonin since its isolation and identification in bovine pineal tissues in 1958 [57] provide convincing rationale for the ubiquitous presence of this ancient molecule, synthesized in all tested eukarya, bacteria, and archaea [58–60]. Nevertheless, proposals presenting melatonin as an effective regulator of phase separation under various contexts including neurodegenerative disorders [61], cancer multidrug resistance [62], viral phase separation [63], dementia [64], cataractogenesis [65], and mitochondrial function [66], offer a novel perspective in the investigation of the underexplored, expansive functions of the indoleamine for more than 2 billion years in Archaea and Bacteria prior to the advent of Eukarya [66].
The first report of intracellular amyloids detected in the domain of Archaea stratifies an additional layer of significance in the synergy between melatonin and adenosine triphosphate (ATP) in the regulation of phase separation and amyloidogenesis that began close to 4 billion years ago (Ga). This review will apply current knowledge to explore potential molecular mechanisms employed by melatonin in synergy with ATP for the regulation of phase separation that can modulate pathological amyloidogenesis promoted by SARS-CoV-2 in Neuro-PASC (Figure 1).
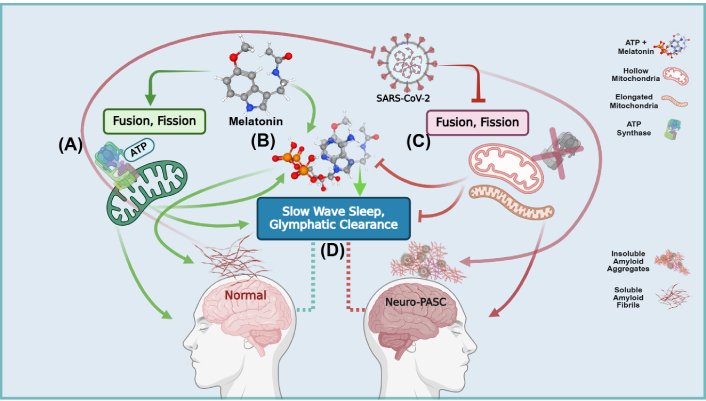
Schematic overview of the synergistic regulation of amyloid solubilization and clearance during slow wave sleep in Neuro-post-acute sequelae of SARS-CoV-2 infection (PASC). (A) During viral infection/persistence, exogenous melatonin enhances the production of adenosine triphosphate (ATP) and synthesis of mitochondrial melatonin, preventing SARS-CoV-2 inhibition of mitochondrial bioenergetics and dynamics; (B) melatonin augments the adenosine moiety effect that promotes the solubilizing effect of ATP to regulate protein phase transitions during aggregation. The hydrating effect of melatonin—facilitated by the indole π clouds and hydrogen bonding with water molecules—combined with ATP, prevents hydrophobic collapse and delays the transitioning into β-sheets during protein folding; (C) the SARS-CoV-2 virus and its structural, non-structural, and accessory proteins facilitate and accelerate the nucleation of physiological amyloid fibrils that have additional antimicrobial protective function into pathological amyloid aggregates. The SARS-CoV-2 virus further exacerbates the accumulation of insoluble amyloid aggregates by diverting nutrients away from mitochondrial oxidative phosphorylation (OXPHOS) ATP production into anabolic glycolytic pathways that support viral replication and maintenance. The suppression of fusion and fission creates dysfunctional hollow mitochondria devoid of the ATP synthase and elongated mitochondria. Unable to differentiate into functional subpopulations under nutrient deficiency, mitochondrial dysfunction further elevates imbalances between energy production and the synthesis of macromolecular precursors; (D) the optimal clearance of soluble amyloids during slow wave sleep by the glymphatic system must be supported by the adequate presence of both melatonin and ATP. The SARS-CoV-2 virus during infection and persistence prevents the effective brain clearance of excess insoluble amyloid aggregates due to deficient ATP and melatonin as a result of mitochondrial dysfunction. Created in BioRender. Loh, D. (2025) https://BioRender.com/b31s634
Amyloids, phase separation, and SARS-CoV-2: the perfect storm for Neuro-PASC
The ~4 Ga relationship between amyloids, phase separation, and viruses
Since its first proposal in 1991 [67], the amyloid hypothesis remains controversial. Notwithstanding the copious experimental evidence correlating the formation of Aβ with cytotoxic effects that may advance the development of NDDs [68–71], the detection of Aβ in cognitively normal individuals presents unresolved questions on the nature and function of Aβ oligomeric assemblies that inevitably increase with the aging process [72–77]. Recent advances in the antimicrobial functions of Aβ that may protect the brain against viral infections begin to reveal the multilayered complexity of amyloidogenesis [78]. Bacteria including Escherichia coli produce protective biofilms strengthened by amyloid fibrils known as curli [79]. Yet curli formation triggers the enhanced production of cytotoxic host Aβ oligomers that suppressed curli synthesis and inhibited E. coli biofilm density and elevated susceptibility to gentamicin antibiotic treatment [80]. The results of this explicit experiment open an unexpected door revealing the ancient, complex roles of amyloids in all three major domains of life where the neurodegenerative effects of aberrant Aβ aggregation may be the result of protective antimicrobial activity [81]. Furthermore, amyloids confer advantageous adaptive fitness via the inheritance of proteinaceous epigenetic memory [82].
The discovery of amyloid-forming, prion-like domains in Archaea consolidates their functional, physiological existence not only in all three major domains of life but also in the last universal common ancestor (LUCA) [83, 84]. Accordingly, amyloids may have played a major role in the origin of life, increasing the stability of RNA in extreme, fluctuating environments by acting as templates and catalysis for chemical reactions that favor replication [85]. While the exact origin of viruses remains enigmatic [86], and the debate on whether giant viruses constitute the fourth domain of life continues [87], the early cellular origin of viruses sharing a common evolutionary path with Archaea and Bacteria since their inception is, nonetheless, supported by phylogenomic data [88] as well as direct and indirect inferences from paleovirology [89]. All evidence examined to date appears to suggest that viruses are ancient and may even predate LUCA [90–92]. Upon invasion by viruses, ancient unicellular living organisms, especially archaea, inevitably formed cytotoxic amyloids as their only course of effective protection [46, 93–95].
Due to the nature of viral replication that is dependent upon phase separation, viral invasions of early organisms can readily transform functional proteins in their soluble monomeric states into insoluble, crystalline, Aβ-sheet assemblies [96]. Viruses facilitate the heterogeneous nucleation and phase transition of amyloids via direct physicochemical mechanisms that act as catalysts, promoting and accelerating the assembly of Aβ-sheets [97, 98]. Despite being potentially protective in nature, the cytotoxicity of amyloid oligomers is a conundrum requiring reconciliation and clarification [99–102].
Phase-separated biomolecular condensates promote amyloidogenesis
Phase separation is an intrinsic feature of nature used by all tested living organisms in the three major domains of life to organize and sustain fundamental biological processes. Spontaneous, nonequilibrium thermodynamic conditions governing the competition between entropy and enthalpy, drive intracellular phase separation in the assembly of membraneless organelles, also known as biomolecular condensates (BCs) [103–105]. BCs are dynamic, micron-scale, fluid compartments that can rapidly organize/reorganize cellular biochemistry to accelerate, slow, promote, or inhibit cellular functions and reactions, by either increasing, decreasing, including, or excluding reactants, enzymes, and substrates according to the fluctuating requirements of the cell [106]. Recent advances link the formation of amyloid fibrils to phase separation of condensates [107], where the interface of condensates promotes the accelerated formation of amyloid fibrils [108]. Viruses employ molecular sorting driven by phase separation and the exploitation of the host phase separation machinery to enable viral replication and persistence [63, 109–113]. Fluctuations in the energy landscape of aggregation pathways occurring at BC interfaces can subsequently become active sites for the heterogeneous nucleation of pathological amyloid fibrils promoted by viral phase separation [114–116].
BCs have been observed to both promote and inhibit the self-assembly of fibrillar proteins [117–120], which is a process commonly associated with NDDs including AD, PD, as well as prion diseases. The self-assembly of functional amyloids, dependent upon intrinsically disordered proteins (IDPs), may have played decisive roles in the origin of life and the evolution of species [121–123]. The formation of amyloid fibrils rich in insoluble β-sheets networks, stabilized by hydrogen (H) bonds acting as chemical adhesives [124], is intrinsically a phase transition of proteins from an amorphous liquid state into a solid, insoluble state via phase separation, nucleation, and growth processes [125–127]. The addition of phase-separated BCs near natively folded protein assemblies can trigger the abrupt acceleration of protein aggregation in the self-assembly of amyloid aggregates [117, 128]. Nevertheless, protein folding—whether self-assembling into stable, highly-ordered, β-sheets or physiological, well-defined, functional structures—is a common feature exhibited by, potentially, all polypeptide sequences, regardless of their native states or localization [129–133]. Perturbation in the energy landscape can directly affect the outcome of protein folding and aggregation. The transitioning into β-sheets during protein folding involves hydrophobic collapse, formation of H-bonds, and intermolecular electrostatic interactions that lower the Gibbs free energy but increase solvent entropy [134, 135].
SARS-CoV-2 escalates the self-assembly of crystalline Aβ-sheets
Viruses can accelerate the phase transition of amorphous amyloids into stable β-sheet aggregates. Acting as chemical catalysts, viruses modulate hydrophobic collapse and the nucleation/condensation of proteins, potentially by altering the activation barrier (ΔG#) for protein folding via a wide array of molecular mechanisms—including phase separation—that will be addressed, as allowed by scope, in this review [97, 98, 134]. Accordingly, abundant in vitro studies report that different structural, non-structural, and accessory proteins of the SAR-CoV-2 virus from wildtype and prominent variants readily form amyloid aggregates that are commonly associated with NDDs. Even SARS-CoV-2 nucleic acids that have been deactivated by UV radiation are able to induce amyloid aggregation in vitro, by directly acting as physicochemical mechanistic catalysts that promote amyloid aggregation via heterogeneous nucleation and phase transition that do not involve templating. The aggregation of disease-associated proteins is often seeded by BCs containing proteins with unfavorable molecular interactions [117]. The enhanced cytotoxicity of ground-state amyloid crystals assembled from SARS-CoV-2 accessory open reading frame 6 (ORF6) protein can increase the level of neuronal apoptosis compared to higher free energy, non-crystalline amyloid assemblies [136–153]. Please see Table 1 for individual study descriptions and corresponding observed effects. The formation of β-sheet aggregates as a result of viral infection is possibly an evolutionarily conserved response in eukaryotes and archaea. As such, it is not unreasonable to expect living organisms to be able to effectively manage the cytotoxic effects via the maintenance of proteostasis.
SARS-CoV-2 accelerates and exacerbates host amyloidogenesis
| SARS-CoV-2 amyloidogenic components | Study type | Observed effects | Ref. |
|---|---|---|---|
| Spike 1058 fragment (1058HGVVFLHVTYV1068) | All-atom discrete molecular dynamics (DMD) simulations | Spike 1058 accelerated amyloid-beta (Aβ)42 cytotoxicity, aggregation, and fibrillization in vitro | [136] |
| Spike532 (532NLVKNKCVNFNFNGLTGTGV551)Spike601 (601GTNTSNQVAVLYQDVNCTEV620) | In vitro conversion assay | Spike532 and 601 selectively seed and accelerate amyloid fibril formation of human prion protein and Aβ1–42, respectively | [137] |
| Synthetic spike (S) protein peptides | In vitro | Endoproteolysis of S protein by the neutrophil elastase protease produced amyloidogenic peptides. Whereas full-length folded S-protein did not form amyloid fibrils | [138] |
| S protein receptor binding domain (RBD) of the Alpha and Omicron variants | Molecular dynamics simulation | Aβ42 amyloid fibrils can bind to the RBDs of the S proteins and diffusively slide on the RBD surfaces | [139] |
| S RBD of the Delta Plus and Omicron variants | In vitro, in silico/experimental analysis | The RBD self-assembles into aggregates with 48.4% β-sheet content. The Omicron variety displays higher amyloidogenic potential due to distinct mutations in the RBD regions | [140] |
| S protein 1 (S1) | In vivo male Sprague-Dawley rat (6 weeks old) model | In the rat brain, a single intranasal dose of S1 (0.5 μg/10 μL) doubled the aggregation of α-synuclein (α-syn) relative to controls | [141] |
| Recombinant SARS-CoV-2 S1, S2, RBD, and nucleocapsid (N) proteins (NCAPs) | In vitro, Escherichia coli, human embryonic kidney (HEK) Expi293F cell line, and other eukaryotic cell cultures | Recombinant S1, S2, RBD, and NCAPs spontaneously self-assemble into nanoparticles/nanofibers forming amyloid-like structures in tested cell lines | [142] |
| S proteins and NCAPs | In vitro HEK293 cells, bioinformatics analysis | S proteins and NCAPs displayed high binding affinity with α-syn. The direct interactions elevated α-syn expression and accelerated aggregation, forming Lewy-like bodies in HEK293 cells | [143] |
| NCAP low complexity domain (LCD) | In vitro | The LCD of NCAP phase separates and forms amyloid-like structures in the presence of RNA | [144] |
| NCAP, S protein | In vitro, neuroblastoma SH-SY5Y cells | NCAP accelerated the aggregation of α-syn into amyloid fibrils via nucleation of multiple protein complexes. Whereas S protein had no effect on α-syn aggregation | [145] |
| Envelope (E) protein C-terminus-derived 54SFYVYSRVK62 (SK9) peptide | In vitro | SK9 not only self-assembles into amyloid fibrils, but also binds to amyloidogenic hotspots in lipid-free serum amyloid A (SAA), promoting SAA fibrillization | [146] |
| E protein SK9 peptide (C-terminus) | Molecular dynamics simulation | The binding of SK9 to SAA promotes amyloidosis by increasing the frequency and stability of amyloid fibrils to favor β-strand formation | [147] |
| Open reading frame 6 (ORF6) | High-speed atomic force microscopy visualization | ORF6 amyloidogenic peptides spontaneously self-assemble into amyloid protofilaments | [148] |
| ORF 6, ORF10 | In vitro, atomic force microscopy, transmission electron microscopy, amyloid prediction algorithms | ORF6 and ORF10 peptide sequences self-assemble into ground state amyloid crystals markedly elevated the rate of apoptosis at low concentrations in a human-derived neuroblastoma cell line (SH-SY5Y) | [149] |
| S protein fusion peptide 1, 2, ORF10, non-structural protein 6 (NSP6), NSP11 | In vitro aggregation study | All structural, accessory, and NSPs studied form amyloid aggregates with high propensity under near-physiological in vitro conditions | [150] |
| NSP3 | Transgenic Alzheimer’s disease (AD) Drosophila melanogaster eye model [glass multiple repeats (GMR) > Aβ42] | NSP3 misexpression in AD fly eye worsens Aβ42-mediated neurodegeneration, increases apoptotic cell death and ROS | [151] |
| UV-inactivated SARS-CoV-2 virus | Ex vivo, healthy human cerebrospinal fluid (CSF) | UV-inactivated nucleic acids act as catalysts promoting amyloid aggregation and markedly depleting soluble proteins in human CSF compared to controls | [152] |
| SARS-CoV-2 S protein | Transnasal infection of 18hACE2 mice | Increased accumulation of phosphorylated tau protein and activation of glycogen synthase kinase 3β in the ventral tegmental region induced neuronal cell death | [153] |
It is almost implausible that an intrinsic, fundamental flaw in the design of the molecular chaperone disaggregase system is responsible for the increase in neurological dysfunctions associated with viral infections in humans, especially when considering the alarmingly high prevalence of Neuro-PASC in infected/exposed individuals. While the emergence of amyloids may have begun as early as LUCA [84], the existence of a proteostasis-defending heat shock response system that arose around the same time implies that this relationship is not only evolutionarily conserved but should also be robust and effective [154]. Yet this dynamic relationship is dependent upon an intricate balance between proteostasis and energy homeostasis [155]. Effective induction of stress-induced chaperones must be supported by the reduction of other housekeeping proteins to manage proteotoxic stress. Aberrant phase separation prevents the requisite assembly of molecular condensates that exert gain-of-function to amplify chaperone synthesis, and loss-of-function to repress expendable cellular functions. Thus, the ineffective shut-down of housekeeping processes during chaperone induction due to macromolecular assembly defects often results in less-than-optimal performance [156]. The successful assembly of BCs is ultimately determined by thermodynamic principles that govern phase separation, and the presence or absence of ATP.
Chaperones: the dilemma of energetic cost in amyloid disaggregation
Humans and mammals expend approximately 20% of resting energy in protein synthesis, while the cost for the degradation of damaged proteins is at least 20% of total energy expenditure [157, 158]. Thus, the destruction and resynthesis of proteins becomes an energetically costly expenditure. Consequently, the ability to salvage and reuse aggregated, unfolded/misfolded proteins and return them to their native folded states at a considerably lower energetic cost [159], allows stress-induced chaperone systems, including heat shock protein 70 (HSP70) [160, 161], to confer living organisms a unique survival advantage during evolution [157]. However, the degradation of mature amyloid fibrils by chaperones may involve trade-offs that are also dependent upon energetic costs. Amyloids were once believed to be indestructible under physiological conditions [162]. Recent advances reveal that the incomplete degradation of mature fibrils resulting in smaller fragments not only increases the cytotoxicity of the fragments over the original longer fibrils, but also seeds the production of additional amyloid aggregates [163–165]. Consequently, chaperone systems including HSP70 and its co-chaperone DNAJB1 [166] engage in “all-or-nothing” switch-like, tactical maneuvers—involving ATP-dependent unzipping and depolymerization—that can effectively target and disassemble short toxic oligomeric α-synuclein (α-syn) fibrils, but are unable to disassemble larger tau fibrils [167].
Ultimately, heat shock protein chaperone systems such as HSP70 achieve their molecular efficiency by consuming ATP energy [168]. Subsequently, the availability of ATP directly correlates with HSP70-mediated disaggregation of oligomeric fibrils [169, 170]. A lack of ATP can result in incomplete degradation, generating potentially toxic intermediate species that can seed additional fibril aggregation [163, 171]. While longer amyloid fibrils will require not only additional ATP but also increased copies of chaperones to achieve ideal chaperone-to-co-chaperone ratios that can efficiently bind and disassemble amyloid aggregates [172]. The increased metabolic cost of generating adequate copies of heat shock chaperones during stress is a considerable, limiting factor on the efficiency of chaperones in vivo. Molar ratios as high as 1:1 (HSP70:α-syn) were required in order to shift the immune landscape of rodent models from a proinflammatory one to an immunomodulatory phenotype [173]. Drosophila melanogaster larvae exposed to 36°C exhibited increased stress tolerance by elevating inducible heat-shock response expression of HSP70, albeit at a high energetic cost at the level of whole-organism metabolic rate. This increase in energetic cost associated with elevated gene expression may in turn impose limiting thresholds on the process itself in order to balance fitness against survival during evolution [174].
While it is unimaginable that the robust chaperone disaggregase system that evolved alongside amyloids since LUCA is incapable of fully disaggregating large amyloid fibrils resulting in proteostasis failure that causes neurological and other health impairments [175–177] under different contexts, especially during aging [178]; it is conceivable that nature never intended chaperones to assume the full responsibility of disaggregating large amyloid fibrils with their multifaceted, protective physiological functions, in addition to their Janus-faced cytotoxic effects. After all, the energy landscapes of protein folding and aggregation are diametrically opposed, competing mechanisms. As such, for chaperones to be able to both refold proteins and disaggregate amyloid fibrils may be a nonviable task. Molecular chaperones facilitate protein refolding into the native state by lowering the free-energy barrier while consuming ATP energy [135, 168]. Whereas, the assembly of aggregates associated with kinetically-trapped intermediates involve intermolecular reactions that drive the aggregates towards the thermodynamic ground state [135, 179]. Although seemingly impossible, living organisms have relied on melatonin, a simple, cost-effective solution to both prevent and disaggregate amyloid fibrils for billions of years.
Melatonin: an indispensable molecule in the glymphatic brain-cleaning system for amyloid aggregates
Cellular stress induces the expression of the HSP70 chaperone [180]. Stress can also modulate the thermodynamic energy landscape to promote phase-separated formation of amyloid fibrils and their subsequent aggregation [181]. Female Wistar Albino rats subjected to chronic stress of cerebral hypoperfusion but treated with 10 mg/kg melatonin via intraperitoneal (IP) injection for 14 days exhibited an impressive 5.94- and 6.05-fold reduction in the expression of HSP70 in their CA1 and CA3 hippocampal neurons, respectively, compared to hypoperfusion controls not treated with melatonin [182]. The fact that melatonin can suppress HSP70 while rescuing neurons from the damaging effect of hypoperfusion in a rodent model implies that melatonin may be highly effective at maintaining proteostasis during stressful conditions, albeit at a lower, more manageable energetic cost. Consequently, a deeper examination of the correlations between melatonin and the glymphatic system that is associated with the clearance and efflux of Aβ in rodents and humans is warranted.
The glymphatic system and the clearance of soluble amyloids in Neuro-PASC
In neurodegenerative disorders, proteostasis failure can be a vicious cycle where reduced protein degradation may be directly responsible for the ineffective clearance of amyloid aggregates [176, 183, 184]. Deficient proteostasis allows the transition of amyloid fibrils from soluble, amorphous states into insoluble, crystalline β-sheet aggregates [185], via breaching solubility limits coupled with a breakdown of supersaturation—promoted by defective clearance—where the solute concentration overwhelms its thermodynamic solubility [186]. Hence, maintaining the solubility of proteins could be one of the most important deciding factors that can ensure the effective clearance and efflux of amyloids from the brain via the glymphatic system to prevent amyloid crystallization [187]. Higher levels of soluble Aβ42 in cerebrospinal fluid (CSF) is associated with better neuropsychological function and increased volume in the hippocampus [188].
The efflux of soluble amyloid fibrils in the brain is facilitated by various clearance mechanisms including the blood brain barrier, blood-CSF barrier, the glymphatic drainage system, and intramural periarterial drainage [189]. Since the twin discoveries of meningeal lymphatics and the glial-lymphatic (glymphatic) system, increased understanding of how the movement of CSF is responsible not only for the delivery of nutrients but also for the clearance of brain waste [190, 191] begins to emerge. The dysfunction of the glymphatic system and meningeal lymphatics [192] is associated with defective clearance of metabolic waste in the CSF and the subsequent deposition of insoluble amyloid crystals resulting in the progression of NDDs [183, 193–196]. The discovery of the extension of the CSF flowing contiguously and continuously from the central nervous system to distal ends of peripheral nerves expands the potential reach and effect of the glymphatic system in the clearance of waste and the delivery of nutrients in health and disease [197]. However, the extent to which glymphatic-lymphatic brain clearance contributes to total brain clearance is currently unknown, and whether yet unidentified direct/indirect drainage routes exist, awaits elucidation [198, 199].
Sleep deprivation impairs glymphatic clearance of Aβ biomarkers
A novel, cross-sectional and longitudinal study, employing the analysis along the perivascular space (ALPS) index, investigated the relationship between whole-brain glymphatic activity and clinical/pathological features in participants with AD dementia (n = 47), mild cognitive impairment (n = 137), and healthy controls (n = 235). Although the ALPS index reflects only global glymphatic activity that may not identify regional glymphatic dysfunction, the results of the study revealed that glymphatic dysfunction precedes amyloid pathology including increased Aβ PET burden [200]. While Aβ42 in the CSF breaching the positive threshold often predicts brain atrophy and cognitive decline [201]. A multi-site randomized crossover clinical trial measured the changes in the plasma levels of Aβ42/Aβ40 and tau peptides from evening to morning in 39 healthy participants (between 50–65 years old) under in-laboratory conditions of both normal and deprived sleep conditions. Results of the study showed sleep-active elements—including higher EEG Delta power and lower brain parenchymal resistance—enhanced the overnight glymphatic clearance of amyloid aggregates; whereas sleep deprivation associated elements reduced the ability of the glymphatic system to clear Aβ biomarkers to plasma [202].
While the glymphatic system is only capable of removing soluble amyloid fibrils [203], the SARS-CoV-2 virus enhances the production of insoluble amyloid crystals, reducing soluble proteins [152] to severely compromise the cerebral efflux of Aβ via the glymphatic system. Consequently, dysfunction of the glymphatic system is often reported in recovered COVID-19 patients with only mild infections [204], but are associated with neurological impairments [205]. It is perhaps not a coincidence that the higher EEG Delta power that is correlated with higher overnight glymphatic clearance of amyloids is associated with deep, restorative, non-REM stage 3 slow wave sleep [206]. During slow wave sleep, melatonin is released from the pineal gland into the circulatory system, where maximum slow wave activity has been reported to coincide with melatonin secretion rhythm, with frequency band crests occurring before the rise of plasma melatonin concentrations [207]. Unexpectedly, fragmentation of stage 3 slow wave sleep in healthy male subjects was found to elevate morning melatonin in saliva by 1.8 times compared to controls [208]. Hypothetically, the increase in saliva melatonin may be associated with the activation of the serotonergic system during slow wave sleep suppression [209]. Alternatively, if melatonin is utilized for the glymphatic clearance of amyloids via solubilizing and preventing crystallization of amyloids, the observed 50% reduction of stage 3 slow wave sleep could severely inhibit glymphatic clearance during active sleep, resulting in higher levels of morning melatonin detected in the saliva as a result of unused melatonin during slow wave sleep glymphatic clearance. The subsequent increase in urinary melatonin excretion in cataract surgery subjects may be another example where the reduced utilization of melatonin for solubilizing and clearing cataractous amyloid aggregates in the crystalline lens via the ocular glymphatic system results in elevated circulatory/excretion melatonin levels [65, 210, 211].
Melatonin facilitates brain clearance of soluble amyloids in Neuro-PASC
Pineal melatonin is released directly into the CSF via the pineal recess of the third ventricle. Ventricular CSF melatonin measured in animals is at least one order of magnitude higher compared to levels detected in the blood of these animals. Whereas the ratio of pineal melatonin released into the CSF and the blood in humans still awaits quantification [212–215]. The indisputable correlation between sleep and the clearance of brain waste [216–218] suggests a definitive, protective role of melatonin in the brain during sleep [212, 219–221]. Postmortem ventricular CSF melatonin was conclusively demonstrated to be negatively correlated with the severity of AD pathology. Aged individuals with early neuropathological changes, including an accumulation of amyloid aggregates, exhibited a significant ~3.5- to ~7-fold decline in their CSF melatonin levels compared to individuals without any amyloid aggregates [222]. Similarly, a staggering 5-fold decline in ventricular postmortem CSF melatonin was identified in 85 AD patients compared to 82 age-matched controls without AD [223].
The adequate release of melatonin from the pineal gland into the CSF during slow wave sleep becomes an important consideration for the clearance of brain waste. An analysis of postmortem human brain material and melatonin levels in postmortem pineal glands obtained from AD subjects and healthy controls revealed that alterations in CSF melatonin levels closely mirrored the changes in the pineal melatonin content [224]. Young adult Sprague-Dawley (SD) rats, when subjected to both pinealectomy and intracerebroventricular (icv) Aβ1–42 infusion, experienced increased cognitive impairment and increased accumulation of Aβ1–42 and γ-secretase compared to pinealectomy-only and icvAβ1–42-only groups. However, 50 mg/kg melatonin supplementation via IP injection for 40 days improved all tested parameters in all groups to closely match control levels [225]. Accordingly, pineal gland dysfunction is believed to be responsible for many NDD-associated symptoms, and that the use of melatonin supplementation is an efficacious remedy for these conditions [226–229]. Importantly, pinealectomy experiments on animals revealed that the reduction in hippocampal melatonin levels is not only exaggerated by treatment with Aβ, but also induced learning and memory deficits [230, 231].
Accordingly, the dysregulation of melatonin from the fragmentation of NREM slow wave sleep resulting in increased amyloid aggregation and related symptoms are increasingly associated with proteostasis failure in neurodegenerative disorders [175, 232]. The insoluble amyloid crystalline assemblies induced by the SARS-CoV-2 virus not only present huge obstacles against clearance by the glymphatic system, but also accelerate and promote the self-assembly of additional insoluble Aβ-sheet-rich, inert, stable structures that encourage sequestration of proteins, promoting loss-of-function toxicity. The lower free energies and reinforced stability of these amyloid crystalline assemblies may be responsible for their increased cytotoxicity as a result of delayed/defective clearance, which is exacerbated by elevated depletion of soluble proteins by SARS-CoV-2 nucleic acids in human CSF compared to uninfected controls [149, 152, 233]. The Omicron variant was associated with sleep deterioration (new onset insomnia or exacerbation of chronic insomnia) together with altered brain structures in infected patients compared to healthy never-infected controls [234]. While asymmetric bilateral glymphatic dysfunction was reported in mild COVID-19 patients, where cognitive abnormalities were more evident in older patients and subjects identified with right-sided glymphatic dysfunction [204, 205].
Nonetheless, at the concentration of ~1.54 mM, melatonin administered for four weeks to chronic unpredictable mild stress (CUMS) mouse model of depression was able to restore glymphatic system function and improve sleep structure [235]. For close to three decades, a wealth of experimental in vivo and in vitro work studied the ability of melatonin to prevent amyloid fibrillization and disassemble amyloid aggregates in neurodegenerative disorders [225, 236–251]. Table 2 is a representative—but not comprehensive—collection, presented in chronological, descending order, of in vivo, ex vivo, and in vitro studies that examined the effects of melatonin on Aβ and phase-separated amyloidogenic aggregates under various contexts.
Melatonin supplementation inhibits amyloidogenesis and attenuates neurodegenerative disorders
| Melatonin dosage/duration | Study design | Results | Ref. |
|---|---|---|---|
| 50 mg/kg body weight (b.w.) via intraperitoneal (IP) injection for 40 days | Young adult male Sprague-Dawley (SD) rat Alzheimer’s disease (AD) model induced by intracerebroventricular (icv) infusion with amyloid-beta (Aβ)1–42 (100 μg) + pinealectomy | Melatonin treatment in icvAβ1–42 + pinealectomy rats, icvAβ1–42-, or pinealectomy-only rats significantly rescued impaired spatial memory and reduced accumulation of Aβ1–42 and γ-secretase | [225] |
| 20 mg/kg b.w. via oral route for 40 days | Male Wistar rat AD model, induced by icv streptozotocin (STZ) injection (3 nmg/kg b.w.) | Melatonin treatment improved cognitive performance of AD rats, reducing amyloid stimulation of microglia activation in hippocampal CA1 and CA3 regions | [236] |
| 10 mg/kg b.w. via IP njection for 30 days | Male Rattus norvegicus Wistar rat AD model, induced by icv-STZ injection (3 nmg/kg b.w.) | Melatonin reversed cognitive impairment in the spatial version Y-maze test and suppressed the accumulation of Aβ induced by icv-STZ injection | [237] |
| 0.5 mg per mouse per day in drinking water for 3 months | APP/PS1 (mutant APP and PS1 transgenic) AD and C57/BL6J mice as controls | Melatonin-treated transgenic mice, compared to controls, exhibited improved cognitive function and reduced deposition of Aβ plaques via suppression of NOD-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome activation and increased transcription factor EB (TFEB) nuclear translocation | [238] |
| 100–5000 μM melatonin | In vitro study—aggregation/disaggregation of repeat domain tau (K18wt) performed in assays without adenosine triphosphate (ATP) | A dose-dependent disaggregation of preformed tau aggregates was observed: 14% with 100 μM, 54% with 5000 μM | [239] |
| 200–5000 μM melatonin | In vitro study—aggregation/disaggregation full-length tau (hTau40wt) by melatonin in Neuro2A cells | Tau treated with 200 μM melatonin showed no change in morphology compared to controls; 5000 μM melatonin treatment did not prevent aggregation but disaggregated tau fibrils into broken filaments | [240] |
| 0.3 mg per mouse in drinking water from day 7 after tauopathy induction to day 28 at termination | 4-month-old C57BL/6J mice injected with human tau mutation P301L (AAV-hTau) | Increased ROS and tau hyperphosphorylation starting at day 7 precedes cognitive decline; melatonin treated animals showed reduced memory impairment, tau hyperphosphorylation, ROS, and neuroinflammation | [241] |
| 10 μM/L melatonin | Ex vivo brain slices from human and 3-month-old SD rats exposed to okadaic acid (OA) to induce tau hyperphosphorylation | Melatonin reduced tau hyperphosphorylation and ROS to control levels in OA-treated human and rat brain slices | [241] |
| ~6 mg melatonin per mouse daily in drinking water starting at age 4 months until euthanasia | Transgenic Tg2576 AD mice, terminated at 4 months 1 week or 15.5 months | The brains of animals treated with melatonin terminated at 15.5 months exhibited impressive declines in oligomeric Aβ40/Aβ42 together with significantly increased lymphatic clearance of soluble monomeric Aβ40 and Aβ42 compared to untreated mice at the same age. Survival rates of melatonin-treated transgenic mice at 15.5 months were similar to wildtype controls | [242] |
| 10 mg/kg (IP) daily for 3 weeks | Male wild-type C57BL/6N mice (8 weeks old) injected with Aβ1–42 peptide | Melatonin treatment reversed Aβ1–42-induced synaptic disorder, memory deficit, and prevented Aβ1–42-induced apoptosis, neurodegeneration, and tau phosphorylation | [243] |
| 25 µM and 250 µM melatonin | In vitro α-synuclein (α-syn) peptide aggregation in primary neuronal cultures | Melatonin blocked α-syn fibril formation and destabilized preformed fibrils in a dose- and time-dependent manner; increased viability of primary mixed neurons treated with α-syn to ~97% in a time-dependent manner | [244] |
| Melatonin (50 mg/kg b.w.) IP injection for 5 days | Arctic mutation (E22G) male Swiss albino AD mice induced by Aβ1–42 synthetic peptides (scrambled and protofibrils) via icv injection | Melatonin treatment in Aβ protofibril-injected mice restored brain glucose levels to protect ATP production; inhibited ROS production and maintained homeostasis of antioxidant enzymes, intracellular calcium and acetylcholine levels in neuronal cells compared to controls and mice injected with scrambled AB1-42 peptides | [245] |
| 10 mg/kg (IP) × 5/day for 2 days, then × 2/day for 5 days | Arsenite-induced oxidative injury in substantial nigra of adult male SD rats | Reduced arsenite-induced α-syn aggregation, lipid peroxidation, and glutathione depletion | [246] |
| ~65 µg melatonin per mouse (2.6 mg/kg b.w.) in drinking water for 10 weeks, starting at age 14 months | Transgenic Tg2576 AD mice | Melatonin treatment failed to reduce brain Aβ levels or even oxidative damage | [247] |
| 40-ppm (w/w) in pelleted minimal basal diet (~5 mg/kg b.w.) for 11 weeks | Male B6C3F1 mice aged 6, 12, and 27 months | Significant reduction of Aβ in brain cortex tissues: 57% in Aβ40 and 73% in Aβ42; increased melatonin levels in cerebral cortex in all 3 treated age groups (12 > 6 > 27 months) compared to untreated | [248] |
| 6 mg melatonin per mouse daily in drinking water starting at age 4 months until euthanasia at 15.5 months | Transgenic Tg2576 AD mice | Increased survival in treated mice (3 deaths, 41 survivals) compared to untreated controls (13 deaths, 31 survivals) | [249] |
| 1.5 mg melatonin per mouse daily in drinking water starting at age 4 months | Transgenic Tg2576 AD mice | Striking reductions in Aβ levels in brain tissues of treated mice at 8, 9.5, 11, and 15.5 months | [249] |
| 0.3 mM melatonin dissolved in 2 mM ammonium acetate | In vitro study—Aβ peptide (1–40) β-sheet/fibril formation | Inhibited β-sheet formation by targeting hydrophobic Aβ-peptide segment (29–40) intermolecular activities | [250] |
| 1 nM–200 µM melatonin | In vitro study—Aβ synthetic peptides (1–40) and (1–42) β-sheet/fibril formation | Progressive reduction of Aβ1–40 β-sheet structures to 24% after 24 h incubation; immediate reduction of Aβ1–42 β-sheet structures from 89% to 65%, decreasing to 59% after 4 h; complete disaggregation into amorphous material after 6 h (100 μM melatonin, 250 μM Aβ1–40 peptides) | [251] |
ppm: parts per million; w/w: weight per weight
Note. Adapted from “Light, Water, and Melatonin: The Synergistic Regulation of Phase Separation in Dementia” by Loh D, Reiter RJ. Int J Mol Sci. 2023;24:5835 (https://doi.org/10.3390/ijms24065835). CC BY.
The conundrum of resolving contradictory results from in vivo and in vitro experiments on melatonin and amyloid disaggregation
The results obtained from in vivo studies delineated a distinct dose- and time-dependent relationship between melatonin supplementation and the prevention of amyloid deposition-induced neurodegenerative symptoms in healthy and diseased animals tested. The highest melatonin supplementation recorded in the experiments in Table 2 was 6 mg/day in drinking water administered to transgenic Tg2576 AD mice for 11.5 months, starting at 4 months of age [242]. Compared to the same Tg2576 mice given ~65 µg melatonin per mouse in drinking water for only 10 weeks [247], the differences were stark. Transgenic AD mice supplemented with high-dose melatonin (HDM) exhibited lower mortality rate similar to non-transgenic mice. The brains of HDM-treated animals showed impressive declines in oligomeric Aβ40 and Aβ42 compared to same-age untreated controls. It is noteworthy that HDM treatment significantly elevated lymphatic clearance of soluble monomeric Aβ40 and Aβ42 peptides in transgenic AD mice compared to non-treated wildtype controls [242]. Tg7256 AD mice supplemented with a 92-fold reduction in melatonin in drinking water, starting 10 months later, for a total duration of only 10 weeks, fared poorly. These mice showed disappointing results where melatonin treatment failed to reduce neither brain Aβ levels nor oxidative damage [247].
Recent advances begin to reveal that the wide range of molecular mechanisms employed by melatonin to improve cognitive function and reverse amyloid-induced cognitive impairment are dependent upon the regulation of phase separation. By modulating BC dynamics, melatonin effectively adjusts the behavior of immune system mediators such as the NOD-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome and transcription factor EB (TFEB) [238, 252, 253]. Melatonin regulation of phase separation processes that drive the fibrillization of amyloids reflects an ancient synergistic relationship dependent upon ATP and other molecular interactions. Consequently, the variation and fluctuation in these conditions may explain the discrepancies and inconsistent results observed in both in vitro and in vivo studies examining the ability of melatonin to prevent and/or disassemble amyloid aggregates.
Melatonin disassembly of amyloid aggregates is dependent upon hydrophobic molecular interactions and ATP
Early in vitro experiments reported that melatonin concentrations as low as 10 μM began to inhibit aggregation of Aβ1–40 synthetic peptides. While at concentrations approaching 100 μM, melatonin incubated with Aβ1–40 synthetic peptides (250 μM) produced soluble, amorphous assemblies within 6 h and immediate reductions of β-sheet structures at time 0. Whereas controls using antioxidants, melatonin analogs, or Aβ1–40 synthetic peptides showed distinct Aβ-sheet fibrillization [251]. In contrast, in vitro experiments using melatonin concentration as high as 5000 μM were only able to produce unimpressive dose-dependent disassembly of preformed tau aggregates (14% with 100 μM, 54% with 5000 μM) [239]. Similarly, 200 μM melatonin failed to elicit morphological changes in preformed, full-length tau (hTau40wt) fibrils, and 5000 μM melatonin was unable to prevent aggregation, but was able to fragment the full-length tau fibrils into small, broken filaments [240]. The conflicting results between the Aβ1–40 synthetic peptide and the preformed tau fibril experiments may reflect the extent that melatonin can modulate the hydrophobic interactions that drive fibrillization during phase-separation and nucleation.
Synthetic amyloid peptides used in the late 1990s are usually monomers that have not undergone primary nucleation processes [254]. Whereas preformed fibril used in later experiments may be derived from synthetic peptides formed via nucleation. Post primary nucleation, preformed fibrils accelerate secondary nucleation events and the formation of cross β-sheet amyloid aggregates [255, 256]. The repeat domain tau (K18wt) and full-length tau were preformed, post-nucleation aggregates [239, 240, 257]. Therefore, whether melatonin is present or absent before the nucleation events can dramatically modify the outcome of aggregation and disaggregation of proteins in experiments.
The removal of water molecules from protein hydration shells drives phase separation and nucleation
Overcrowding, hydrophobic, and electrostatic interactions are understood to drive phase separation of Aβ peptides, where solid-liquid interfaces serve as sites for heterogeneous primary and accelerated secondary nucleation events [115, 255]. In particular, dehydration—the removal of water molecules from protein hydration shells into bulk water—is the central player in the entropy tug-of-war in phase separation thermodynamics during condensate formation, where released water gains entropy and confined water molecules in the dense phase are penalized entropically [258–260]. In dehydration, the resultant positive change in water entropy promotes nucleation where amyloid aggregation is accomplished via the release of water molecules surrounding hydrophobic, monomeric amyloid peptides [261, 262]. Accordingly, the hydrophobic collapse [134, 263] of amyloid protofilaments as a result of the release of water molecules leads to amyloid aggregation, and the increase in ion concentration that augments the salting-out effect also produces similar aggregation effects [264, 265]. The phase separation of tau droplets into gel-like states before transitioning into cytotoxic fibrils that are associated with NDDs is also fueled by the dehydration of hydrophobic protein residues [266, 267].
Therefore, the presence of melatonin in the critical stage where water is expelled from hydrophobic residues may be a deciding factor that can prevent further aggregation and nucleation of amyloids. As such, the ability of melatonin to disassemble preformed, post-nucleation fibrils may reflect an important step that is present in in vivo conditions, but not necessarily in experimental setups using preformed fibrils. Unexpectedly, when preformed α-syn fibrils [268] were treated with 25 μM melatonin for 6 h, there was an obvious destabilization and reduction in fibril aggregates and the formation of shorter fibrils. At compound:peptide ratios of 5:14 and 2:14, 25 μM and 10 μM melatonin completed blocked α-syn oligomerization, respectively. Whereas, a mere 2.5 μM concentration of melatonin at compound:peptide ratio of 1:28 was able to exert an inhibitory effect on α-syn oligomerization [244]. The effectiveness of melatonin in the disaggregation of preformed α-syn and the blocking of further oligomerization underscores a unique synergy between melatonin and ATP in the regulation of phase separation by maintaining the hydration of proteins to prevent aberrant aggregation.
The ability of melatonin to disaggregate preformed amyloids is dependent upon mitochondrial ATP production
Importantly, the differences in neuronal cell lines used and the amount of fetal bovine serum (FBS) incubated with the cells may dramatically alter the amount of ATP that may be available to interact with melatonin synergistically to solubilize and disassemble preformed fibrils. Commercially available fetal bovine sera from four different sources (Gibco Thermo Fisher, Euroclone, Microgem, and Sigma-Aldrich) were found to contain a wide range of ATP from as high as 1300 pmol/L (Gibco) to as low as 250 pmol/L (Sigma) [269]. Thus, the amount of FBS added to the testing medium may affect total ATP available. In addition, the synthesis of ATP by different cell lines including primary neurons and Neuro2A (N2a) cells under different culture conditions may also be different, leading to different experimental outcomes reported in literature [270]. Mixed neuronal cultures of mesencephalon and neostriatum were used to study the effect of melatonin on preformed α-syn fibrils. These cells were incubated in 10% FBS for 48 h and switched to 5% calf serum for the assay. No glucose was used [244]. Most commercially available FBS is collected from bovine fetuses from pregnant cows during slaughter; whereas fetal calf serum (FCS) is collected from calves under 12 months old, and may contain higher protein than FBS. The level of ATP in commercially available FCS has not been determined. Conversely, neuroblastoma N2a cells were used to study the effect of melatonin on preformed full-length tau, using 0.5% FBS and without any glucose added during experiments [240].
Primary neuron cells, but not N2a, produce ATP under glucose depleted conditions
Primary neuronal cells, including pheochromocytoma of the rat adrenal medulla (PC12) cells (cultured in 5% FBS and 4.65 mM glucose), have been reported to produce 5 mM to 150 mM of ATP under normoxic, steady-state conditions. Upon glucose depletion, the mitochondria of primary neurons have been demonstrated to continue to produce a staggering 500 mM ATP as a result of the proposed astrocyte-neuron lactate shuttle (ANLSH) [271]. Whereas, N2a cells can produce ~37 mM of ATP per mg protein when supplemented with 5% FBS and 25 mM glucose [272]. The need for a high level of glucose is potentially due to the fact that under lower FBS concentrations, N2a cells must switch to an anaerobic glycolytic phenotype that requires glucose for the continued production of ATP. When FCS was reduced from 5% to 0.1% in 8.5 mM glucose, mitochondrial ATP production in N2a cells was completely shut down, with zero ATP production. However, total ATP production in these N2a cells was basically unaltered due to the activation of anaerobic glycolysis as a compensatory mechanism [273].
The N2a cells used in the full-length tau study were treated with only 0.5% FBS [240] instead of the standard 5–10% FBS used in experiments. Furthermore, without glucose in the medium, N2a cells under 0.5% FBS may not be able to produce any ATP via anaerobic glycolysis upon total inhibition of mitochondria. Whereas, primary neurons used in the α-syn study were treated first with 10% FBS followed by 5% FCS [244]. Although the medium also lacked glucose, mitochondria of primary neurons may be able to continue the production of ATP due to the proposed ANLSH effect [271]. At the very least, merely the discrepancy in FBS used in the studies may account for a reduction of as much as 90% ATP in the tau study. On the contrary, a doubling or even tripling of ATP in the α-syn study may be possible from the high amount of FBS and FCS employed [269]. These differences in ATP available to interact with melatonin may account for the contradictory effects observed where a very low concentration of melatonin was able to effectively disaggregate preformed α-syn fibrils post-nucleation; while the opposite was observed in the disaggregation of preformed tau full-length fibrils.
Transgenic Tg2576 mice have dysfunctional mitochondria
The fact that primary neurons from transgenic Tg2576 AD mice have defective mitochondrial function with diminished capacity for maximal ATP production capacity [274] also explains why 267 mg/kg body weight (b.w.; 6 mg per mouse at 22.5 g b.w.) was a necessary, effective dose to suppress the progression of AD in transgenic Tg2576 mice and extend their survival by reducing oligomeric Aβ40/Aβ42 [242, 249]. Whereas, the supplementation with only 40 ppm melatonin in diet pellets (equivalent to ~5 mg/kg b.w.) produced impressive reductions of Aβ40 (57%) and Aβ42 (73%) in the brain cortex tissues of normal aging B6C3F1 mice with functional mitochondria [248]. This distinct and potentially compensatory relationship between melatonin and mitochondrial ATP production is evident in Lingulodinium polyedra (L. polyedra)—ancient unicellular organisms with reduced mitochondrial genomes—that produced as much as ~20 mM melatonin under stress to regulate the phase separation of guanine crystals responsible for their bioluminescence [66].
Melatonin, ATP, and the adenosine moiety effect: the energy cost-effective solution for maintaining solubility of condensates
ATP: the biphasic, universal cosolute of BCs
In 1952, ATP was first reported to function as a classic hydrotrope that can dissolve insoluble aggregates [275]. This role was independently verified in different experiments decades later [276, 277]. Recent advances reveal the unique molecular structure of ATP—comprising a high-energy, hydrophilic, negatively charge triphosphate moiety, and a hydrophobic adenine nucleobase attached to a hydrophobic ribosyl group—is the primary reason why ATP is selected as the preferred energy currency used by all living organisms to regulate phase separation [278, 279]. The emergence of life required an efficient energy transfer system. Nature selected ATP as the ultimate energy transfer system for kinetically stable and thermodynamically active phosphates despite the availability of other viable phosphate carriers [280]. In the context of phase separation, phosphate carriers release high-energy phosphates to phosphorylate peptides to form and stabilize membraneless condensates. The high negative charge of phosphates can suppress charge-charge repulsion to facilitate and initiate phase separation [281].
Both ATP and high energy phosphate carriers can engage proteins via nonspecific weak interactions that promote phase separation by displacing water H-bonds, favoring the folded state by substituting water H-bonds with triphosphate H-bonds. Nevertheless, the highly negatively charged triphosphates continue to interact with hydrophobic protein patches to cause severe aggregation [282, 283]. The triphosphate moiety of ATP has been demonstrated to enhance the fibrillization of tau K18 fibrils in vitro [284]. Whereas, the stoichiometric incorporation of ATP (0.45 mM) with oligo-lysine segments containing amyloidogenic residues drives aberrant phase separation towards the self-assembly of fibrils [285]. Yet, the attachment of the adenosine moiety distinguishes ATP from other high-energy phosphate carriers. The hydrating, aromatic adenosine moiety can shield the aggregating effects of the triphosphate moiety, solubilizing solvent-exposed hydrophobic patches via π-π, π-cation and van der Waals interactions [279, 283, 286].
Consequently, a biphasic effect—where low levels of ATP promote phase separation and aggregation, but high levels have the opposite effect—has been extensively observed and reported [278, 287]. The addition of 5 mM ATP promoted phase separation of IDP-rich HNRNPG protein via charge neutralization and conformational compaction. Whereas at levels above 24 mM, ATP reduced intermolecular contacts to dissolve condensates [288]. While the addition of merely 0.25 mM ATP to wild-type α-syn monomers was adequate to begin the induction of fibril aggregation in a dose-dependent manner. However, the continued increase of ATP results in the inhibition of α-syn aggregation. In the presence of 10 mM ATP, a significant loss in late-stage fibril aggregation was clearly indicated by a reduction of Thioflavine T (ThT) fluorescence, confirming the biphasic effect of ATP in α-syn aggregation [289].
The solubilizing effect of ATP is dependent upon the adenosine moiety
The recognition that ATP may facilitate the transition of a droplet into a fibril and back into a droplet [285, 290] supports previous computational and experimental studies that showed that at physiological levels between 0.5 mM to 10 mM, ATP prevents Aβ misfolding via stabilization through the triphosphate moiety and the solubilizing effect of the adenosine moiety [291]. Even though ATP can displace water H-bonds, altering protein hydration, leading to protein folding, the solubilizing effect of the adenosine moiety is highly effective in preventing or delaying further transitioning into insoluble aggregates. Despite the fact that hydrophobic adenosine easily binds to accessible hydrophobic sites in unfolded proteins, its solubility in water is surprisingly low [292]. However, when adenosine is combined with the hydrophilic triphosphate moiety that is surrounded by three–four layers of hypermobile water [293], its solubility is greatly enhanced. This ingenious combination results in, at minimum, an order of magnitude increase in anti-fibrillation performance compared to classic hydrotropes [292, 294].
ATP prevents amyloid fibrillization via adenosine van der Waals energy
ATP, at 10 mM concentration, delays the formation of Aβ-sheets, preventing fibrillization by inducing the formation of amorphous aggregates instead [295]. Recent advances determined the anti-fibrillation suppressive effect of ATP stems not from the high energy phosphate moiety, but rather the hydrophobic adenine moiety that increases solubility via enhanced van der Waals energies [286]. On its own, the triphosphate moiety is unable to prevent fibrillation of Aβ1–42 measured by ThT assays [292]. Whereas an analysis of individual contributions of the triphosphate, adenine, and ribose moieties of ATP towards changes in the free energy of solvation for different aggregation states of the Aβ16–22 peptide indicated that the suppressive effects of ATP on amyloid fibrillation are derived largely from adenine van der Waals interactions, followed by ribose. The ribosyl group in the adenosine moiety of ATP is attached to adenine. Together the adenosine moiety contributes ~10.0 kcal mol–1 towards van der Waals energy during aggregation studies, compared to ~3 kcal mol–1 for phosphates. Although triphosphate electrostatic interactions provided inconsequential contributions towards changes in solvation energies in aggregation studies for Aβ16–22, when triphosphates are attached to adenine and ribose, the combined suppressive effect of the triphosphate and adenosine moieties on fibrillation is the greatest [286].
Nucleation changes the course of fibril aggregation and the subsequent dissolution requirement for ATP
Nevertheless, experiments employing all-atom molecular dynamics for different force fields revealed that ATP, at 150 mM concentration, can destabilize and inhibit the aggregation of preformed Aβ16–22 peptides via π-π stacking, NH-π interactions, and dose-dependent increases in H bonding [296]. Although primary neurons are capable of producing 150 mM ATP under glucose replete conditions [271], the tested concentration of ATP in 136 organ, tissue, and cell sources obtained from the three major domains of life averaged only ~4.41 mM [297]. Consequently, the use of ATP at concentrations between 100–500 mM to dose-dependently inhibit the aggregation and dissolve preformed Aβ40 fibrils in complementary wet-lab experiment and molecular simulation may require justification [298]. It is unclear why a wide range of ATP, from 10 mM to 500 mM, is required to inhibit and dissolve amyloid aggregates. The fact that wild-type α-syn monomers require only 10 mM ATP, but Aβ16–22 prefibrils and Aβ40 preformed aggregates require 150 mM and 500 mM, respectively, to dissolve aggregates reveals the important step in nucleation that changes the course of fibril aggregation. Viruses including SARS-CoV-2 are convenient catalysts that promote aggregation of amyloids via heterogeneous nucleation [152]. Consequently, the ancient synergistic relationship between melatonin and ATP becomes even more critical in the prevention and inhibition of amyloid aggregation as the result of viral infections and viral persistence.
Melatonin compensates for low ATP availability
The potential requirement for exceptionally high concentrations of ATP to inhibit and dissolve aggregates post nucleation events could present living organisms, especially early unicellular organisms without mitochondria, with an energetic cost challenge that is detrimental to survival. The efficient management of proteostatic energetic cost confers significant survival advantages during evolution [157]. This advantage is further enhanced when smaller proteins with low production and maintenance cost, including melatonin, are utilized. It is not a coincidence that the molecular mass of the penultimate, rate-limiting enzyme in the melatonin biosynthetic pathway—serotonin N-acetyltransferase (SNAT)—is only ~15-, ~17-, and 23-kDa in archaea, cyanobacteria, and humans [arylalkylamine N-acetyltransferase (AANAT)], respectively [60, 299, 300]. In comparison, the molecular mass of the ATPase enzymes responsible for the production of ATP is ~730-, ~545-, and ~592-kDa in archaea, bacteria (E. coli), and humans, respectively [301–303].
Unicellular dinoflagellates, including L. polyedra, have significantly reduced mitochondrial genomes, comprising only three protein-coding genes, while the majority of 45 other genes are permanently lost and the remainder transferred to the nucleus [304]. Under duress, L. polyedra may not be able to elevate ATP synthesis to regulate the phase separation of guanine crystals that control their bioluminescence. Instead, L. polyedra increases melatonin synthesis from ~1 μM to staggering levels of as much as ~20 mM (4.61 μg/mg protein) [66, 305]. The synthesis of extreme levels of melatonin by L. polyedra to regulate the phase separation of guanine crystals is highly reminiscent of in vivo studies that administered HDM (267 mg/kg b.w.) to transgenic AD mice with dysfunctional mitochondria [242]. Despite the potential effects of reduced ATP concentration in neurons of transgenic mice [306], HDM was able to suppress the formation of oligomeric Aβ40/Aβ42 and markedly increase the glymphatic clearance of soluble Aβ40 and Aβ42 compared to untreated mice at the same age [242].
Melatonin forms H-bonds with water and stacks with the adenine moiety
Melatonin is able to compensate for lower ATP concentration and reduced solubilizing capacity, by enhancing the adenosine moiety. Melatonin is an amphiphile with hydrophobic parts that change the conformation of melatonin in the presence of water molecules as a result of the hydrophobic effect [307]. Even though melatonin has low solubility in water, it can form stable H-bonds with water. Car-Parrinello molecular dynamics and ultraviolet/infrared spectroscopy studies indicate that melatonin possesses five distinct H-bond sites for water. Melatonin’s amide NH and indole NH groups are H-bond donors to water molecules. Conversely, the amide carbonyl, methoxy oxygen, and indole π cloud of melatonin accept H-bonds from water molecules. Helmholtz free energy studies reveal that water hydrogen molecules when coordinated with the O of the amide group can exhibit an infinite average residence time due to the high degree of stability [308, 309]. Just one single water molecule attached to melatonin changes its conformational preference, where strong H-bonds produce substantial electronic frequency shifts [309] that can affect how melatonin reacts with other molecules, especially free radicals [310].
The adenosine moiety of ATP has a high structural homology to the indole moiety of melatonin (Figure 2). The aromatic indole side chain of tryptophan has been clearly demonstrated to stack comfortably with the aromatic ring systems of adenine in experiments testing molecular interactions between ATP and homopeptides [292]. The dominant π-π-stacking interactions observed between the adenosine moiety and indoles [281] may explain the synergistic association between melatonin and ATP in the regulation of phase separation that began close to 4 Ga [66]. The homologous affinity permits effortless stacking of the indole ring in melatonin with the adenine nucleobase in ATP. Melatonin is not only hydrophobic, but is also capable of binding water molecules that can enhance the solubilizing effect of the hydrophobic adenosine moiety. The stacking of aromatic side chains between tryptophan and adenine in ATP can elevate the van der Waals attraction energy to 83 kJ/mol [292]. The delocalized π-systems of melatonin not only confer increased stability, but also greatly enhance the anti-fibrillation suppressive effects of the adenosine moiety of ATP (Figure 2) [292, 311].
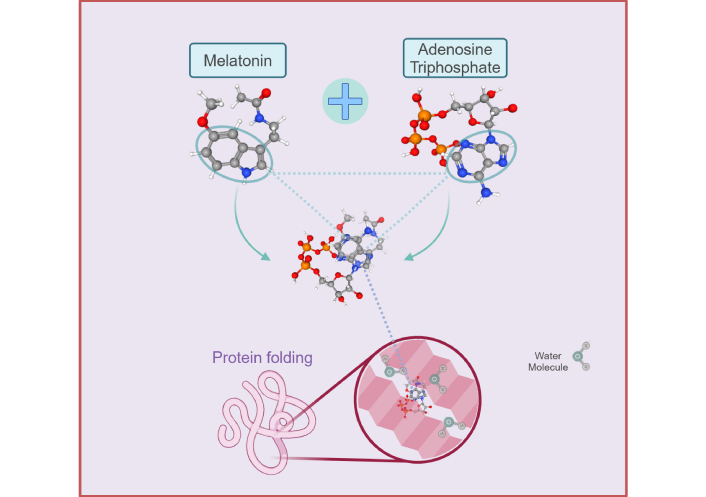
Diagram depicting the homologous molecular structures between melatonin [312] and adenosine triphosphate (ATP) [313] that augment the adenosine moiety effect of ATP in the regulation of protein folding and phase transitions during phase separation. The hydrophobic adenine significantly enhances the solubilizing effect of ATP to prevent aggregation of phase-separated proteins as a result of hydrophobic interactions with the triphosphate moiety. The aromatic indole ring of melatonin can stack comfortably with the adenosine moiety to further promote its hydrating effect via van de Waals interactions that effectively prevent the aggregation of solvent-exposed hydrophobic patches induced by the triphosphate moiety of ATP. Created in BioRender. Loh, D. (2025) https://BioRender.com/t35s942
Therefore, increased melatonin in the vicinity of ATP may significantly elevate the solubilizing effect without the need to increase total ATP concentration. Early organisms may have depended upon this synergy to manage proteostatic energy costs and enhance survival advantage. Conversely, an absence or reduction in melatonin would require extremely high levels of ATP to inhibit further progression of amyloid aggregates post nucleation. Untargeted metabolomic analyses measured both ketone bodies and metabolites derived from the glycolytic and pentose phosphate energy metabolism pathways collected from postmortem prefrontal cortex brain tissues from AD and subjects without clinical AD diagnosis. The results obtained revealed a significant deficit in global energy metabolism in the brains of AD patients [314]. The dramatic reduction in pyruvate essentially suppresses mitochondrial oxidative phosphorylation (OXPHOS). Without the chemical reduction of pyruvate, nicotinamide adenine dinucleotide (NAD+) cannot be regenerated to fuel continued glycolysis that supplies the reduced form of NAD+ (NADH)—the essential source of reducing equivalents—for the mitochondrial tricarboxylic cycle and the electron transport chain during OXPHOS [315, 316].
Reduced production of ATP suppresses slow wave sleep and impedes effective clearance of amyloid aggregates
The strong correlation between sleep and the clearance of brain waste has been clearly identified in literature [216–218]. The exact molecular mechanism responsible for the evidence reported remains elusive. In order to sustain synaptic transmission, neurons require approximately 40% of total ATP produced in the cortex [317]. Whereas during slow wave sleep, the surge of ATP from the reduction of neuronal activity [318] may be critical in facilitating the solubilization of amyloid crystals for removal by the CSF and associated waste-clearing systems. Since the adenosine moiety of ATP is largely responsible for the solubilizing effect of ATP as a hydrotrope, it is not surprising that increased adenosine levels in the CSF enhances both glymphatic flow and clearance of wastes in rodent models [319]. Conversely, fragmentation of slow wave sleep may lead to insufficient ATP in the CSF that not only prevents the effective solubilization and clearance of amyloid aggregates, but also the ability to utilize melatonin for the enhancement of the solubilizing effect of the adenosine moiety. Thus, the puzzling phenomenon of increased circulating melatonin associated with slow wave sleep fragmentation may merely reflect a deficiency in ATP during slow wave sleep [208].
Recent advances reveal both positive and negative feedback relationships between ATP and sleep. Adequate ATP is required for the induction of sleep, while slow wave sleep increases the amount of ATP to promote sleep. Whereas, sleep deprivation not only reduces the amount of ATP available for use by the CSF but also inhibits slow wave sleep. Experimental work studying the slow wave sleep-promoting neurons innervating the dorsal fan-shaped body (dFBN) from Drosophila brains reported that mitochondrial fusion and fission of dFBNs regulate sleep by altering ATP surplus and deficits, respectively. Increased ATP production in dFBNs induces sleep, whereas mitochondria fragmentation that reduces ATP production in dFBNs inhibits sleep induction [320, 321]. This distinct dualistic feedback cycle underscores the relevance of melatonin, which can enhance mitochondrial ATP production, and the SARS-CoV-2 virus, which can cause mitochondrial dysfunction. The multidirectional association between mitochondrial dysfunction, including reduced ATP synthesis, resulting in failure of glymphatic clearance of brain waste [322] emphasizes the relevance of the sleep-promoting effects of melatonin [323] and the dysregulation of sleep by SARS-CoV-2 [234]. Sleep dysregulation suppresses the effective, timely clearance of brain waste (Figure 1). While the SARS-CoV-2 is essentially a natural magnet for amyloid aggregation that can significantly elevate the risk for development and progression of Neuro-PASC, unexpected challenges complicate the targeting of amyloid aggregation in Neuro-PASC and other viral infections.
Janus-faced amyloids: protectors and perpetrators in Neuro-PASC
Controversial and disappointing results from two phase 3 trials of the monoclonal antibody, aducanumab that targets the reduction of insoluble Aβ [324–327], implies that merely suppressing or inhibiting the formation of insoluble Aβ may not fully address the complex, multi-faceted functions of amyloid aggregation that drive the progression of AD and other neurodegenerative disorders. The cytotoxic effects of Aβ oligomers associated with NDDs are extensively documented in literature [328–330]. Nevertheless, when the functional nature of amyloids, including pigmentation of melanosomes [331] and the storage of pituitary hormone peptides [332] are taken into consideration, a different perspective on amyloids emerges. Increasingly, during viral infection and viral persistence, the aggregation of amyloids is understood to be a protective, functional, defensive response to contain pathogenic infection [80, 132]. The perplexing conundrum in the attempt to unravel the true nature and molecular mechanisms propelling amyloid aggregation in health and disease is the fact that both pathological protein misfolding and functional assemblies form the classic, unmistakable cross-β sheets [129–131, 333]. During viral infection and persistence, amyloids can assume antimicrobial activities, acting as formidable biosensors of the immune system [133]. Whereas antimicrobial peptides (AMPs) form amyloids in order to execute host defense mechanisms against invading pathogens. The structural similarity between AMPs and amyloids and their common features of self-assembly, the aggregation of β-sheet-rich structures, and the membrane-disrupting mechanisms of small, soluble, transient oligomers confer AMPs and amyloids their antimicrobial function and cytotoxic amyloidogenic properties [99, 100, 334–338].
During Salmonella typhimurium bacterial infections, the expression and marked acceleration of Aβ oligomer deposition in the brains of transgenic 5XFAD AD mice protected against S. typhimurium meningitis. In comparison, higher mortality is observed in APP knockout mice that are unable to express high levels of Aβ during bacterial infection. In transgenic Caenorhabditis elegans, the expression of human Aβ1–42 significantly enhanced worm survival during Candida and S. typhimurium infections. Whereas the aggregation of Aβ oligomers in cultured human brain neuroglioma and Chinese hamster ovary cells increased host cell resistance to infection by Candida [339]. Nevertheless, the entrapment of microbes and other antimicrobial properties is facilitated by the same qualities of Aβ peptide oligomerization and fibrillization that contribute to the pathophysiology of NDDs. Ultimately, the oligomerization and fibrillization of Aβ, if not effectively cleared in a timely manner, inevitably increase the risk of developing neurodegenerative pathologies associated with amyloidosis, including AD.
The herpes simplex virus 1 (HSV1) is believed to play major roles in the development of AD [340–342]. In transgenic 5XFAD AD mice, HSV1 infection led to the rapid fibrillization and oligomerization of Aβ, dramatically worsening the acceleration of amyloidosis in AD mice. However, these same mechanisms also allowed the Aβ oligomers to bind and entrap the HSV1 virus, inhibiting infection while increasing survival (P = 0.045) and protecting AD mice from the development of acute encephalitis compared to wild-type, non-AD, HSV1-infected controls [343]. While a large retrospective cohort study in Taiwan, China (33448 subjects) that analyzed the association between HSV infections and dementia reported that HSV patients exhibited a 2.56-fold elevated risk (95% CI: 2.351–2.795, P < 0.001) for developing dementia compared to the non-HSV cohort. Additionally, the use of anti-herpetic medication in the treatment of HSV infections was associated with a reduced risk of dementia [344]. Accordingly, the generation of amyloids during SARS-CoV-2 and other viral infections and persistence may confer an antimicrobial, protective effect. Notwithstanding, the increased production of transient, soluble, cytotoxic, oligomeric amyloids during viral infection and persistence requires effective clearance to prevent oligomers maturation forming insoluble crystals that require higher proteostatic energetic cost to eliminate. The increasing prevalence of Neuro-PASC may underscore intrinsic barriers to overcoming challenges in cellular metabolism presented by the SAR-CoV-2 virus.
Phase separation: wielding the double-edged sword in amyloidosis and viral infection and persistence
Recent advances indicate that phase separation is the molecular mechanism that confers antimicrobial properties to AMPs, allowing AMPs to inhibit the transcription and translation of viruses and bacteria [345, 346]. AMPs bind and sequester nucleic acids of pathogens, forming condensates with pathogen RNAs, thus, preventing normal transcription and translation required for replication [345]. In order to facilitate viral replication in host cells, the N protein of SARS-CoV-2 must undergo phase separation. The successful formation of viral replication-competent condensates requires the presence of non-specific RNAs [347]. These multivalent RNA-protein and protein-protein interactions between the N protein and RNAs generate structured or dynamic droplets that are suitable for nucleocapsid assembly or genome processing compartmentalization, respectively [348–352]. In addition to RNA, the phase behavior of N protein BCs is tuned by classic mediators of phase separation, including pH and salt [347, 353]. What is unexpected is the ease at which the N protein can partition into BCs formed by human ribonucleoproteins including fused in sarcoma (FUS) and TAR DNA-binding protein 43 (TDP-43) [347].
SARS-CoV-2 alters the molecular grammar of host physiological condensates
The phase separation of FUS and TDP-43 forms condensates comprising a diverse range of material properties in both health and disease. The aberrant aggregation of FUS and TDP-43 changes the molecular grammar of these condensates that drive the progression of NDDs including ALS, and frontotemporal dementia (FTD) [354]. Consequently, the partitioning of N proteins into host condensates may alter the molecular grammar of functional condensates, turning them into pathological aggregates that are associated with disease-states. The ultimate irony is that host physiological condensates are hijacked by viruses to serve as ready scaffolds that promote the formation of RNA condensates that support and accelerate viral replication. Nonetheless, the double-edged sword of phase separation cuts both ways. Recent experiments reported that AMPs can partition into N protein condensates with a range of different results, from the reduction of condensate dynamics to the formation of fibrillar structures that impair the normal function of RNA transcription and virion assembly in N protein condensates [355].
The aberrant aggregation of α-syn transforms functional, physiological oligomers and protofibrils into pathogenic, fibrous filamentous Lewy body assemblies that are responsible for promoting a range of α-synucleinopathies including PD, dementia with Lewy bodies, and multiple system atrophy (MSA). The SARS-CoV-2 virus is believed to cause α-synucleinopathies associated with NDDs by intensifying the propensity for α-syn aberrant aggregation [356]. In vitro experiments using neuroblastoma SH-SY5Y cells reported observing direct interactions between the N protein, but not the S protein, of SAR-CoV-2 and α-syn. The interactions result in the formation of complexes where the N protein acts as a critical nucleus that can significantly speed up the formation and aggregation of α-syn amyloid fibrils [145]. Conversely, bioinformatics analysis that was experimentally verified in human embryonic kidney (HEK) 293 cells demonstrated high affinity binding of α-syn to both the S and the N protein of SARS-CoV-2. Furthermore, both the S and N protein induced upregulated expression of α-syn and accelerated its aggregation, causing the formation of Lewy-like bodies (Table 1) [143].
SARS-CoV2 exploits the biphasic features of ATP to enhance phase separation for replication and maintenance
Phase-separated condensates, regardless of origin, promote the generation of amyloid fibrils at BC interfaces [108]. Recombinant SARS-CoV-2 proteins—S protein 1, 2 (S1, S2), receptor binding domain (RBD), and N—can spontaneously self assemble into nanoparticles and nanofibrils to form amyloid-like structures in receptor specific and non-specific mammalian cell lines [142]. The low complexity domain (LCD) of the SARS-CoV-2 N protein phase separates with RNA. Although the formation of amyloid fibrils by LCD condensates is not dependent on RNA, the maturation and the stacking of stable, Aβ-sheets can be modulated by RNA interactions (Table 1) [144]. Notwithstanding, it is determined that phase separation of SARS-CoV-2 N protein is maximally optimized by the presence of oligonucleotides at a ratio of 1:1. Whereas, phase separation of the N protein can be dramatically terminated by just a slight increase in either N protein or nucleic acid [111].
The hydrophobic adenosine moiety that is attached to the ATP nucleotide comprises the nucleobase adenine. Therefore, the concentration of ATP, as a result of the solubilizing effect of adenosine, regulates the phase separation propensity of SARS-CoV-2 in a biphasic manner [357]. In vitro experiments reported that at a molar ratio of 1:500 (N protein:ATP), all phase-separated N protein droplets formed at a lower ratio (up to 1:200) were dissolved [287]. Unexpectedly, ATP was observed to displace nucleic acids such as arginine that bind to the N protein to induce phase separation. By replacing the displaced nucleic acid, ATP can then proceed to solubilize and dissolve the phase-separated droplets formed by N protein and other nucleic acids [358]. Therefore, as part of an effective hijacking strategy that is evolutionarily conserved, viruses including SAR-CoV-2 must be able to hijack and control mitochondria—capable of large-scale ATP production—that can derail and terminate viral replication and maintenance.
SARS-CoV-2 reprograms host mitochondrial metabolism and epigenetics to promote viral replication and maintenance of viral reservoirs
SARS-CoV-2 reprograms host metabolism, impairing mitochondrial respiration and fatty acid oxidation capacity to cause metabolic disturbances by altering mitochondrial cristae structure and downregulating core mitochondrial genes [359–363]. Data analysis performed on post-mortem brains of patients infected with SARS-CoV-2 employing single-nucleus RNA sequencing (snRNA-seq) revealed that cognitive impairment post COVID-19 infection is associated with the dysregulation of mitochondrial genes that collectively impairs OXPHOS capacity of mitochondrial complexes I, II, III, and IV [364]. Furthermore, LUC7L2, a gene which suppresses OXPHOS but promotes glycolysis [365], is the only gene upregulated by SARS-CoV-2 [364]. Syrian hamsters infected with the SARS-CoV-2 virus exhibited suppression of energy metabolism without evidence of direct viral invasion of skeletal muscles. Nonetheless, the continuous downregulation of genes regulating protein translation in the cytoplasm and mitochondria, as well as enzyme genes associated with fatty acid β-oxidation and the tricarboxylic acid (TCA) cycle in skeletal muscle mitochondria, were observed in these infected hamsters, resulting in muscle fatigue and atrophy of skeletal muscle fibers [361]. Whereas, a single-center prospective cohort study of post-COVID-19 patients reported persistent mitochondrial dysfunction that impaired fatty acid oxidation capacity, especially in subjects hospitalized with acute infections [362].
An analysis of the plasma proteome obtained from > 7000 subjects revealed that symptoms of Neuro-PASC were associated with changes not only in inflammatory proteins but also mitochondrial proteins associated with OXPHOS [366]. Mitochondria from 3D cultured PC12 neuron-like cells exhibited dose-dependent changes in distribution, morphology, and function when exposed to SARS-CoV-2 peptides [367]. SARS-CoV-2 accessory proteins were demonstrated to reprogram mitochondrial metabolism in A549 lung epithelial cells—favoring glycolysis over OXPHOS and reducing ATP production—by altering mitochondrial cristae structure, mitochondrial bioenergetics, and mitochondrial dynamics [359]. Suppression of mitochondrial OXPHOS supports the effective hijacking of host metabolism, redirecting carbon energy substrates away from mitochondrial OXPHOS towards glycolysis that can fuel the non-oxidative arm of the pentose phosphate pathway (PPP) [368]. The multi-pronged approach in the upregulation of glycolysis and the PPP enhances SARS-CoV-2 replication and persistence by altering cellular metabolism that supports viral anabolic metabolism [369, 370]. The suppression of OXPHOS also reduces ATP concentration in the cytoplasm, preventing the biphasic effect of ATP in dissolving phase-separated droplets that can effectively inhibit the formation of viral factories that support viral replication and persistence [371].
In addition, changes in epigenetic patterns showing enhanced epigenetic drift and accelerated aging were reported in a genome-wide DNA methylation study on COVID-19 patients, 6 months post-infection, compared to healthy controls [372]. It may not be a coincidence that aberrant methylation disrupts the formation of BCs [373] resulting in genome instability, promoting abnormal gene expression that accelerates aging. Age-related neurodegenerative disorders are believed to be caused by mitochondria dysfunction that can result in significant reductions in ATP energy production [374–376]. A study integrating spatial transcriptomics data and high-dimensional weighted gene co-expression network analysis examined multiple cortical and subcortical regions in postmortem brains of subjects with COVID-19 and controls with matched pulmonary pathology. The brain-wide dysregulation of mitochondrial and synaptic pathways identified in COVID-19 brains overlapped considerably with alterations commonly observed in age-related NDDs including AD and PD. Intriguingly, antiviral responses were identified in regions absent direct, SARS-CoV-2 infection [377].
In the study of Neuro-PASC, an analysis of the nuclear encoded mitochondrial gene signatures from two cohorts of long COVID and COVID convalescent patients at 6-months post infection, revealed stark differences in mitochondria gene expression compared to non-symptomatic controls. The results imply that COVID patient recovery is mediated by mitochondria repair. Conversely, continued mitochondrial dysfunction and dysregulated mitochondrial gene expression may drive symptoms associated with Neuro-PASC [378]. Viruses including SARS-CoV-2 must hijack host resources to facilitate viral replication and maintenance. The modulation of host metabolic pathways that switches ATP production from OXPHOS to glycolysis has important ramifications.
Diversion of carbon energy substrates to glycolysis suppresses endogenous melatonin production
The faster generation of a lower concentration of ATP via glycolysis favors the production of viral factories via phase separation due to the biphasic effect of ATP that stabilizes condensates at lower concentrations. Consequently, the dysregulation of metabolic flexibility [379] and the disturbance of the delicate balance between ATP production rate and yield that maintains energy homeostasis [380] may prevent the timely delivery and utilization of ATP. Reduced ATP impacts not only neurogenesis [381] but also causes neurological dysfunction [382] as a result of impaired, timely clearance of amyloid aggregates by CSF brain waste clearance systems during slow wave sleep that is dependent upon high concentrations of ATP and melatonin released from the pineal gland.
SARS-CoV-2 redirects carbon substrates to the PPP that does not produce pyruvate
The effectiveness of brain clearance may be further impaired by mitochondria dysfunction that can dramatically reduce endogenous melatonin synthesis in mitochondria of pinealocytes and all affected brain tissues [58, 383–385]. Acetyl-CoA is a metabolite of the TCA that is an important cofactor utilized by the rate-limiting enzyme during melatonin synthesis in mitochondria [386]. Despite the fact that SARS-CoV-2 rewires host metabolism away from OXPHOS to favor glycolysis, which can theoretically generate enough pyruvate for acetyl-CoA production in TCA cycles, SARS-CoV-2 actually redirects carbon energy substrates to fuel the anabolic, non-oxidative arm of the PPP instead. The PPP is an alternative glycolytic pathway which does not produce pyruvate as a metabolite, but generates 5-carbon sugars that support the synthesis of viral components [368, 387]. Furthermore, SARS-CoV-2 is reported to alter mitochondrial metabolism, shifting carbon uptake, and using fatty acids to generate acetyl-CoA for use in de novo lipid synthesis to support the production of the membrane coat and double membrane replication centers [368, 388, 389]. Therefore, mitochondrial dysfunction during Neuro-PASC may substantially reduce the mitochondrial production of melatonin not only in the pineal gland, but potentially in all affected organs and tissues. Inadequate melatonin synthesis in mitochondria exerts a negative feedback reaction where the indoleamine is prevented from safeguarding mitochondrial functionality including ATP synthesis and reducing oxidative stress. The diversion of nutrients towards anabolic pathways during viral infections can further depress mitochondrial OXPHOS ATP synthesis via a novel, recently discovered mechanism.
SARS-CoV-2 promotes distinct mitochondrial subpopulations that lack cristae or the ability to produce ATP
Recent advances in mitochondrial bioenergetics reveal that mitochondria dynamically respond to nutrient deficiency by forming distinct subpopulations that either specialize in the production of ATP or macromolecular precursors that support cellular growth [390]. Optimal OXPHOS is dependent upon the availability of metabolites from the TCA cycle. Whereas, the production of important macromolecular precursors—proline and ornithine—is dependent upon reductive synthesis that must be maintained by a high mitochondrial membrane potential (ΔΨm) that is traditionally associated with OXPHOS ATP synthesis [390–392]. Under nutrient replete conditions, mitochondria can produce a high level of ATP via OXPHOS and the excess nutrients are used to produce macromolecular precursors vital for sustaining cell growth and other physiological functions including collagen synthesis [393]. Conversely, when nutrient supply is deficient, such as in the case of viral infection and persistence where nutrients are diverted to anabolic pathways that support viral replication and maintenance, mitochondria are demonstrated—in both human and mouse cell lines—to efficaciously differentiate into two distinct subpopulations. One subpopulation specializes in the production of ATP, while the other is only able to support the reductive synthesis of macromolecular precursors due to the absence of both cristae and ATP synthase. More importantly, the successful subdivision into distinct subpopulations is dependent upon mitochondrial fusion and fission. When mitochondrial dynamics are impaired, mitochondria with deficient nutrient supply are unable to differentiate into subpopulations. Undifferentiated mitochondria under nutrient deprivation assume distorted, elongated morphologies, and are unable to produce both ATP and perform reductive biosynthesis. During nutrient deficiency, mitochondria with impaired fusion and fission capacity are forced to abandon either OXPHOS ATP production or the reductive synthesis of macromolecule precursors in order to survive [390].
Fusion and fission regulate the proper differentiation of mitochondrial subpopulations
During nutrient deficiency, the disturbance of mitochondrial dynamics that suppress fusion or fission can derail the formation of mitochondria enriched in ATP synthase and cristae and others that are only enriched in the enzyme, pyrroline-5-carboxylate synthase (P5CS), responsible for reductive synthesis of macromolecular precursors. The inhibition of either mitofusin (Mfn, responsible for fusion) or dynamin-like-protein-1 (Drp1, responsible for fission), forces the selection between the preservation of either OXPHOS or reductive synthesis of macromolecular precursors (Figure 1) [390]. Knocking out Mfn1/2 in mouse embryonic fibroblasts (MEFs) rendered the cells incapable of separating into subpopulations with distinct functions, under both nutrient replete and deficient conditions. While mitochondria from Drp1–/– MEFs cultured under nutrient-deficient conditions not only could not subdivide but also exhibited morphological changes including elongation and bulb-like enlargement [390].
SARS-CoV-2 dysregulation of mitochondrial dynamics produces dysfunctional, hollow, elongated mitochondria with high membrane potential
A transcriptomic and metabolomic data analysis of the changes in mitochondrial morphology and function induced by SARS-CoV-2 accessory proteins in A549 human lung carcinoma cells reported the detection of two distinct subpopulations of mitochondria with high and low activity. Hollow mitochondria devoid of cristae exhibited lower membrane potential and activity level. These effects were associated with the downregulation of optic atrophy 1 (OPA1) responsible for maintaining cristae morphology [359, 394]. Whereas, mitochondrial subpopulations with higher activity and membrane potential still exhibited diminished ATP production capacity [359], supporting the finds of Ryu et al. [390] (2024).
The high-curvature apex at cristae invaginations of the inner mitochondrial membranes are exclusive anchors for ATP synthase dimers. Accordingly, ATP output during OXPHOS can be dramatically impacted by dysregulation in cristae morphology, or worse, the complete absence of cristae invaginations [395, 396]. Intriguingly, the suppression of fission 1 (Fis1), the primary recruiter of Drp1, did not cause elongation of mitochondria by SARS-CoV-2 accessory proteins [359]. Conversely, an in vitro study employing HEK293 cells infected with the SAR-CoV-2 virus reported the suppression of Drp1 expression resulted in the aberrant elongation of mitochondria with high ΔΨm when exposed to N protein-RNA clusters, but not single RNAs from SARS-CoV-2 (Figure 1) [397].
The aberrant elongation of mitochondria may reflect the inability to differentiate into optimal subpopulations. Whereas the high ΔΨm observed in recent studies is controversial. Under physiological conditions of nutrient deficiency, a high ΔΨm in mitochondrial subpopulations can imply the presence of elevated reductive biosynthesis that correlates with a high NAD(P)H/NAD(P)+ ratio [where NAD(P)H and NAD(P)+ represent the reduced and oxidized forms of nicotinamide adenine dinucleotide phosphate, respectively] [390]. Whereas under pathological virus-induced nutrient deficiency conditions, a high ΔΨm observed with elongated mitochondrial morphologies may imply dysregulated mitochondrial dynamics that resulted in mitochondria capable of only ATP production, explaining the elevated ATP levels when compared to uninfected controls [397]. It would not be surprising that if the production of macromolecular precursors by elongated mitochondria were also measured, their levels may also exceed those of uninfected controls. Therefore, more studies may be necessary to elucidate how the SARS-CoV-2 virus modulates mitochondrial dynamics and their ability to differentiate into subpopulations that can respond to nutrient deficiency efficiently. Ultimately, the disturbance of mitochondrial dynamics leading to mitochondrial dysfunction and deficiency of ATP and melatonin is indirectly responsible for exacerbating amyloidogenesis in Neuro-PASC. Supplementation with melatonin is a time-tested solution that can overcome these challenges by protecting mitochondrial dynamics and elevating ATP synthesis [398].
Melatonin outmaneuvers amyloidogenesis by increasing mitochondrial ATP production and enhancing mitochondrial function
The increase in amyloid aggregation as a function of antimicrobial response and the increased generation/aggregation of amyloid fibrils at condensate interfaces, accelerated by heterogeneous nucleation facilitated by viruses, including SARS-CoV-2, require the timely and effective clearance by the CSF during slow wave sleep. The use of exogenous melatonin not only can elevate endogenous production [399, 400], but also can effectively reverse mitochondrial dysfunction and elevate ATP production, overcoming the challenges associated with reduced ATP production during viral infection and persistence and the development of Neuro-PASC and other aging-related neurodegenerative disorders.
Melatonin regulates fusion and fission to elevate ATP production
Melatonin is extensively reported in literature to protect mitochondrial dynamics, enhancing ATP production under different contexts by maintaining membrane potential and balancing mitochondrial fusion and fission [401, 402]. By enhancing the expression of OPA1, melatonin normalizes mitochondrial fusion and fission, elevating ATP production to protect against cardiac ischemia/reperfusion (I/R) injury in vivo and hypoxia reoxygenation injury in vitro [403]. Similarly, melatonin normalized mitochondrial function, structural integrity, and ATP production by enhancing the expression of Opa1 and Mfn1/2 in SD rat aorta vascular smooth muscle cells treated with beta-glycerophosphate to induce calcification [404]. Conversely, in red vastus lateralis muscles of Zücker diabetic fatty rats, melatonin restored ATP production to control levels by downregulating fusion proteins Opa1 and Mfn2 while upregulating fission proteins Drp1 and Fis1 (for additional details on study design and effects, please see Table 3) [405].
Whereas, in a streptozotocin (STZ)-induced diabetic mouse model, daily melatonin supplementation at 10 mg/kg b.w. rescued neural retinal dysfunction and prevented retinal microvascular permeability via Mfn2 upregulation that rebalanced fusion and fission in retinal mitochondria of diabetic mice compared to healthy controls [406]. Notwithstanding, 10 mg/kg was inadequate for normalizing hyperglycemia in STZ diabetic mice. While both 10 mg/kg and 30 mg/kg upregulated the expression of Opa1 and downregulated Drp1 in the hearts of rats subjected to non-lethal mechanical trauma (MT)-induced myocardial injury, only treatment with 30 mg/kg melatonin reduced apoptosis rates by > 50%, compared to < 10% in rats treated with 10 mg/kg melatonin [407]. While H9c2 cardiomyoblasts pretreated with 100 μM melatonin before incubation with 20% traumatic plasma for 12 h were able to restore ∆Ψm loss and maintained ATP production similar to control levels (for additional details on study design and effects, please see Table 3) [407].
Melatonin’s impressive array of additional ATP-elevating mechanisms
Since the early 1990s, melatonin has been demonstrated to utilize an extensive range of distinct mechanisms to increase mitochondrial ATP synthesis in in vivo, in vitro, and in situ experiments that studied different organs, tissues, and cell lines. In addition to modulating mitochondrial dynamics, these diverse mechanisms include elevating the expression of OXPHOS enzymes in skeletal muscles and bone marrow mesenchymal stem cells in a dose-dependent manner [408, 409], and reducing state 4 respiration proton leak and mitochondrial permeability transition pore activity in brown adipose tissue [410]. Melatonin increased the lifespans of both aged senescent and senescent-resistant mice by increasing ATP production and restoring optimal mitochondrial function [411]. Melatonin treatment in septic mice reversed damage to mitochondrial proteins that increased ATP production and normalized ATP/ADP ratios compared to controls [412]. In rat liver cold-storage models, both in vitro [413] and in situ [414] experiments verified that melatonin added to perfusates during reperfusion restores liver morphology and function, mitigating I/R injury by elevating ATP production (please refer to Table 3 for additional details on study designs and effects reported in the aforementioned studies).
Mitochondria from normal mouse liver cells treated with 1 nM to 1 mM melatonin exhibited different levels of activity enhancement in OXPHOS complexes in a dose-specific manner. Melatonin at 1 nM in complex I, 1 nM to 1 μM in complex II, 1 mM in complex III, and 1 mM in complex IV produced 56%, > 31%, 30%, and a staggering 609% increase in activity levels, respectively [415]. Although melatonin treatment during reperfusion in cold-storage livers was unable to completely restore the loss of ATP production to control levels [414], melatonin pretreatment in male Wistar rats subjected to ruthenium red-induced mitochondrial damage not only prevented the reduction in complex I and IV activity in a time-dependent manner, but also increased complex I and IV activities in the liver mitochondria by 33% and 233%, respectively, compared to controls. Male Wistar rats not subjected to ruthenium red toxicity (60 μg/kg, IP) but treated with 10 mg/kg (IP) all showed significant increases of up to 166% in complex I and IV activity levels in the liver and brain mitochondria in a time-dependent manner. Importantly, the additive effect of melatonin on brain and liver mitochondria OXPHOS activity completely vanishes starting at 120 min for complex I in the brain and complex IV in the liver mitochondria (please refer to Table 3 for additional details on study designs and effects reported in the aforementioned studies) [416].
Melatonin regulates sirtuins to elevate ATP synthesis in mitochondria
Sirtuins (SIRTs) are NAD+-dependent class III histone deacetylases that can tune the assembly and disassembly of BCs by acetylation and deacetylation of regions within IDPs [417–419]. A substantial amount of evidence in literature supports a definitive, dependent relationship between melatonin and SIRTs, especially SIRT1 and the mitochondrial SIRT3 [420], in enhancing mitochondrial function and energy homeostasis. In vivo melatonin treatment enhanced mitochondrial function in STZ-induced diabetic rats subjected to myocardial I/R (MI/R) injury. Melatonin reduced MI/R injury by elevating activity levels in complexes II, III, and IV to increase ATP production in an AMPK-PGC-1α-SIRT3 signaling-dependent manner [421]. Oocytes from aging female mice treated with 1 µM melatonin increased mitochondrial ATP production by 54% compared to controls. The addition of melatonin to the terminal culture medium elevated the expression of Sirt1 and Sirt3 to improve oocyte maturation, fertilization, and blastocyst formation [422]. In vitro porcine oocytes supplemented with 500 nM melatonin were able to abolish rotenone-induced mitochondrial dysfunction causing embryo development impairment. Melatonin increased SIRT1 expression that not only promoted mitochondrial biogenesis but also elevated the expression of mitochondrial OXPHOS enzymes complex I and V to return the 50% reduction of ATP production back to 75% of control levels (please refer to Table 3 for additional details on study designs and effects reported in the aforementioned studies) [423].
It is noteworthy that both cancer cells and viruses divert nutrients away from mitochondrial OXPHOS into the oxidative and non-oxidative branches of the PPP to produce metabolites that fuel reductive biosynthesis instead of producing ATP [368, 424]. Consequently, by redirecting carbon flux back to mitochondrial OXPHOS and increasing ATP production, melatonin has been shown to cause cancer cell apoptosis. Lung tumor tissue samples from patients who took melatonin (5 mg/day) for one month showed a 183% increase in SIRT3 expression. Whereas Lewis lung cancer (LLC) mice that were treated with 10 mg/kg b.w. (IP) also showed enhanced expression of SIRT3 that significantly elevated ATP production by more than 320%. In vivo melatonin treatment in LLC mice restored ΔΨm, and increased mitochondrial complex I and IV activity levels by 1.75- and > 14.5-fold, respectively. The redirection of glycolytic intermediates from anabolic pathways to mitochondrial OXPHOS resulted in an impressive 5-fold reduction in tumor size at 16 days of treatment (Table 3) [425].
Melatonin supplementation enhances mitochondrial dynamics and bioenergetics to support and elevate ATP production
| Melatonin dosage/duration | Study design | Results | Ref. |
|---|---|---|---|
| 20 mg/kg b.w. (IP injection) 24 h before cardiac I/R injury | In vivo cardiac I/R injury C57BL6 mouse model | Melatonin reduced cardiac I/R stress and I/R injury biomarkers, supporting myocardial function via enhancing Opa1 expression | [403] |
| 5 μM 12 h prior to HR treatment | In vitro HR injury treatment using C57BL6 ESCs | Melatonin blocked apoptosis, normalized membrane potential and mitochondrial fusion/fission via Opa1, restoring ATP production to control levels. Opa1 knockout abolished all beneficial effects in cardiomyocytes | [403] |
| 5 μM prior to calcification induction; total 14 days duration | In vitro SD rats aorta VSMCs treated with 10 mM β-GP for 14 days to induce calcification | Melatonin treatment decreased β-GP-induced calcification in VSMCs, normalized ΔΨm dissipation, enhanced expression of Opa1, Mfn1/2, protected mitochondrial structural integrity, reduced fragmentation, and maintained ATP production levels similar to controls by enhancing expression of OXPHOS enzymes | [404] |
| Oral 10 mg/kg b.w., daily for 12 weeks | In vivo ZDF rats, RVL muscles | Long-term melatonin supplementation in ZDF rats enhanced oxidative phenotype, elevating ATP production and increasing antioxidant enzymes by upregulating Drp1, Fis1 and downregulating Opa1, Mfn2, compared to controls | [405] |
| 10 mg/kg b.w. daily in water by oral gavage, starting 7 days post STZ injection, for 3 months | In vivo: male C57BL/6J injected with STZ, 100 mg/kg b.w. to induce type 1 diabetes | Melatonin treatment restored STZ-induced inhibition of Mfn2 to rebalance fusion and fission in diabetic retina | [406] |
| 10 mg/kg or 30 mg/kg, IP injection, 5 min after trauma | In vivo: SD rats subjected to non-lethal MT-induced myocardial injury | 30 mg/kg melatonin reduced myocardial apoptosis by 50% by enhancing protein expression of Opa1, Mfn2 and suppressing mitochondrial fragmentation via downregulating Drp1 in the rats subjected to MT | [407] |
| Pretreatment—100 μM/L melatonin for 2 h | In vitro H9c2 cardiomyoblasts cultured in 20% traumatic plasma for 12 h | Melatonin pretreatment restored ∆Ψm and ATP production to control levels by elevating activity of mitochondrial OXPHOS enzymes complex I, II, III, and IV | [407] |
| 0.01 μM, 0.1 μM, and 1 μM (30 min for ATP) | Murine C2C12 myoblasts | Dose-dependent enhancement of ATP production at 8.6%, 30.8%, and 45.6% (0.01 μM, 0.1 μM, and 1 μM, respectively), 1 μM melatonin elevated the gene expression of OXPHOS enzymes Nd4, Sdha, and Atp5a by 262.7%, 85.9%, and 311.3% respectively | [408] |
| 5 mg (0.5% melatonin) and 50 mg (5% melatonin) | In vitro: SD rat BMMSCs treated with melatonin-loaded interconnected electrospun nanofiber three-dimensional scaffold | Dose-dependent elevation of ATP (29.1%, 59.9%) and membrane potential (130%, 200%) for the respective 0.5% and 5% melatonin scaffolds compared to saline controls. 5% melatonin scaffold-treated BMMSCs increased protein expression of OXPHOS enzymes Atp5, Nd4, and Sdha by 90.5%, 120%, and 80.4%, respectively, compared to controls | [409] |
| 10 mg/kg b.w. diluted in drinking water, for 6 weeks | In vivo: mitochondrial functions in interscapular brown adipose tissue of ZDF rats | Enhanced mitochondrial functionality, increasing ATP production by reducing proton leak and mitochondrial permeability transition pore activity, while enhancing antioxidant superoxide dismutase activity | [410] |
| 10 mg/kg b.w. in drinking water from 1–5 months and 1–10 months of age | In vivo: diaphragmatic mitochondria from female senescent prone (SAMP8) and senescent resistant (SAMR1) mice | Melatonin treatment in drinking water for 9 months extended lifespans of all treated subjects and rescued age-dependent mitochondrial dysfunction in SAMP8 mice, increasing ATP production by > 125% and > 70% compared to SAMP8 and SAMR1 vehicle controls, respectively | [411] |
| 30 mg/kg b.w. in 4 separate doses: IP injections-30 min before CLP, 30 min and 4 h after CLP. Subcutaneous injection, 8 h after CLP | C57/BL/6 mice septic mice induced by CLP with iNOS–/– and iNOS+/+ as wild type controls | Melatonin reversed sepsis-induced damage to mitochondrial OXPHOS proteins and normalized ATP/ADP ratio in iNOS+/+ wild type mice. The increase in ATP production was even higher than melatonin-treated iNOS–/– septic mice | [412] |
| 25–200 µM tested for dose-dependent effects. 100 µM added to reperfusion perfusate | In vitro: male Wistar rat liver cold-storage I/R model | Melatonin improved liver function after cold storage and reperfusion, indicated by significant elevations in bile and bilirubin production in a dose-dependent manner. Treatment with 100 µM melatonin during reperfusion increased ATP production by > 88% compared to untreated controls | [413] |
| 100 µM added to the perfusate solution of KHB and glucose | In situ perfusion: male Wistar rat liver cold-storage I/R model | Reperfusion with melatonin rescued the dramatic 7-fold loss of ATP production in cold-storage livers compared to normal controls. However, ATP levels in melatonin treated livers were still 4–5 times lower in comparison to normal livers not subjected to cold storage | [414] |
| 1 nM to 1 mM | In vitro high-resolution respirometry, fluorometry and spectrophotometry study of mitochondria from normal mouse liver cells | Melatonin increased ATP synthesis by improving respiratory efficiency via reduced ΔΨm and oxygen flux; and enhancing activity of OXPHOS complexes at different doses: complex I (56% at 1 nM), complex II (> 31% from 1 nM to 1 μM), complex III (30% at 1 mM), and complex IV with the most significant increases (609% at 1 mM) | [415] |
| 10 mg/kg b.w. IP injection, 10 min before ruthenium treatment | In vivo male Wistar rats with mitochondrial damage induced by ruthenium red (60 μg/kg b.w., IP injection) | Melatonin IP injection increased activity levels of complex I and IV in the liver and brain of both ruthenium red-treated and untreated subjects. Normal subjects displayed increases in activity of 76% within 30 min and 10% within 60 min for complex I in liver and brain, respectively; these effects were diminished completely at 120 min (brain), with a 75% reduction in activity (liver) at 180 min. Activity increases of 150–166% within 30–90 min and 25% within 30 min for complex IV were observed in the liver and brain, respectively. These increases totally vanished at 120 min (liver) and 180 min (brain) after injection | [416] |
| Oral 10 mg/kg b.w./day × 5 before MI/R; IP injection 10 min before the reperfusion | In vivo MI/R injury study using STZ-induced type 1 diabetic rat model (8-week-old male SD rats); in vitro confirmation experiment using H9c2 cardiomyoblasts | Melatonin reduced MI/R injury by reducing oxidative stress and enhancing mitochondrial biogenesis, while elevating activity levels in complexes II, III, and IV to increase ATP production in an AMPK-PGC-1α-SIRT3 signaling-dependent manner | [421] |
| 100 nM, 1 µM, and 100 µM added in terminal culture medium | In vitro fertilization of oocytes using aged mice. Females aged 10–12 months, males aged 8–12 weeks | Melatonin elevated mRNA expression levels of antioxidant-related genes Sirt1, Sirt3, Gpx4, Sod1, and Sod2, improving oocyte maturation, fertilization and blastocyst formation in aged oocytes compared to controls, with 1 µM achieving the best results, increasing ATP production by 56% compared to controls | [422] |
| 500 nM | In vitro porcine oocytes treated with 600 nM rotenone to impair development via mitochondrial deficiency | Melatonin rescued porcine oocytes from rotenone-induced impairment of early embryo development in a receptor-and SIRT1-dependent manner. By enhancing mitochondrial biogenesis, restoring ΔΨm, and elevating protein expression of OXPHOS enzymes complex I and V melatonin rescued 50% ATP depletion by rotenone treatment to 75% of control levels | [423] |
| 10 mg/kg b.w. IP injection every other day (mice)5 mg/day × 30 days (lung cancer patients) | In vivo: LLC mouse model (male C57 mice); human lung cancer patients | Melatonin enhanced the expression of SIRT3 in humans and mice. In LLC mice, melatonin significantly elevated ATP production by > 3.2-fold, restoring mitochondrial function by elevating membrane potential and increasing the activities of OXPHOS complexes I (1.75-fold) and IV (> 14.5-fold), causing a 5-fold reduction in tumor size at 16 days | [425] |
β-GP: beta-glycerophosphate; AMPK: AMP-activated protein kinase; ATP: adenosine triphosphate; Atp5a: ATP synthase F1 subunit alpha; BMMSCs: bone marrow mesenchymal stem cells; b.w.: body weight; CLP: cecal ligation and puncture; Drp1: dynamin-like-protein-1; ΔΨm: mitochondrial membrane potential; ESCs: embryonic stem cells; Fis1: fission 1; Gpx4: glutathione peroxidase 4; HR: hypoxia-reoxygenation; iNOS: inducible nitric oxide synthase; IP: intraperitoneal; I/R: ischemia/reperfusion; KHB: Krebs-Henseleit bicarbonate; LLC: Lewis lung cancer; Mfn: mitofusin; MI/R: myocardial I/R; MT: mechanical trauma; Nd4: NADH dehydrogenase subunit 4; Opa1: optic atrophy 1; OXPHOS: oxidative phosphorylation; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; RVL: red vastus lateralis; SAMP8: senescent accelerated prone 8; SAMR1: senescence-accelerated mouse resistant 1; SD: Sprague-Dawley; Sdha: succinate dehydrogenase complex flavoprotein subunit A; SIRT3: sirtuin 3; Sod1: superoxide dismutase 1; STZ: streptozotocin; VSMCs: vascular smooth muscle cells; ZDF: Zücker diabetic fatty
Conclusions
The unknown long-term health and social-economic impact of Neuro-PASC remains a major concern and potential burden for society in the post-pandemic era. It is believed that more than half the world’s population has been exposed to SARS-CoV-2. Unaddressed, the disproportionate prevalence of Neuro-PASC in younger and middle-aged individuals can critically reduce workforce productivity and innovation. The increased disability and morbidity of individuals in their prime may lead to crises in health care systems, economic downturns, and eventual societal collapse under the worst-case scenario. The formation and aggregation of amyloids as a natural, protective response to viral presence poses a formidable barrier against solutions that directly target the inhibition and disaggregation of amyloids. While the enslavement of mitochondria into organelles that support reductive biosynthesis and the acceleration of heterogeneous nucleation that substantially increases amyloid aggregates compound the difficulty in finding effective, safe remedies for Neuro-PASC. There is an urgent need to clarify the effect of the SARS-CoV-2 virus on mitochondrial function. The reduction in ATP and melatonin synthesis due to mitochondrial dysfunctions further decimates evolutionarily-conserved synergistic mechanisms that provide significant proteostatic cost advantage favored by living organisms in all three major domains of life in the regulation of phase-separated amyloid aggregates. Absent accessible, viable remedies, the thorough examination and exploration of the full range of benefits in the appropriate clinical application of melatonin in Neuro-PASC are duly warranted.
Abbreviations
| AD: | Alzheimer’s disease |
| AMPs: | antimicrobial peptides |
| ATP: | adenosine triphosphate |
| Aβ: | amyloid-beta |
| b.w.: | body weight |
| BCs: | biomolecular condensates |
| CSF: | cerebrospinal fluid |
| dFBN: | dorsal fan-shaped body |
| Drp1: | dynamin-like-protein-1 |
| EEG: | electroencephalogram |
| FBS: | fetal bovine serum |
| FCS: | fetal calf serum |
| FUS: | fused in sarcoma |
| Ga: | billion years ago |
| H: | hydrogen |
| HDM: | high-dose melatonin |
| HSP70: | heat shock protein 70 |
| HSV1: | herpes simplex virus 1 |
| I/R: | ischemia/reperfusion |
| IDPs: | intrinsically disordered proteins |
| IP: | intraperitoneal |
| L. polyedra: | Lingulodinium polyedra |
| LUCA: | last universal common ancestor |
| Mfn: | mitofusin |
| N: | nucleocapsid |
| N2a: | Neuro2A |
| NAD+: | nicotinamide adenine dinucleotide |
| NDDs: | neurodegenerative diseases |
| NNP: | non-hospitalized Neuro-post-acute sequelae of SARS-CoV-2 infection |
| OPA1: | optic atrophy 1 |
| OXPHOS: | oxidative phosphorylation |
| PASC: | post-acute sequelae of SARS-CoV-2 infection |
| PD: | Parkinson’s disease |
| PNP: | post-hospitalization Neuro-post-acute sequelae of SARS-CoV-2 infection |
| PPP: | pentose phosphate pathway |
| S: | spike |
| SIRTs: | sirtuins |
| STZ: | streptozotocin |
| TCA: | tricarboxylic acid |
| TDP-43: | TAR DNA-binding protein 43 |
| α-syn: | α-synuclein |
| ΔΨm: | mitochondrial membrane potential |
Declarations
Acknowledgments
Special thanks to Daniel Matrone for technical assistance. Figures 1 and 2 created in https://BioRender.com.
Author contributions
DL: Conceptualization, Writing—original draft, Writing—review & editing, Visualization. RJR: Writing—review & editing. Both authors read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2025.
Publisher’s note
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.