
Abstract
Exposure to stressful conditions plays a critical role in brain processes, including neural plasticity, synaptic transmission, and cognitive functions. Since memory-related brain regions, the hippocampus (Hip), the amygdala, and the prefrontal cortex, express high glucocorticoid receptors (GRs), these areas are the potential targets of stress hormones. Stress affects memory encoding, consolidation, and retrieval, which may depend on many factors such as the type, duration, the intensity of the stressor or the brain region. Here, this review mainly focused on the mechanisms involved in stress-induced memory impairment. Acute/chronic stress induces structural and functional changes in neurons and glial cells. Dendritic arborization, reduction of dendritic spine density, and alteration in glutamatergic-mediated synaptic transmission via N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors are mechanisms that stress affect long-term memory formation. Exposure to acute or chronic stress could interplay with multiple neurotransmitter signaling, modulating the neuronal circuits involved in memory impairment or state-dependent learning. Stress hormones also modulate the expression of microRNAs in the specific brain regions responsible for stress-induced behaviors. Because of expressing GRs in astrocytes and microglial cells, stress could affect the morphology, structure, and functions of these glial cells in memory-related brain regions. Astrocytes play a crucial role in stress-induced aversive or fear memory formation. Over-activation of the microglial cells enhances the release of inflammatory cytokines, which results in neuronal injury. Stress has a prominent role in cognitive decline to induces memory problems, particularly in older adults. Due to the issue’s importance, here the provided overview attempted to address the question of how stress alters neuronal epigenetic regulators, synaptic transmissions, and glial activity in the brain.
Keywords
Stress, memory impairment, neurotransmitters, astrocytes, microglia, microRNAsIntroduction
Stress is an adaptive neurobiological response that enables the body to adjust to internal and external environmental challenges [1]. Stress-responsive system is a complex adaptive system that seems to be highly conserved across species [2]. It is well established that two different systems but interconnected, including the hypothalamic-pituitary-adrenocortical (HPA) and the sympathetic-adrenomedullary (SAM), have significant roles in the coordination of both physical and psychological responses to stress situations [3]. The SAM system is a component of the sympathetic division of the autonomic nervous system. It is responsible for the first phase of stress response which leads to a short-term reaction to provide appropriate responses [4]. Following the activation of the SAM system, the adrenal gland center releases epinephrine (adrenaline) into the blood circulation. Then, the increased circulating epinephrine facilitates a rapid mobilization of metabolic resources to induce fight/flight fast responses [5, 6]. Exposure to a stressor increases the secretion of corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) from the paraventricular nucleus (PVN) of the hypothalamus to stimulate the anterior pituitary gland to secrete adrenocorticotropic hormone (ACTH) into the blood circulation [7, 8]. The ACTH, in turn, induces glucocorticoid synthesis and release from the cortex of adrenal glands, which are cortisol in humans and corticosterone in rodents [9]. Evidence suggests that endogenous synthesis of corticosteroids is not limited to the adrenal cortex. Using messenger RNA (mRNA) and immunogold electron microscopic analysis, it has been shown that the hippocampal neurons express the specific enzymes, including P450 (c21), P450 (2D4), P450 (11β1), and 3β-hydroxysteroid dehydrogenase to produce a low nanomolar level of corticosterone in rats [10].
The glucocorticoids are responsible for long-term responses and help the body adapt to stress. Circulating glucocorticoids seems necessary for brain development, neuronal survival and neurogenesis, psychophysiological adaptation to stress conditions, and adaptive immune responses [11, 12]. The secretion of glucocorticoids from the HPA axis is tightly regulated by endocrine and neuronal systems [13]. The rate of glucocorticoid release and the level of glucocorticoid release could determine ‘fast’ and ‘delayed’ negative feedback regulation, respectively, to protect the body from prolonged activities [14]. Glucocorticoid negative feedback may be mediated at multiple levels via glucocorticoid receptors (GRs) located in the hypothalamus and the anterior pituitary gland. The HPA axis is also modulated through a glucocorticoid-independent neuronal mechanism [13]. Both direct and indirect pathways have a central role in controlling the end product of the HPA axis (Figure 1) [15].
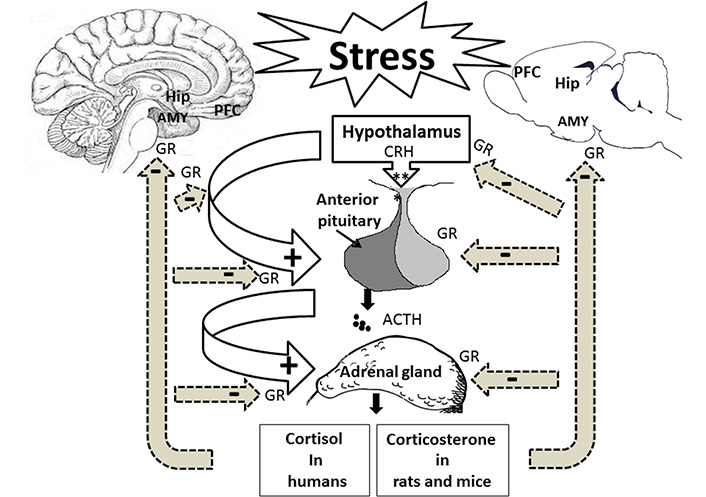
Stress affects the hippocampus (Hip), the amygdala (AMY), the prefrontal cortex (PFC), and the HPA axis. Following stress exposure, the hypothalamus is activated to release CRH into the bloodstream to stimulate the pituitary gland for producing ACTH. The bloodstream delivers ACTH to the adrenal glands to release cortisol in humans and corticosterone in rodents. Stress hormones via GRs affect memory-related brain regions. +: stimulation; –: inhibition
The limbic brain structures, including the Hip, the AMY, and the PFC, control the PVN activity via the glutamatergic and gamma aminobutyric acidergic (GABAergic) innervations [16]. Glutamatergic projections of the Hip and the PFC indirectly inhibit the PVN neurons in the hypothalamus. Thus, they play an essential role in terminating the HPA axis-induced stress response and have a negative feedback modulation on this axis [17, 18]. Neuroimaging studies have shown that the decrease in the anterior cingulate cortex volume could be associated with the dysregulation of HPA activity [19, 20]. Electrical stimulation of the Hip decreased glucocorticoid secretion [21], whereas the lesion of this brain region caused the stress response [22]. In contrast, the AMY GABAergic projection disinhibits the hypothalamic PVN neurons resulting in positive regulation of the HPA activity [23]. AMY activation is associated with stress-related behaviors in an emotional state, such as fear [24].
Corticosteroid hormones affect neurons and glia through two intracellular receptors: type 1 mineralocorticoid (MRs) and type 2 GRs. They are members of the nuclear receptor superfamily of ligand-dependent transcription factors [25]. Since MRs have a higher affinity to glucocorticoids than GRs, it is likely that type 1 receptors are primarily occupied in basal/non-stressful conditions to maintain stress responses. On the other hand, the activation of GRs through a high level of glucocorticoids following stress exposure is associated with the neuroendocrine stress response to restore homeostasis by a negative feedback loop [26]. The different distribution of GRs and MRs in the brain regions, including the hypothalamus [27], the PFC [28], the Hip [11], and the AMY [29] have directly been targeted by psychogenic and physical stressors. Interestingly, stress exposure alters mRNA expressions of both MRs and GRs in the brain regions, including the Hip and the AMY. The exposure to single prolonged stress (SPS) caused an early decrease in GR and MR mRNAs and protein expressions [29]. Evidence suggests that both acute and chronic stress could alter the expression levels of GR in the PFC, the Hip, and the hypothalamus to modulate GR phosphorylation [30]. Hence, these brain regions may be targeted directly by psychogenic and physical stressors.
Exposure to stressful conditions has a significant influence on learning and memory processes. Memory formation is associated with neuronal synaptic changes [31], significantly strengthening existing synapses by increasing the number and size of the dendritic spine with concomitant changes in synaptic markers. Although the general outcome of stress is memory impairment [32], some studies have reported the neutral [33] or even the facilitative role of stress on memory formation [34]. It seems that depending on the type, intensity, and duration of stressors, the stress has a variety of effects on memory formation. In this review, the main focus was on the mechanisms involved in stress-induced memory impairment.
Effects of stress on memory
Stress impairs memory formation
The end product of neuroendocrine stress cascades, glucocorticoids, plays a critical role in brain processes, including neural plasticity, synaptic transmission, and cognitive functions such as learning and memory. Glucocorticoids exert their cellular effects by acting on MRs, and GRs in the brain regions, which are essential for memory performance [35, 36]. The elevation of cortisol under a stressful situation or exogenous glucocorticoid administration affects memory consolidation. The exposure to the acute stressful stimulus affected the consolidation of object recognition task memory in mice and resulted in long-term memory impairment without any adverse effect on short-term memory. Blocking cannabinoid receptors (CBs) of adrenergic neurons improved stress-induced memory loss [37], indicating the role of the cannabinoid system in the modulation of cognitive function. In line with these findings, human research has shown that acute stress in pre-encoding information enhances the recall of emotional memories [38]. Moreover, an investigation has revealed an inverted U-shape curve relation between stress intensity and memory acquisition and reconsolidation [39]. On the other hand, memory impairment is dependent on cortisol level, for example, exposure to acute stress before long-term retrieval leads to loss of memory recall at the lowest cortisol level. In contrast, a concurrent rise in cortisol levels enhanced long-term memory [40]. Long-term memory was negatively affected by acute stress during memory consolidation and before memory reactivation. In a study conducted by Sardari et al. [32], it has been shown that exposure to inescapable stress before the retention trial of a passive avoidance task induces memory retrieval impairment. Furthermore, post-training exposure to stress also decreased memory retrieval on the test day [34, 41]. Interestingly, before being exposed to the retention stage, the same stressful situation completely reversed stress-induced memory impairment and showed a typical memory performance [34]. This result indicated that recalling a memory under stress is state-dependent, and memory enhancement occurs in a similar stressful state that happens during the consolidation of information.
Stress-induced robust changes in structural and functional neuronal plasticity [42] eventually altered glutamatergic-mediated synaptic transmission via N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors, which are essential components in synaptic plasticity, therefore, they could affect memory storage [43]. Experiments prove that stress changes dendritic arborization and alters dendritic spine density in different brain regions. Chronic stress results in spine density loss and retraction of apical dendritic branches in the hippocampal CA1 and CA3 regions [44]. This effect was reversed by the CRH receptor type 1 (CRHR1) antagonist [44, 45]. The activation of NMDA receptor-mediated CRH-induced dendritic spine loss through calpain activation was shown previously [46]. Furthermore, the PFC, a cortical area important for working memory, executive functioning, and goal-directed behaviors, has a most vulnerable dendritic domain when interacting with stressful events. Moreover, acute stress potentiates glutamatergic transmission in the PFC [47], facilitating working memory through glucocorticoid-inducible kinase signaling [48]. In contrast, chronic stress causes the impairment of prefrontal-dependent memory formation [49] by acting on suppressing glutamatergic transmission. The effects of stress on dendritic remodeling are region-specific. Inactivation of the basolateral AMY (BLA) before chronic stressors prevents volume reduction of the PFC [50].
Stress produces state-dependent learning
State-dependent learning refers to a special kind of learning in which the retrieval of an event requires the animal to be in a similar state of acquired information [51]. Our previous study has indicated that exposure to acute elevated platform stress has an amnesic effect on memory consolidation and retrieval in the passive avoidance task. Our results also indicated that pre-test exposure to stress reversed the post-training stress-induced memory impairment. Since the information retrieval was performed if the animal was in the same stress state during the encoding phase, our data indicated that acute stress produces state-dependent memory retrieval (Figure 2) [34]. An autoradiographic study also showed that re-exposure to acute restraint stress enhanced the AMY NMDA receptor activity [52]. Intra-BLA microinjection of a selective antagonist of 5-hydroxytryptamine 1A receptor (5-HT1A) receptors inhibited state-dependent learning under stress in rats. Several mechanisms seem involved in stress-induced state-dependent learning, and further studies should figure out the mechanisms.
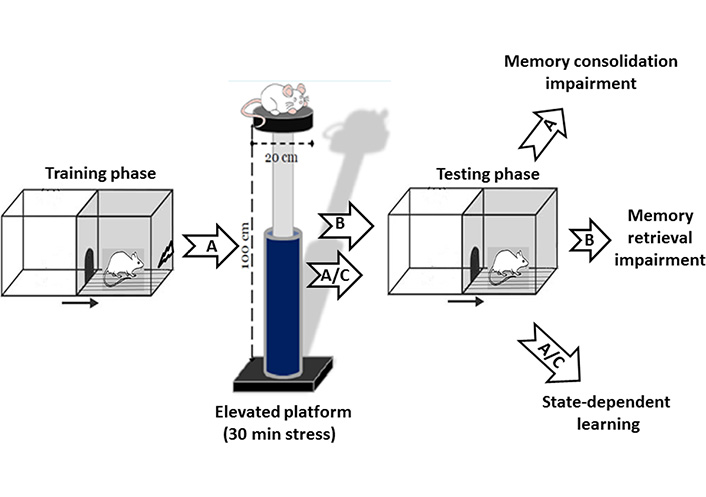
Effect of stress on passive avoidance memory. A: Post-training exposure to acute elevated platform stress (30 min) impaired memory consolidation in rats. B: Pre-test exposure to the same acute stress-induced memory retrieval impairment. A/C: The memory impairment induced by post-training exposure to stress was restored in the animals that received same pre-test acute stress exposure named stress-induced state-dependent memory retrieval
Stress affects long-term potentiation and long-term depression
The activation of the HPA axis and SAM system following exposure to a stressful event could modulate cognitive and emotional memory through changes in synaptic strength [53]. Some studies reported that stress induces alterations in synaptic plasticity through cellular mechanisms, including long-term potentiation (LTP) and long-term depression (LTD). Electrophysiological recordings were implicit that stress has a distinct effect on the efficacy of glutamatergic synaptic transmission in the Hip, the PFC, and the AMY [54]. For example, high glucocorticoid levels induced NMDA-dependent LTP impairment in the CA1 region and did not affect the dentate gyrus (DG). Acute stress attenuated LTP formation in the dorsal Hip, while it enhanced LTP in the ventral Hip [55–57]. Acute stress impaired mGlu3-LTD in the specific synapse between the BLA and the PFC, which may be correlated with the cognitive dysfunction of the PFC during stress-related disorders [58]. Unpredictable shock stress has been shown to have no significant effect on spine morphology but could enhance synaptic plasticity in principal the BLA neurons [59]. A CB1 receptor antagonist could recover chronic stress-induced LTP impairment, furthermore, repeated stress could alter muscarinic LTP in the hippocampal slices [60]. Moreover, chronic stress concurrent with exercise could improve the dorsal hippocampal LTP associated with an increased expression of BDNF to enhance memory [61].
The neuro-cognitive processes contain multicomponent stages, including encoding, consolidation, and retrieval. The most intensive studies showed that stress might affect all three specific phases involved in memory formation and directly is dependent on many factors such as the stressor’s timing, which in turn, cause improvement or impairment of memory performance [35]. The animal examination has indicated that elevation of cortisol under stressful situations or exogenous glucocorticoid administration affects memory consolidation. The exposure to the acute stressful stimulus affected the consolidation of object recognition memory in mice and resulted in long-term memory impairment without any adverse effect on short-term memory. Interestingly stress-induced memory loss is reversed by blocking CBs of adrenergic neurons [37], indicating the role of the cannabinoid system in the modulation of cognitive function. In line with these findings, human research has shown that acute stress in pre-encoding information enhances the recall of emotional memories [38]. An inverted U-shape curve relation exists between stress intensity and memory acquisition and reconsolidation [39]. Memory impairment may depend on cortisol level. For example, the exposure to acute stress before the long-term retrieval led to the loss of memory recall in the lowest cortisol level, while long-term memory was enhanced with a concurrent rise in cortisol level [40].
Long-term memory was negatively affected by acute stress during memory consolidation and before memory reactivation. Sardari et al. [32] showed that exposure to inescapable stress before the retention trial of a passive avoidance task induces memory retrieval impairment. Furthermore, post-training exposure to stress decreased memory retrieval on the test day [34, 41]. The same stress situation just before exposure to the retention stage completely reversed stress-induced memory impairment [34]. These findings indicated that memory recall is state-dependent, and memory enhancement occurs in a similar stressful state during the consolidation of information.
Neurotransmitters are involved in the effects of stress on memory
Different stressors affect various neurotransmitter systems’ molecular and cellular functions, including GABAergic, cholinergic, glutamatergic, serotonergic, dopaminergic, and endocannabinoid system (ECS). The changes in neurotransmitters’ release and synaptic concentrations are associated with the type of stress and brain regions [62]. A large body of evidence considers the effects of stress on neurotransmitter systems in learning and memory processes. Here, we provide an overview of how stress-induced neurotransmission changes may have happened during the encoding and retaining of new information in memory-related brain regions.
GABAergic system
GABAergic dysfunction leads to neurological and mental diseases [63, 64]. Gamma-aminobutyric acid (GABA) receptors, including GABA subtype A (GABAA), GABA subtype B (GABAB), and GABA subtype C (GABAC), are ionotropic receptors that control and manage cognitive functions broadly expressed in the central nervous system (CNS). Metabotropic GABAB receptors bind to G protein inhibitors (Gi/o) to mediate prolonged slow inhibitory action. Both pre- and post-synaptic GABAA and GABAB receptors inhibit the excitatory and inhibitory neuronal functions. Hence, activation of these receptors modulates LTP in the different brain regions [65, 66]. Stressful conditions alter the GABAergic transmission to change and adapt emotional and behavioral responses. For example, exposure to new environmental stressors or swimming stress increased GABA release in the Hip of mice [67]. Acute restraint stress increased GABA efflux in the BLA. In contrast, the same type of stress did not affect the central AMY efflux using an in vivo microdialysis technique [68].
The GABAergic system modulates the HPA axis mainly through GABAA receptors. In normal conditions, CRH neurons of the PVN are under tonic inhibition of GABA transmission. Stressful situations enhance the release of GABA and activation of AMY GABAA receptors, eliminating the tonic inhibition of GABA to increase the release rate of stress hormones [69]. Our previous results showed that inhibiting GABAA receptors in the BLA via microinjection of muscimol, a GABAA receptor agonist, increased the response of ineffective acute stress to impair memory retrieval in a passive avoidance paradigm [32]. Interestingly, prenatal stress decreased the hippocampal spine densities and impaired spatial memory formation in adult offspring by enhancing the GABA transmission in the brain’s developmental stage [70]. Therefore, GABA receptors may play a critical role in regulating the activity of the HPA axis to mediate memory formation under stress.
Dopaminergic system
Dopamine-induced effects are mediated via G-protein coupled receptors classified into two subclasses: D1- and D2-like receptors. Stimulation of dopaminergic D1 or D2 receptors enhances or inhibits the adenylyl cyclase activity to change cyclic adenosine monophosphate (cAMP) concentration, respectively [71]. Dopamine receptors are highly expressed in different brain areas, including the ventral tegmental area (VTA), the nucleus accumbens (NAc), the substantia nigra, the Hip, and the AMY [72]. Dopamine receptors are involved in the formation of different types of learning and memory, such as passive avoidance learning [73], spatial memory [74], and reward-related learning and memory [75].
Exposure to acute or chronic stress increases dopamine release in the NAc, the VTA, and the PFC [76]. In general, acute stress increases the amount of dopamine in the brain to increase the risk of an adult’s desire to use addictive substances [77]. Blocking the PFC D1 receptors attenuated the conditioned place preference (CPP) induced by immobility stress in rats [78]. Additionally, inhibition of the PFC D1 receptors reversed the stress-induced working memory impairment [79]. There is increasing concern that the activation of these receptors may reduce the glutamate release and thereby induces the reduction of neuronal firing rate in the PFC [80]. Systemic administration of sulpiride, a selective D2 receptor antagonist, attenuated glucocorticoid-induced memory retrieval impairment [81]. In contrast, our previous study also showed that inactivation of the BLA D1 or D2 receptors inhibited the improving effects of nicotine on stress-induced memory retention impairment (Figure 3) [82]. These findings suggest that the dopamine receptors alone or in cooperation with other neurotransmitters modulate the impressive effects of stress hormones on memory formation.
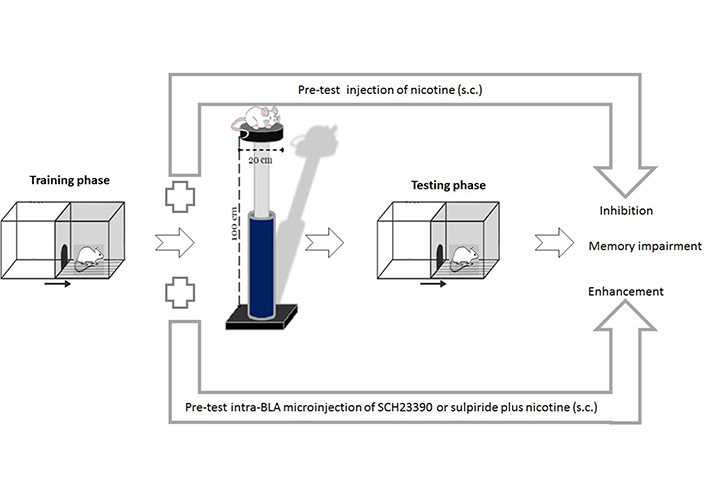
Effects of pre-test intra-BLA microinjections of D1 or D2 receptors antagonists on nicotine-induced improvement of memory retrieval impairment under stress. Pre-test exposure to acute elevated platform stress (30 min) impaired memory retrieval in rats. Nicotine administration improved stress-induced memory retrieval impairment. Pre-test intra-BLA microinjection of SCH23390 (a selective dopamine D1 receptor antagonist) or sulpiride (a selective dopamine D2 receptor antagonist) inhibited nicotine-induced improvement of the stress amnesic effect. s.c.: subcutaneous
Serotonergic system
Serotonin is mainly produced in the raphe nuclei of the brain stem. There are seven types of serotonergic receptors. Based on the chemical structure of serotonin, these receptors are called 5-hydroxytryptamine (5-HT). The serotonergic receptors are a combination of metabotropic and ionotropic receptors that have excitatory and inhibitory roles in the CNS [83]. The 5-HT receptors regulate the release of different neurotransmitters, including glutamate, GABA, and acetylcholine. The serotonergic heteroreceptors are therapeutic targets for treating depression, Alzheimer’s diseases (AD), and Parkinson’s diseases [84]. DNA microarray analysis was used to show that memory formation increases the differential gene expression of 5-HT receptors. For example, there is an enhancement of gene expression of 5-HT1A-1F, 5-HT2A, and 5-HT5A receptors during the passive avoidance memory formation. By contrast, spatial memory requires the gene expression of 5-HT2C, 5-HT3A, and 5-HT6 receptors in the Morris water maze [85].
Evidence suggests that exposure to chronic stress leads to depression and anxiety disorders [86, 87]. Chronic foot shock stress caused depression, which was reversed by injecting antidepressants and increasing serotonin levels in the mice’s brains [88]. Antidepressants attenuate stress-related mood disorders and efficiently improve stress-induced amnesia. Recently, Léa Blondelle et al. [89] showed that exposure to mild unpredictable stress impaired spatial memory formation while using Bombax costatum as an antidepressant attenuated the amnesic effect of stress. Since serotonin plays a role in chronic stress-induced cognitive dysfunction, Natarajan and coworkers [90] found that exposure to chronic stress caused cell death in the interfascicular nucleus of the dorsal raphe, which results in decreased serotonergic innervation of the medial PFC (mPFC). However, they showed that the treatment with MK801, a competitive NMDA receptor antagonist, blocked stress-induced deficits in memory recall. Thus, one may suggest that chronic stress may increase glutamate release, which results in serotonergic neuronal injury in raphe nuclei [90]. Exposure to chronic stress enhanced the AMY sensitivity to serotonin, and the blockade of 5-HT2C receptors attenuated the stress-related fear memory in mice [91]. Inactivation of the ventral hippocampal 5-HT7 receptors also reversed the stress-induced freezing behavior and fear memory formation [92]. Our previous study showed that intra-BLA microinjection of (S)-WAY-100135, a selective antagonist of 5-HT1A receptors, prevented the impairing effect of stress on memory consolidation and retrieval in the passive avoidance learning task [34].
ECS
The ECS comprises a neuromodulatory system to mediate synaptic plasticity and neurogenesis, which are essential for memory formation. Anandamide and 2-arachidonoylglycerol (2-AG) as arachidonic acid (AA) derivatives bind to the CBs, which are densely expressed throughout our brains and bodies. CBs are metabotropic receptors that bind to inhibitory G-protein, and activating these receptors inhibits neuronal adenyl cyclase activity. Three types of CBs have been identified in the brain known as CB1, CB2, and G protein-coupled receptor 55 (GRP55). CB1 receptors are abundantly expressed in the CNS, including the AMY and the Hip. They are mainly responsible for mediating the effects of endocannabinoids in the brain [93]. Due to the high expression of CB1 receptors on presynaptic terminals, these receptors act as neuromodulators to inhibit neurotransmitter release [94]. Generally, activation of CB1 receptors leads to the impairment of hippocampal-related memory [95, 96]. Consumption of marijuana impairs the acquisition, consolidation, and retrieval of memory formation in humans [97]. On the other hand, activating the BLA CB1 receptors facilitates fear memory formation [98]. Cannabinoids have facilitating or inhibitory effects on memory formation based on the type of memory, brain regions, and memory stages [99, 100]. Notably, under pathological conditions, activation of the CBs has neuroprotective effects [101]. For instants, the activation of the CB1 receptor improved memory formation in AD animal models with memory deficits and cognitive disorders [102, 103]. Regarding stress-related disorders, it seems that cannabinoids improve stress-induced anxiety [99, 104]. Since chronic stress impairs cognitive function, the ECS may have a modulatory role in glucocorticoid-mediated outcomes [105]. Exposure to acute stress after memory consolidation leads to impairment of long-term object recognition. Intra-hippocampal administration of a CB antagonist prevented memory deficit [37]. Moreover, following footshock stress, activation of CBs abolished memory loss and facilitated LTP in the Hip [106]. Enhancement of cannabinoid signaling through local administration of a CB1/CB2 receptors agonist, WIN 55,212-2, into the dorsal striatum increased memory consolidation in a passive avoidance task. Moreover, central or peripheral blockade of CB1 receptor signaling eliminated the effect of acute stress-induced memory enhancement [107]. Collectively, these findings demonstrated that cannabinoid signaling pathways might serve as a potential therapeutic target to regulate glucocorticoid-mediated stress memory performance.
microRNAs mediate memory formation under stress
In 2001, the presence of microRNAs (miRNAs, small endogenous RNAs) in the mammalian system was reported for the first time. Currently, it is well established that these non-coding RNAs are involved in the various fundamental biological processes, including development concerning their role in fine-tuning gene expression based on post-transcriptional and translational regulation [108, 109]. Therefore, the function of miRNAs might correspond to the onset and pathophysiology of various neurobiological diseases [110].
More recently, the alternative expression levels of miRNAs in the brain regions were suggested to be associated with the behavioral stress response. In a study conducted by Volk et al. [111], it has been shown that chronic stress increases the level of miR-15a in the AMY, which in turn decreases FK506-binding protein 51 (FKBP51) as a specific transcription factor for GR activity. Therefore, one may suggest that miR-15a is essential for stress adaptation in chronic stress. Another experiment implicated that foot shock stress could upregulate miR-34 in the adolescent rat hypothalamus and correlated with the decrease of CRF receptor type 1 (CRFR1) expression as a validated target for miR-34 [112, 113]. Moreover, the upregulation of miR-34 inhibits dendritic spine growth, which is associated with memory loss [114]. Furthermore, anxiety-like behavior in response to acute stress was reduced in miR-34 knockout mice. It seems that a lack of miR-34, a critical modulator of the stress response, could protect from the aversive stress effect [115].
Other evidence supports that exposure to early life stress induces a change in the expression of clusters of miRNAs which contribute to memory impairment. Liu et al. [116] determined that spatial memory was impaired in response to early stress, and the expression patterns of miR-135a and miR-16 were changed in rats. They reported that the miR-135a level was reduced in the PFC, while miR-16 was upregulated in the rat Hip [116]. Both miRNAs have been associated with the regulation of serotonin levels in the synaptic cleft. For instance, the downregulation of miR-135 leads to declining serotonin levels and corresponds to stress-induced behavior [117]. Moreover, acute stress downregulates brain-specific miR-135a and miR-124 in the mice AMY and positively increases the expression of the protein level of the GR (MR), therefore acts as a mediator of stress response in the AMY [118].
Recent studies revealed that early transient changes of the miRNA hippocampal expressions without concomitant alteration on the AMY miRNAs are necessary for Hip-dependent fear memory. Fear memory is closely related to stress and affects the brain structure related to learning and memory processes. Moreover, early up-regulation of miR-181a [119] and miR-151 [120] in the dorsal Hip induces long-term hippocampal memory through down-regulation of hippocampal protein levels of protein kinase AMP-activated catalytic subunit alpha 1 (PRKAA1) and anterior pharynx defective 1a (APH1a) receptively following fear conditioning. Moreover, an early decrease in the expression of miR-187-3p was observed in the dorsal Hip following a contextual fear-conditioning paradigm in mice. Interestingly, the downregulation of miR-187-3p is associated with the decrease of stabilin-2 (stab2) protein level [121]. It seems that stress exposure with activating the HPA axis induces modification on neuronal epigenetic regulators to change some hippocampal miRNA-dependent pathways for regulating memory formation. Thus, miRNA expression changes are potentially being used as a novel target for memory dysfunction.
Stress changes the number and structure of astrocytes
Astrocytes are the most abundant glial cells in the CNS. They contribute to the blood-brain barrier formation [122], neurotrophin secretion [123], neurotransmitter recycling [124], and regulation of neuronal synaptogenesis [125]. Astrocytes participate in information processing, neuronal plasticity, and LTP via releasing the neuroactive compounds known as gliotransmitters. Astrocytes release D-serine as an endogenous co-agonist of NMDA receptors to enhance the occurrence of NMDA-dependent LTP in nearby excitatory synapses [126]. Additionally, the activation of the astrocytic cAMP enhances the lactate shuttle from astrocytes to neurons, serving as energy for synaptic plasticity and memory formation processes [127]. Interestingly, astrocytes undergo structural plasticity during memory formation. Ostroff and co-workers [128] have shown that astrocytes of the lateral AMY play a critical role in the morphological remodeling of the synapses during implicit memory consolidation. Hence, astrocytes are involved in forming new memories and induction of LTP, and any structural and functional changes will affect the memory formation processes.
Like neurons and microglia cells, the astrocytes express the GRs and MRs [129]. With the elevation of the stress hormones and extracellular glutamate after HPA axis activation, astrocyte is one of the critical cells that respond to stress’s physiological consequences and are influenced structurally and functionally by stress conditions. Stress induces the hypertrophy of astrocytes and a reduction in gap junction coupling between cells in the Hip and the neocortex. The latter effects reduce functional coupling between hippocampal and neocortex astrocytes which is associated with the attenuation of astrocyte’s capacity to supply neurons with L-lactate. Moreover, the disruption of intracellular networks between the astrocytes is associated with decreased hippocampal LTP and spatial memory impairment [130]. Exposure to chronic stress also reduces the volume fraction of fine astrocytic protrusions and the number of astrocytes in the AMY, which may affect AMY-related memory formation [131]. Astrocytes’ specific elimination of GRs in mice results in impairment of fear memory and aversive memory formation, which suggests that the signaling of the astrocytic GRs regulates stress-induced-aversive memory formation [132]. In general, it can be suggested that astrocytes should be considered a cellular target for therapies for stress-induced memory impairment.
Stress-induced significant changes in microglia may be involved in memory impairment
Exposure to stressful conditions and activation of signaling pathways of stress receptors leads to the inhibition of the production of proinflammatory cytokines such as interleukin-1β (IL-1β), IL-18, IL-6, or tumor necrosis factor-α (TNF-α) in the CNS [133]. However, the immune system function disrupts and leads to the overactivation of inflammatory pathways under severely stressful conditions. Microglia cells, the CNS macrophage cells, express the stress hormone receptors, including the glucocorticoid (GC), MR, and norepinephrine (NE) receptors. Therefore, these cells are one kind of the key cells in regulating stress-associated outcomes [134, 135]. Severe and intense stress over-activates the microglia cells and thereby enhances the release of inflammatory cytokines, leading to neuroinflammation. The morphology of microglial cells changes due to stress, and they show fewer branches and an enlarged soma called ameboid microglia [136]. Besides, as a danger signal, stress triggers the formation and activation of microglia inflammasomes. The inflammasomes are intracellular protein structures that involve in the formation of proinflammatory cytokines in cells and play a critical role in innate immunity [137]. The perturbations in neuroimmune function can lead to impaired function of neuronal synapses and deficits of synaptic plasticity that underlie cognitive dysfunction.
Microglia cells have a pivotal role in memory formation processes by monitoring the neuronal microenvironment, controlling neuronal activity, and neurotransmitter release, maintenance of dendritic spine densities, and mediating the forgetting of remote memories [138]. Because of their role as synaptic sensors that control synaptic development and function, these cells are considered the fourth component of the “quad-partite synapse” in addition to the pre-and postsynaptic terminals and the astrocytes [139]. Previous studies have confirmed that stress could affect memory formation via microglia cells. For example, exposure to chronic restraint stress increases the expression of hippocampal microglia inflammasomes such as Nod-like receptor protein 3 (NLRP3), leading to neuronal injury and memory impairment [140]. The hippocampal expression of NLRP3, IL-1β, and IL-18 was increased, followed by cognitive impairment in socially isolated mice [141]. The microglia cells affect the neurogenesis and maturation of neuronal synaptic in the brain of a mouse model of early life stress. Exposure to stressors dysregulates the microglia function, negatively affecting the neurogenesis and neuronal functions, and may have long-lasting consequences over the lifespan [142].
Memory impairments associated with stress-related disorders
A human feels stress from time to time (in everyday life). Among the most potent stressors are psychological and psychosocial stressors, which have unhealthy consequences and adverse effects on the mind and body [143]. Although normal activation of the HPA axis is required for stress response adaption, excessive activation of the HPA axis seems to have detrimental outcomes and is a risk factor for predisposition to several diseases [144, 145]. There is an unanswered question about how stress may affect an individual’s health. Evidence implicated that stress-related neurological responses are closely linked to anxiety disorders such as post-traumatic stress disorders (PTSD) and mood disorders, including major depressive disorder (MDD). Moreover, stress increases the risk of psychiatric disorders such as bipolar and schizophrenia [146, 147]. Besides the negative impact of stress on an individual’s mental illness, long-term exposure to stressors is associated with cognitive dysfunction. It has a prominent role in cognitive decline, leading to amnestic mild cognitive impairment [148] to induce memory problems, particularly in older adults [149].
Glucocorticoids, as end products of stress-related responses, play a critical role in memory formation via binding to their specific receptors [150–152]. Stress hormones interplay between multiple neurotransmitter signaling pathways in the various brain areas. These interactions play a modulatory role in stress circuits, mainly neural circuitry involved in memory performance [152]. It should be noted that the ECS has a modulatory role in glucocorticoid-mediated outcomes to impair cognitive functions during chronic stress [105]. Several observations demonstrated the enhanced endocannabinoid signaling pathways via inhibiting fatty acid amide hydrolase (FAAH), an enzyme responsible for endocannabinoid degradation, to reverse the negative impact on the chronic stress-exposed animals in an object recognition task [153]. Acute stress following memory consolidation led to long-term object recognition memory impairment, while intra-hippocampal microinjection of CB antagonists prevented the memory deficit [37]. The activation of CBs improved memory loss by facilitating the hippocampal LTP [106]. Pre-test intra-dorsal striatal microinjection of WIN 55,212-2, an agonist of CBs, increased memory formation in the passive avoidance task [107]. This study indicated that the central or peripheral blockade of CB1 receptors eliminated the effect of acute stress on passive avoidance memory. These findings suggest that the ECS may be a potential therapeutic target to regulate glucocorticoid-mediated stress memory performance. Furthermore, activating the hippocampal cholinergic system via acetylcholine nicotine receptor signaling pathways is critical for preventing cognitive decline [154]. Keshavarzian et al. [82] reported that pre-test nicotine administration improved acute stress-induced memory impairment. They also showed that the BLA activation of dopamine receptors before memory consolidation reversed the impairing effect of stress on memory retention. The interaction between stress hormones and the dopaminergic system can be suggested to regulate memory formation [155, 156].
Approximately 30% of stroke patients develop long-term memory impairment within one year of onset [157]. Notably, after AD, vascular dementia is the leading cause of dementia in the world that occurs due to insufficient and impaired blood flow in the brain [158]. On the other hand, stroke is a physiological stressor that activates the HPA axis. Chronic activation of the axis exacerbates stroke outcomes [159]. Clinical studies have shown that chronic stroke patients’ psychological symptoms, such as anxiety or depression, correlate with cognitive dysfunctions [160]. Animal studies have also shown that pre-stroke exposure to psychological stress increases infarct volume and neurological deficits and has detrimental effects on cognitive function [161]. Activation of the HPA axis during the stroke affects the lesion area (mainly the cerebral cortex and striatum) and the contralateral side of the lesion area. Activation of the hippocampal GC receptors in the contralateral side of the ischemic brain leads to hippocampal neuronal damage, which is accomplished by cognitive and psychiatric disturbances that include delayed consequences of stroke [162]. Thus, it can be suggested that targeting stress hormones and their receptors may be a therapeutic approach to ischemia-related cognitive impairments.
Conclusions
Stress regulates multiple CNS functions, including memory formation, mood, emotional behaviors, and reward. Evidence suggests that acute and chronic stress change synaptic transmission, neuronal epigenetic modulators, and glial activity in memory-related brain regions. It is important to note that stress develops neurodegenerative disorders with cognitive dysfunctions. Multiple neurotransmission changes happen during memory consolidation, retention, and retrieval under stress. Besides, exposure to severe stressful conditions changes the activity and morphology of astrocytes and microglial cells, which in turn increases the neuroinflammatory cascades and induces neuronal injury. Changing the CNS miRNAs expressions, developing neurodegenerative diseases and cognitive dysfunctions are stress-related responses in humans and laboratory animals. Although various stress mechanisms have been studied in memory impairment following neurodegenerative diseases, some questions remain that are essential to answer for finding appropriate therapies. Therefore, further studies should investigate precise targets of stress signaling pathways that contribute to memory formation. We hope this review will draw more attention to developing treatment strategies for stress-related disorders.
Abbreviations
| 5-HT: | 5-hydroxytryptamine |
| 5-HT1A: | 5-hydroxytryptamine 1A receptor |
| ACTH: | adrenocorticotropic hormone |
| AMY: | amygdala |
| BLA: | basolateral amygdala |
| CBs: | cannabinoid receptors |
| CNS: | central nervous system |
| CRH: | corticotropin-releasing hormone |
| ECS: | endocannabinoid system |
| GABA: | gamma-aminobutyric acid |
| GABAA: | gamma-aminobutyric acid subtype A |
| GABAB: | gamma-aminobutyric acid subtype B |
| GABAergic: | gamma aminobutyric acidergic |
| GRs: | glucocorticoid receptors |
| Hip: | hippocampus |
| HPA: | hypothalamic-pituitary-adrenocortical |
| IL-1β: | interleukin-1β |
| LTP: | long-term potentiation |
| miRNAs: | microRNAs |
| mRNA: | messenger RNA |
| MRs: | mineralocorticoid |
| NMDA: | N-methyl-D-aspartate |
| PFC: | prefrontal cortex |
| PVN: | paraventricular nucleus |
| SAM: | sympathetic-adrenomedullary |
Declarations
Author contributions
AR: Writing—original draft, Designing the figures, Writing—review & editing. MS: Writing—original draft, Designing the figures. SH: Writing—original draft.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2022.