 Review
Review

Affiliation:
1Rutgers Cancer Institute of New Jersey, New Brunswick, NJ 08903, United States
Email: pengyo@cinj.rutgers.edu
ORCID: https://orcid.org/0000-0003-4797-995X
Affiliation:
2Intra-Cellular Therapies, Inc., New York, NY 10016, United States
Affiliation:
3Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ 08903, United States
Explor Neurosci. 2025;4:100680 DOl: https://doi.org/10.37349/en.2025.100680
Received: January 01, 2025 Accepted: March 04, 2025 Published: April 01, 2025
Academic Editor: Dirk M. Hermann, University of Duisburg-Essen, Germany; Giustino Varrassi, President of the Paolo Procacci Foundation, Italy
The article belongs to the special issue Neuropathic Pain
Neuropathic pain (NP) is a significant global health challenge, affecting an estimated 7–10% of the population. Painful diabetic neuropathy (PDN), a severe complication of diabetes, impacts approximately one in every three diabetic patients. With the rising global prevalence of diabetes, PDN is projected to become an increasingly urgent health concern. Current treatments for PDN often provide inadequate pain relief and are associated with adverse side effects, emphasizing the need for safe and effective therapeutic options. This review examines the limitations of existing pharmacological therapies for PDN and presents the sigma-1 receptor (S1R) as a promising therapeutic target. We explore the biological role of S1R, its implication in NP and PDN, its structural biology, and the expanding preclinical and clinical evidence supporting its potential. Furthermore, we present evidence for various S1R antagonists in addressing NP and PDN, with a particular focus on E-52862 and [18F]FTC-146. These compounds represent first-in-class ligands for therapeutic and diagnostic applications, respectively, marking significant advances in the development of S1R antagonists. This review underscores the potential of S1R antagonism as a strategy for developing more effective treatments for PDN, with the ability to significantly improve patient outcomes.
Neuropathic pain (NP) is a major global health challenge, affecting an estimated 7–10% of the world population [1]. It is frequently associated with a wide range of neurological and systemic disorders, including diabetes, postherpetic neuralgia, spinal cord injuries, and multiple sclerosis [2]. The global NP therapeutics market was valued at over $7 billion in 2023 and is projected to grow at a compound annual growth rate of approximately 8% between 2024 and 2032 [3]. This expansion is fueled by the increasing incidence of diabetes and other chronic conditions, as well as greater awareness among healthcare providers and patients regarding the importance of effective treatment strategies.
NP arises from damage or dysfunction within the somatosensory nervous system, leading to abnormal pain signaling and a spectrum of distressing symptoms [4]. Its pathophysiology involves several key mechanisms, encompassing diabetes, peripheral nerve injury, central nervous system (CNS) damage, infections and autoimmune conditions, and chemotherapy-induced peripheral neuropathy [5].
Among the various types of NP, painful diabetic neuropathy (PDN) is one of the most prevalent and debilitating [3]. PDN emerges as one of the most severe complications of diabetes, a chronic metabolic disorder characterized by sustained hyperglycemia due to impaired insulin production or utilization. Chronic hyperglycemia leads to widespread complications affecting multiple organs, including the peripheral nerve systems [4]. According to the International Diabetes Federation, 537 million adults were living with diabetes in 2021, a figure expected to rise to 783 million by 2045 if current trends persist [6]. Approximately one of every three diabetic patients develop PDN, which is driven by a multifactorial pathophysiology involving metabolic, vascular, and inflammatory mechanisms that cause peripheral nerve damage [4, 7, 8]. Symptoms such as tingling and burning sensations, allodynia, hyperalgesia, and sharp pain, severely affect patients’ quality of life, reduce longevity, and increase healthcare burdens [8].
The complex pathology of PDN, coupled with the frequent presence of comorbidities in patients, makes its treatment quite challenging. At present, there are no therapies capable of completely curing the condition or reversing the underlying nerve damage [9]. The current management of PDN involves lifestyle changes to control blood glucose levels and pharmacological interventions for symptomatic pain relief [4, 10, 11]. This review explores the pharmacological landscape for PDN, examining their mechanisms of action, therapeutic potential, and limitations, and further present the sigma-1 receptor (S1R) as a promising target for developing novel and effective therapeutics for PDN.
Current pharmacological interventions for PDN include antidepressants, anticonvulsants, opioid analgesics, and other topical agents. While only three oral medications and one topical agent have been approved by the FDA specifically for PDN, a broader array of therapeutic options is available in other regions worldwide [11, 12] (Figure 1).
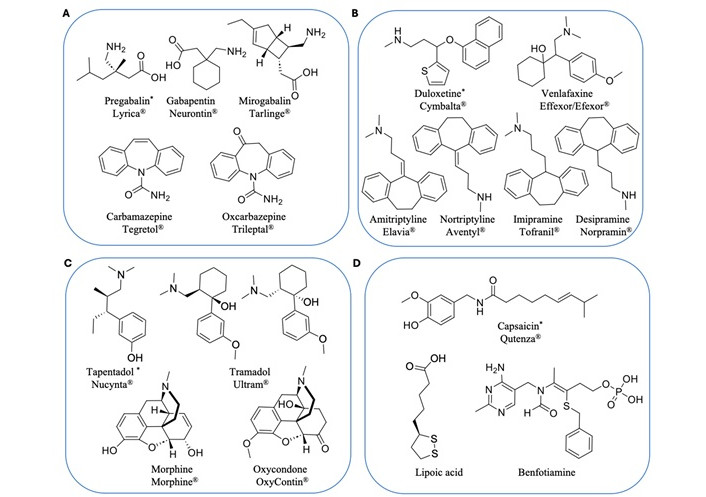
Current pharmacological treatments for PDN. (A) Ion channel blocker anticonvulsants; (B) serotonin-norepinephrine reuptake inhibitors and tricyclic antidepressants; (C) mu-opioid receptor agonists; (D) capsaicinoid topical anesthetics and others. * Approved by the FDA for PDN
Anticonvulsants modulate neuronal activity by acting on calcium and sodium channels and are among the most prescribed pharmacological treatments for PDN. Gabapentinoids, including pregabalin and gabapentin, are widely used as first-line treatments for PDN. These drugs act on the α2δ subunit of voltage-gated calcium channels to reduce neurotransmitter release, calming overactive nerves that contribute to NP [13]. Pregabalin is particularly effective in patients with multiple NP syndromes and improves associated sleep disturbances [14, 15]. Mirogabalin, a newer drug, demonstrates higher selectivity for the α2δ-1 subunit, with improved efficacy and fewer side effects [16]. However, gabapentinoids are associated with adverse side effects such as drowsiness, dizziness, weight gain, and peripheral edema [17, 18]. In addition, there is a risk of abuse, especially with pregabalin [19].
Sodium channel blockers, such as carbamazepine and oxcarbazepine, are a family of epilepsy medications sometimes prescribed for PDN. They inhibit voltage-gated sodium channels to reduce nerve excitability and alleviate pain. Carbamazepine is effective but has black box warnings for severe dermatological reactions, agranulocytosis and aplastic anemia [20]. Oxcarbazepine exhibits its analgesic effect mostly through its active metabolite licarbazepine, and offers a safer profile with fewer side effects, which makes it a preferable choice in this class [21]. However, these sodium channel blockers also carry with many side effects such as drowsiness, dizziness, headaches, nausea, vomiting, and double vision [22–24].
Serotonin-norepinephrine reuptake inhibitors (SNRIs) are a class of antidepressant medications used to treat major depressive disorder, anxiety disorders, chronic NP, and other medical diseases [12]. SNRIs such as duloxetine and venlafaxine are very effective for PDN and are particularly valuable for patients with comorbid depression or anxiety. Duloxetine, an FDA-approved first-line treatment, inhibits serotonin and norepinephrine reuptake to modulate pain signaling in the CNS. It often provides rapid pain relief, sometimes within the first week of treatment [25], with fewer side effects compared to older medications like tricyclic antidepressants (TCAs) [26–28]. Venlafaxine has a similar mechanism of action but with lower binding affinity to serotonin and norepinephrine transporters [29]. SNRIs are generally well-tolerated, though side effects such as nausea, dry mouth, and dizziness are possible [30, 31]. However, the potential for developing serotonin syndrome, a potentially life-threatening condition caused by an excessive accumulation of serotonin, requires careful monitoring [32].
TCAs are a broad class of medications that were first discovered in the early 1950s. Common examples include amitriptyline, nortriptyline, and imipramine, which remain another option for treating PDN. They exert their analgesic effect by inhibiting serotonin and norepinephrine reuptake and targeting additional receptors, such as opioid receptors and ion channels [33, 34]. TCAs are highly effective for NP and can improve associated sleep disturbances. However, their complex pharmacology results in many side effects, including dry mouth, constipation, urinary retention, drowsiness, and memory impairment, making them less favorable compared to SNRIs [35–38].
Several opioid drugs are clinically used for managing moderate to severe PDN, including tapentadol, tramadol, morphine and oxycodone. Tapentadol is approved by the FDA as first-line use [39]. Morphine, a potent μ-opioid receptor (MOR) agonist, provides effective relief for acute pain [40], while oxycodone demonstrates similar potency with active metabolites that enhance its analgesic effects [41]. Tapentadol and tramadol uniquely combine MOR agonism with norepinephrine reuptake inhibition, offering comparable efficacy to oxycodone but with fewer gastrointestinal side effects, such as constipation [42, 43]. Although opioids are effective for some patients to manage PDN, they come with significant risks, such as tolerance, dependence, and respiratory depression, which need cautious use and close monitoring [44–48].
Capsaicin, a plant-derived alkaloid from chili peppers, provides localized pain relief by binding transient receptor potential vanilloid 1 (TRPV1) receptors on nociceptive fibers [49]. The FDA-approved 8% capsaicin patch is applied to affected areas, such as the feet, and offers significant pain reduction in certain patient subpopulations with minimal systemic side effects. However, burning sensations at the application site are a common adverse reaction [50]. Overall, capsaicin has very few reported side effects beyond this localized discomfort. Additionally, antioxidants such as alpha-lipoic acid and benfotiamine supplement are also used to treat PDN in some countries [12, 51–53]. But none of these nutritional supplements have been approved by the FDA for use in the United States.
In summary, while current medications for PDN offer some relief, they are often associated with a range of side effects that limit their therapeutic effectiveness and patient compliance. Alarmingly, fewer than one-third of PDN patients achieve satisfactory pain relief with existing therapies, underscoring the urgent need for innovative treatments and the identification of new therapeutic targets.
It is noteworthy that several TCAs used for PDN treatment also bind to S1R, but with much lower affinity than their primary pharmacological targets. As a result, S1R modulation may play a secondary or minimal role in the analgesic effects of these drugs. Recent research suggests that directly targeting S1R offers a novel mechanism for alleviating NP across various etiologies, including PDN. As explored in this review, S1R represents a promising therapeutic target that could lead to more effective and mechanistically distinct treatments for PDN.
The sigma receptor was initially identified as a subtype (σ) of opioid receptors in the 1970s [54]. However, subsequent research using radioligand binding assays showed that the sigma receptor exhibited very different binding profiles to classical opioids such as naloxone, suggesting that the sigma receptor might possess unique ligand binding sites distinct from other opioid receptors [55]. The development of a benzomorphan-based specific radioligand ([3H]-(+)-pentazocine), which exhibited high affinity and selectivity for S1R, enabled the discovery of two distinct sigma receptors: S1R and S2R. S1R binds to benzomorphans with high affinity and corresponds to the classical σ opioid receptor first described by Su [55], whereas S2R does not bind to benzomorphans but has a high affinity for haloperidol and ditolylguanidine [56]. Moreover, both sigma receptors have been successfully cloned [57, 58], and their crystal structures have been resolved [59–63]. S1R is encoded by the SIGMAR1 gene with 223 amino acids [57], while S2R is encoded by the TMEM97 gene with 176 amino acids [58]. Despite their shared classification as sigma receptors, S1R and S2R exhibit limited structural similarity, with only 23% identity in their primary sequences. Both receptors have been shown to involve in the pathology of NP [62, 64]. However, this review will focus on the S1R only: its biology, function, structure, and pharmacological significance in the context of NP.
S1R does not share homology with any other mammalian proteins, but is highly conserved across species [65]. Interestingly, its closest homolog is the yeast C8-C7 sterol isomerase, though S1R itself has no sterol isomerase activity [57]. The identification of endogenous ligands for S1R remained a longstanding challenge. S1R has no definitively identified endogenous ligands, although some neurosteroids such as progesterone and dehydroepiandrosterone (DHEA) [66], hallucinogen N,N-dimethyltryptamine [67], sphingolipids [68], and choline [69], have been proposed to be S1R endogenous ligands. Importantly, the first S1R knockout (KO) mouse model was developed in 2003, providing a valuable tool for exploring the role of S1R in both normal and pathological physiology [70].
S1R is found in multiple organs, including the heart, gastrointestinal tract, liver, muscle in human body, as well as the nervous system [71, 72]. S1R is highly expressed in both the central and peripheral nervous systems [73, 74]. In the brain, S1R is widely distributed in pain-modulating regions including the dorsal spinal cord, dorsal root ganglion neurons, the periaqueductal gray matter, rostroventral medulla, and locus coeruleus, making it an attractive candidate for pain management [73, 75].
S1R is now widely recognized as a ligand-operated and Ca2+ sensitive chaperone protein that is primarily located at the mitochondria-associated membrane of the endoplasmic reticulum (ER) [71]. Under basal conditions, S1R forms a complex with the binding immunoglobulin protein (BiP) in the ER, essential for proper protein synthesis and the degradation of misfolded proteins [76]. Upon agonist binding or during ER stress, S1R dissociates from BiP and interacts with other proteins in the ER or other organelles, such as the inositol 1,4,5-trisphosphate receptor (IP3R), to regulate Ca2+ signaling and maintain cellular homeostasis and managing stress responses [77]. Meanwhile, S1R can also translocate from the ER membrane to the nuclear and plasma membranes, where it interacts with diverse pharmacological targets, including ion channels and G-protein coupled receptors (GPCRs) [78–83]. Acting as a regulatory subunit of these proteins, S1R can produce a profound impact on signal transduction and transmission [84, 85]. To date, over 50 client proteins with highly divergent sequences and structures have been identified for S1R, underscoring its broad functional repertoire [86]. Consequently, S1R has been considered a potential therapeutic target for neurological diseases including pain, depression and anxiety, memory and learning disorders, and substance disorders [86–88].
The X-ray crystallographic structures of the human S1R in complex with several known agonists and antagonists were successfully resolved at high resolution, offering significant insights into its molecular architecture and ligand interactions. The first breakthrough came in 2016 with the disclosure of human S1R crystal structures in complex with an antagonist (PD144418; Figure 2 and Figure 3) and an ambiguous ligand (4-IBP) [63]. Later in 2018, additional cocrystal structures of S1R with one agonist ((+)-pentazocine), two antagonists (haloperidol and NE-100; Figure 2) were published [61]. As illustrated in Figure 3, these crystal structures reveal S1R adopts a homotrimeric overall architecture, with each protomer containing a single transmembrane domain (Figure 3A). The ligand binding pocket in the S1R structure is located in the cytosolic domain within a β-barrel region, which is predominantly hydrophobic and occluded by the solvent. Key molecular interactions between the receptor and its ligands have been identified based on these crystal structures. Specifically, the anionic sidechain of Glu172 forms a salt-bridge with the cationic amine of the ligands, while the protonated Asp126 forms a hydrogen bond with Glu172 to retain the interaction network. Tyr103 interacts with ligands through π−π stacking and forms a hydrogen bond with Glu172 to stabilize its orientation. Additionally, several hydrophobic residues (Trp89, Leu95, Tyr103, Phe107, and Trp164) are involved in extensive nonpolar interactions with the hydrophobic or aromatic regions of the ligands (Figure 3B).
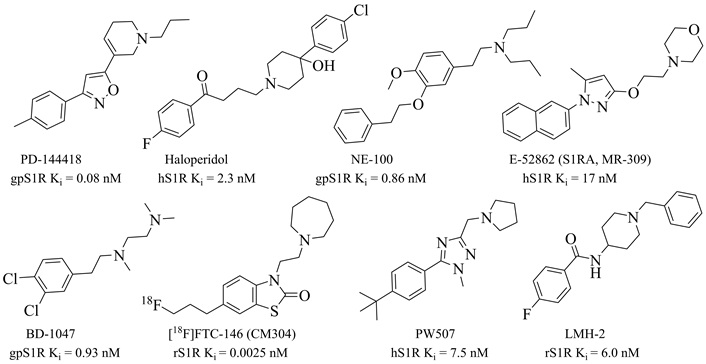
Structures and binding affinities of representative S1R antagonists. Subscripts (h, r, and gp) before S1R indicate different species: human, rat, and guinea pig
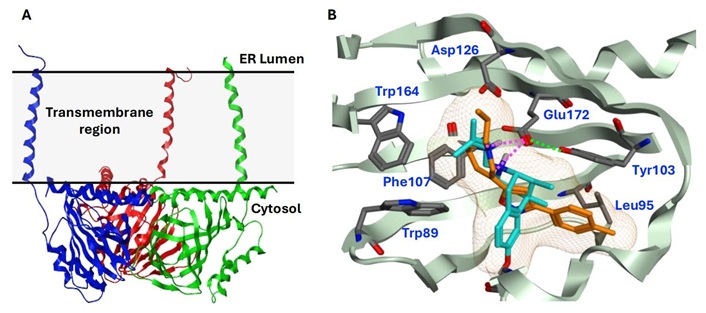
Crystal structures of human S1R homotrimer and ligand-binding sites. (A) Homotrimer structure of human S1R (PDB 5HK1); (B) Superimposition of cocrystal structures of human S1R bound to the antagonist PD144418 (PDB 5HK1; orange) and the agonist (+)-pentazocine (PDB 6DK1; cyan). The secondary structures and residues of S1R are colored in green and gray, respectively. The ligand-binding site is shown in a yellow mesh surface. The salt bridges between Glu172 and ligands are shown in magenta dash lines, while the hydrogen bond between Glu172 and Tyr103 is in a green dash line. ER: endoplasmic reticulum
Given the highly occluded nature of the binding pocket revealed by the original crystal structures, the mechanisms of ligand enter and exit for S1R remain an interesting topic of study. In 2022, the crystal structures of Xenopus laevis S1R in complex with PRE084 (an agonist) and E-52862 (an antagonist) suggest that ligands likely access the binding site via conformational changes in the carboxy-terminal two-helix bundle, rather than the cupin-fold domain [60]. More recently in 2024, several cocrystal structures of Xenopus laevis with neurosteroid ligands, progesterone and DHEA sulfate, were resolved [59]. These findings highlight distinct binding modes of endogenous steroids and underscore the versatility of S1R’s ligand interactions.
Beyond static crystal structures, biochemical and cellular studies have demonstrated that S1R exhibits dynamic ligand-dependent oligomerization: antagonists stabilizing high-molecular-mass oligomers and agonists promoting their dissociation into monomers or dimers [89, 90]. However, X-ray crystallography studies have consistently revealed a homotrimeric architecture for S1R [59–61, 63]. This discrepancy may arise due to the stabilizing conditions used in crystallization, which can favor specific oligomeric conformations. The trimeric arrangement observed in X-ray structures might represent a stable intermediate or a functionally relevant oligomeric form captured in the absence of membrane dynamics and cellular signaling. These findings highlight the importance of studying S1R in diverse experimental conditions to fully understand its conformational flexibility and functional implications. Understanding the conformational changes induced by different ligand classes can aid in the rational design of novel compounds with optimized binding properties and functional outcomes. These advances in S1R structural biology have not only significantly enhanced our understanding of its ligand recognition mechanisms but also provides a solid foundation for the structure-based design of novel S1R-targeting compounds.
S1R interacts with several ion channels involved in pain modulation, including voltage-gated ion channels (e.g., Nav1.5, Kv1.2, and Cav2.2), calcium-activated potassium channels (e.g., SK3), and ligand-gated ion channels [e.g., N-methyl-d-aspartate (NMDA) receptors] [78, 81, 82]. Notably, S1R has been shown to bind directly to the GluN1 subunit of the NMDA receptors [79, 82]. The GPCR nociceptors binding to S1R include cannabinoid CB1 and MOR [80, 83]. A direct interaction between MOR and S1R has also be described in transfected human embryonic kidney (HEK) cells [83]. Importantly, S1R antagonists have been shown to potentiate opioids’ (e.g., morphine, oxycodone) analgesia without exacerbating their side effects, such as dependence, tolerance, and constipation [91–93]. Studies using S1R KO mice and selective antagonists have revealed S1R’s critical roles in the modulation of pain of different etiologies, including chemical-induced hyperalgesia (formalin and capsaicin tests) [94, 95], central and peripheral neuropathy [64, 96, 97], osteoarthritis [98], and cancer pain [99]. In this review, we focus on NP, with a particular emphasis on PDN.
As summarized in Table 1, S1R has been extensively studied in preclinical NP models of multiple modalities. For instance, in a contusion spinal cord injury (SCI) model of central NP, S1R KO didn’t develop mechanical allodynia and thermal hyperalgesia in mice, while E-52862 (aka S1RA or MR309; Figure 2) alleviated mechanical allodynia and thermal hyperalgesia after both acute and prolonged treatments [100]. Similarly, S1R KO mice showed resistance to the development of mechanical allodynia and hyperalgesia in peripheral nerve injury models, such as paclitaxel-induced NP and partial sciatic nerve ligation (PSNL) [101, 102]. Selective S1R antagonists (e.g., BD-1047, S1RA, CM304, and PW507; Figure 2) have demonstrated analgesic efficacy in attenuating mechanical allodynia and hyperalgesia in a range of models, including chemotherapy (paclitaxel, cisplatin, and oxaliplatin) induced neuropathy [97, 101, 103], PSNL [102, 104, 105], and chronic constriction injury (CCI) [97, 106–108]. Furthermore, in clinical trials, E-52862 demonstrated significant efficacy to mitigate chronic oxaliplatin-induced peripheral neuropathy [109, 110]. These findings underscore S1R’s essential roles in amplifying pain responses across diverse pain conditions.
Summary of preclinical studies that describe S1R KO or antagonists in different NP models
| Pain type | Pain model | Specie | Sex | Ligand | Route | Effect on pain | References |
|---|---|---|---|---|---|---|---|
| Central neuropathic pain | Spinal cord contusion | Mouse | F | E-52862 | i.p. 7 days | ↓ mechanical allodynia and thermal hyperalgesia | [98] |
| Mouse | F | E-52862 | i.p. | ↓ mechanical allodynia and thermal hyperalgesia | [100] | ||
| Mouse | F | KO | No mechanical allodynia and thermal hyperalgesia | [100] | |||
| Peripheral neuropathic pain | Streptozotocin-induced | Rat | M | E-52862 | i.p., acute, b.i.d. 7 days | ↓ mechanical hyperalgesia | [97] |
| Rat | M | PW507 | i.p., acute, b.i.d. 14 days | ↓ mechanical allodynia and thermal hyperalgesia | [111] | ||
| Mouse | M | BD-1047 | i.t. 7 days | ↓ mechanical allodynia and thermal hyperalgesia | [112] | ||
| Nicotinamide-streptozotocin induced | Mouse | M | Haloperidol | s.c. | ↓ mechanical allodynia and hyperalgesia | [113] | |
| Mouse | M | LMH-2 | s.c. | ↓ mechanical allodynia and hyperalgesia | [113] | ||
| Zucker diabetic fatty | Rat | M | E-52862 | i.p., acute, b.i.d. 14 days | ↓ mechanical allodynia and thermal hyperalgesia | [114] | |
| High-fat diet | Mouse | M | KO | No mechanical allodynia and thermal hyperalgesia | [115] | ||
| Mouse | M | BD-1047 | i.t. | ↓ mechanical allodynia and thermal hyperalgesia | [115] | ||
| Oxaliplatin-induced | Rat | M | E-52862 | i.p., acute, b.i.d. 7 days | ↓ cold allodynia | [97] | |
| Cisplatin-induced | Mouse | M | CM304 | i.p. | ↓ mechanical allodynia | [116] | |
| Paclitaxel-induced | Mouse | F | E-52862 | s.c. | ↓ mechanical allodynia and cold hyperalgesia | [101] | |
| Mouse | F | BD-1047 | s.c. | ↓ mechanical allodynia and cold hyperalgesia | [101] | ||
| Mouse | F | KO | No mechanical allodynia and cold hyperalgesia | [101] | |||
| Rat | M | PW507 | i.p. | ↓ cold hyperalgesia | [103] | ||
| Partial sciatic nerve ligation | Mouse | M | E-52862 | i.p., acute, b.i.d. 21 days | ↓ mechanical allodynia and thermal hyperalgesia | [104] | |
| Mouse | M | E-52862 | i.v.10 days | ↓ mechanical allodynia and thermal hyperalgesia | [105] | ||
| Mouse | M | KO | No mechanical allodynia and thermal hyperalgesia | [102] | |||
| Chronic constriction injury | Rat | M | BD-1047 | i.t. | ↓ mechanical allodynia | [106] | |
| Rat | M | BD-1047 | p.o. | ↓ mechanical allodynia and cold hyperalgesia | [107] | ||
| Mouse | M | BD-1047 | i.t. | ↓ mechanical allodynia | [108] | ||
| Rat | M | E-52862 | i.p., acute, b.i.d. 7 days | ↓ mechanical allodynia | [97] | ||
| Mouse | M | CM304 | i.p. | ↓ mechanical allodynia | [116] |
i.p.: intraperitoneal; i.pl.: intraplantar; i.t.: intrathecal; i.v.: intravenous; p.o.: oral; s.c.: subcutaneous; b.i.d.: twice daily; KO: knockout
Several preclinical models of diabetes have been used to investigate S1R’s roles in the development of PDN, including chemical-induced, genetically modified, and diet-induced neuropathy models.
Streptozotocin (STZ), a toxin causing selective pancreatic β-cell destruction, induces physiological and behavioral changes resembling type 1 diabetes in humans [117–119]. In STZ-induced diabetic models, S1R has been found to be overexpressed in dorsal root ganglion, implicating its role in the development and maintenance of PDN [112]. BD-1047 inhibited STZ-induced PDN, such as mechanical allodynia and thermal hyperalgesia, after treatment for 7 days [intrathecal (i.t.)] [112]. Similarly, E-52862 significantly reduced mechanical allodynia both after acute [intraperitoneal (i.p.)] and 7-day [i.p., twice daily (b.i.d.)] treatments [97]. Furthermore, another potent and selective S1R antagonist, PW507, demonstrated robust efficacy in alleviating both mechanical allodynia and thermal hyperalgesia after acute (i.p.) and 14-day treatments (i.p., b.i.d.) [111].
In the nicotinamide (NA)-STZ model, which resembles type 2 diabetes by combining β-cell damage and insulin resistance, haloperidol and its analog LMH-2 (Figure 2) exhibited efficacy in alleviating PDN symptoms [subcutaneous (s.c.)], such as mechanical allodynia and thermal hyperalgesia [113].
In the Zucker diabetic fatty (ZDF) rat model, a genetic model of type 2 diabetes characterized by obesity and insulin resistance, E-52862 treatment produced significant reductions in mechanical allodynia and thermal hyperalgesia. These effects were observed after both acute (i.p.) administration and a sustained 14-day twice-daily (b.i.d.) regimen, emphasizing the compound’s robust therapeutic efficacy in this context [114].
Meanwhile, the high-fat diet (HFD)-induced neuropathy model, which simulates the metabolic dysfunction associated with diet-induced obesity, provided additional insights into S1R’s role in PDN. In this model, S1R KO mice did not develop mechanical allodynia and thermal hyperalgesia. Complementing these findings, BD-1047 (i.t.) effectively reduced both mechanical allodynia and thermal hyperalgesia in the same model [115].
Together, these findings across diverse preclinical studies reinforce the therapeutic promise of S1R antagonists in mitigating PDN, offering hope for improved management of this debilitating condition in diabetic patients. However, the precise mechanisms by which S1R antagonists alleviate PDN remain unclear. S1R has been reported to interact with the NR1 subunit of the NMDA receptor, a key player in the development of NP [82, 120–122]. In diabetic neuropathy models induced by STZ, ZDF, or HFD, nerve injuries activate the NMDA receptor, contributing to central sensitization and heightened pain sensitivity. Antagonizing S1R disrupts its interaction with NR1, thereby reducing NMDA receptor activity and dampening pain signaling and amplification [79]. However, other potential mechanisms should not be overlooked. For instance, S1R antagonists have been shown to mitigate STZ-induced peripheral neuropathy by downregulating high-mobility group box 1 (HMGB1), a DNA-binding protein and a proinflammatory modulator for chronic pain [112, 123]. Further research is necessary to elucidate the full scope of pharmacological mechanisms underlying S1R antagonist efficacy.
Initially, S1R was mistakenly categorized as a subtype of opioid receptors, leading to the belief that its binding was off-target and undesirable. For example, haloperidol (Figure 2), a butyrophenone-based antipsychotics, demonstrates potent binding affinity for S1R (Ki = 2.3 nM) and has served as a reference S1R antagonist for over four decades. However, haloperidol preferentially binds to dopamine receptors with sub-nano molar binding affinity [103, 124]. Early selective S1R antagonists, such as NE-100 and BD-1047, also exhibit potent S1R binding affinity (Ki < 10 nM) and have been widely used as research probes [125, 126]. Following the reclassification of S1R as a distinct receptor in the 1990s and the advancements in S1R structural biology in 2010s, increasing interests have emerged in developing potent and selective S1R ligands for multiple diseases. Over the years, a variety of S1R antagonists with various chemical structures have been developed, including pyrrolidine-ones, substituted piperazines, benzothiazole-ones, spirocyclics, di-substituted pyrazoles and triazoles (Figure 2) [104, 127–132]. Several of these S1R antagonists (BD-1047, E-52862, PW507, CM304 and its radio-tracer [18F]FTC-146) have been extensively studied in preclinical and clinical pain research.
E-52862, developed by Esteve Research and Development in Spain, is a potent and selective S1R antagonist that has advanced to clinical development. It demonstrates high binding potency for S1R (Ki = 17 nM) and remarkable selectivity over S2R (Ki > 1,000 nM), as well as a panel of 170 additional targets, including receptors, transporters, ion channels, and enzymes (Ki or IC50 > 1,000 nM) [104]. Among these targets, only human 5HT2A exhibited a moderate binding (Ki = 328 nM), but with weak functional antagonism (IC50 = 4,700 nM) [104]. Structural insights into the binding conformations of E-52862 to S1R, revealed through X-ray crystallography, have enhanced our understanding of ligand-receptor interactions and guided the design of novel S1R ligands [60].
In preclinical studies, E-52862 has exhibited significant analgesic effects across various pain models of different etiologies, including inflammatory pain (induced by formalin, carrageenan, or capsaicin) [96, 104, 133], central NP (SCI) [98], STZ-induced diabetic neuropathy [97, 114], chemotherapy-induced peripheral neuropathy (caused by paclitaxel, cisplatin, or oxaliplatin) [97, 101, 103], and peripheral neuropathy due to nerve injuries (e.g., PSNL, CCI, spared nerve injury) [93, 96, 97, 104]. These studies, conducted in mice and rats, also confirmed a strong safety and efficacy profile, enabling E-52862’s rapid progression to clinical trials.
In phase 1 trials in healthy volunteers, E-52862 demonstrated excellent safety, tolerability, and pharmacokinetics, prompting its further development in various pain conditions [134, 135]. In a phase 2 trial for oxaliplatin-induced peripheral neuropathy in elderly colorectal cancer patients in stages I-IV, E-52862 effectively alleviated acute NP symptoms, such as cold pain and motor impairments, while being well tolerated even under high oxaliplatin exposure [110]. In another phase 2 clinical trial evaluating post-surgical NP occurring more than 3 months post-surgery, E-52862 provided clinically significant pain relief, with fewer patients requiring rescue medicines (opioids or other analgesics) compared to the placebo group throughout the 28-day treatment period [400 mg, once daily (q.d.), oral (p.o.)] [109].
Furthermore, in a phase 2 clinical trial involving patients with moderate-to-severe PDN, E-52862 exhibited a safe profile and promising results [109]. Throughout the 4-week study period (400 mg, p.o., q.d.), E-52862 consistently reduced patients’ pain intensity and Neuropathic Pain Symptom Inventory scores. While statistical significance was not achieved due to high placebo responses, there was a clear trend toward greater pain relief in the E-52862 group compared to placebo. Moreover, a higher proportion of patients receiving E-52862 reported meaningful improvements in their overall condition, as reflected in the Patient Global Impression of Change scale. In addition, E-52862 was well tolerated, with a safety profile comparable to placebo and no increase in the need for rescue medication.
Another noteworthy S1R antagonist is the radio-tracer [18F]FTC-146, the most potent S1R antagonist so far with a binding affinity (Ki) of 0.0025 nM in rat brain homogenates (Figure 2) and > 10,000-fold selectivity over S2R and additional 59 targets [130]. It has been studied in phase 1 clinical trials as a diagnostic agent for PET/MRI imaging to identify sites of nerve damage in patients with chronic NP [136]. Its non-radiolabeled analog, CM304, showed significant analgesic effects in preclinical models, including reducing mechanical allodynia in CCI and cisplatin-induced NP, and inflammatory pain in the formalin test [116]. However, CM304’s pharmacokinetics—characterized by a short elimination half-life (115 min) and moderate clearance (33 mL/min/kg)—limited its progression into clinical development [137]. Nevertheless, CM304’s rapid clearance makes it ideal as a precursor for imaging agents like [18F]FTC-146 [130, 138, 139].
In a phase 1 clinical trial in healthy participants, [18F]FTC-146 demonstrated high uptake in S1R-rich organs such as the pancreas, spleen, and thyroid, moderate uptake in the brain and myocardium, and low uptake in bones and muscles [136, 140]. The total radiation exposure (9.1 mSv) is well within the acceptable range for [18F]-labeled radiopharmaceuticals and below the FDA limit of 30 mSv. Importantly, all participants tolerated the tracer without significant adverse effects [136, 140].
In subsequent clinical trials, [18F]FTC-146 was used to image patients with complex regional pain syndrome and sciatica, enabling precise visualization of nerve damage and effectively monitoring changes in S1R expression [141]. A noteworthy case study involved a female patient with chronic knee pain unresponsive to multiple treatments over several years. PET/MRI imaging with [18F]FTC-146 revealed high radioligand uptake in the intercondylar notch, leading to the surgical removal of an inflamed lipoma and completely resolving her pain [141]. This emphasizes the potential of [18F]FTC-146 to localize pain sources across diverse pathologies. [18F]FTC-146 is currently undergoing more clinical trials, including studies on chronic pelvic pain and pediatric pain, further supporting its potential as a PET/MRI imaging agent for pain diagnosis and management [142].
While demonstrating proof-of-concept efficacy in phase 2 clinical trials for various pain conditions, the clinical development of E-52862 was halted, potentially due to concerns regarding its oral bioavailability and/or efficacy. Notably, clinical trial participants required very high daily oral doses (400 mg, p.o., q.d.), despite E-52862’s potent binding affinity for S1R (Ki = 17 nM) [104]. This underscores a critical need for the development of S1R antagonists with enhanced oral bioavailability and absorption, distribution, metabolism and excretion (ADME) properties, and greater efficacy.
In response to these limitations, our laboratory developed PW507, a potent and selective S1R antagonist with enhanced pharmacokinetic and safety profiles [103]. PW507 demonstrated potent S1R binding affinity (Ki = 7.5 nM), high selectivity over S2R (Ki > 1,000 nM) and a broad panel of 87 targets (Ki > 1,000 nM, SafetyScreen87TM) [103]. In preclinical studies, PW507 exhibited promising in vitro and in vivo ADME and pharmacokinetic properties, minimal toxicity, and significant efficacy to relieve paclitaxel-induced NP and formalin-induced inflammatory pain [103, 111]. In rat models of STZ-induced diabetic neuropathy, PW507 significantly alleviated mechanical allodynia and thermal hyperalgesia following both acute (i.p.) and chronic (i.p., b.i.d. for 14 days) administration, without inducing tolerance and toxicity [111].
Importantly, PW507 represents an improved S1R antagonist compared to E-52862, with superior ADME properties. PW507 demonstrated much lower plasma protein binding than E-52862 (66% vs. 96%), which indicates a greater amount of unbound PW507 available for cellular action, potentially enhancing efficacy. Furthermore, PW507 exhibited a higher oral bioavailability (F = 28% vs. 15%) and approximately 4 times greater brain exposure (B/P = 12 vs. 2.7) than E-52862, providing higher brain exposure [143]. Additionally, PW507 showed preclinical efficacy at a lower dose (20 mg/kg, i.p.) than E-52862 (40 mg/kg, i.p.). All these findings underscore PW507’s potential as a more effective therapeutic option for NP and PDN. PW507 is currently undergoing further preclinical efficacy studies in various pain models using the oral administration route, with plans to advance to clinical trials.
PDN remains a significant clinical challenge due to its complex pathophysiology and limited efficacy of current treatment options. While conventional therapies may offer partial relief for some patients, they are often associated with adverse effects and carry risks of tolerance and dependence. This underscores an urgent need to identify novel therapeutic targets that address the underlying mechanisms of PDN and provide effective symptom relief.
Strong evidence supports S1R as a promising therapeutic target for PDN and other NP conditions. The S1R biological system, through its modulatory role in calcium signaling, plays a crucial role in pain processing. Advances in structural biology have provided detailed insights into the ligand-receptor interaction patterns, facilitating the rational design of novel selective antagonists. Over the past decade, substantial progress has been made in understanding S1R’s role and developing selective S1R antagonists. The promising results of E-52862 in clinical trials has demonstrated the potential of S1R antagonists in effectively alleviating pain. Moreover, the PET/MRI ligand [18F]FTC-146 has emerged as a valuable tool, offering critical insights into S1R expression and occupancy. It plays a pivotal role in refining patient diagnosis and improving clinical trial design, ultimately enhancing the translational success of new pain medicines. However, significant interspecies differences in the pharmacokinetics of S1R antagonists have been observed, which emphasize the necessity of refining preclinical models to enhance translational accuracy and predictive value. To advance this field further, the discovery and development of next-generation S1R antagonists are essential to deliver novel and effective treatments for PDN and NP.
BiP: binding immunoglobulin protein
CCI: chronic constriction injury
CNS: central nervous system
DHEA: dehydroepiandrosterone
ER: endoplasmic reticulum
GPCRs: G-protein coupled receptors
HFD: high-fat diet
KO: knockout
MOR: μ-opioid receptor
NP: neuropathic pain
PDN: painful diabetic neuropathy
PSNL: partial sciatic nerve ligation
S1R: sigma-1 receptor
SCI: spinal cord injury
SNRIs: serotonin-norepinephrine reuptake inhibitors
STZ: streptozotocin
TCAs: tricyclic antidepressants
ZDF: Zucker diabetic fatty
The views expressed in the submitted article are the author’s own and not official positions of the author’s institutions (Rutgers Cancer Institute of New Jersey, Intra-Cellular Therapies, Inc., and Rutgers Robert Wood Johnson Medical School).
YP: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Writing—original draft, Writing—review & editing. QZ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing—original draft, Writing—review & editing, Visualization. SC: Conceptualization, Writing—review & editing.
YP is an inventor on PCT/US2019/066048, which describes PW507 and its analogs as novel S1R ligands. These compounds are also discussed in this manuscript. The author declares that this patent may be relevant to the content of this study, but no financial interests are currently associated with it.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
YP is sponsored by Rutgers Cancer Institute of New Jersey Biomedical Informatics Shared Resource [NCI-CCSG P30CA072720-5917]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Hannah G. Matejowsky ... Alan D. Kaye
Sergei Kozlovskiy ... Dmitry Aminin
Howan Leung ... Rainer Freynhagen
Bushra Yasin ... Alex Bekker
Tom Berfelo ... Jan R. Buitenweg
Laura Demartini, Cesare Bonezzi
Giustino Varrassi ... Matteo L.G. Leoni
