
Abstract
Similar to other psychiatric disorders, drug addiction is linked to changes in neuronal activity within the mesolimbic system, which consists of dopamine (DA) neurons of the ventral tegmental area projecting to the ventral part of the striatum, the nucleus accumbens (NAc). All drugs of abuse indeed artificially increase DA concentration in the NAc, which hijacks the reward system and triggers lasting behavioral alterations, including compulsive drug-seeking and drug-taking behavior despite negative consequences and a high rate of relapse after abstinence. DA chiefly signals through DA receptor (DAR) type 1 (D1R) and type 2 (D2R), which are G protein-coupled receptor (GPCR) that are positively and negatively coupled to adenyl cyclase, respectively. Multiple evidence indicates that the potent modulatory roles of DA on other neurotransmitters and neuromodulator systems implicate the direct physical interactions (i.e., heteromerization) of DAR with other receptors. DAR heteromerization, which is increased in several preclinical models of psychiatric disorders, leads to a reciprocal and fine-tuned modulation of DAR and partner receptors, therefore suggesting that targeting DAR heteromerization may contribute to the development of clinically relevant strategies. Herein, we provide an overview of current methodologies used for detecting receptor heteromers both in heterologous systems and in situ in the brain and discuss their respective advantages and limitations. We also argue that D1R and D2R have been shown to form heteromers with multiple partner receptors in heterologous systems but only few studies were able to a provide proof of their existence in the brain or establish their biological roles. This review will emphasize on studies describing the modulation and functions of DAR heteromerization in the brain in preclinical models of psychiatric disorders, with a particular focus on addiction, a field in which those heteromerization processes have been the most extensively studied.
Keywords
Nucleus accumbens, dopamine, receptor heteromerization, signaling, psychiatric disordersIntroduction
Substance use disorder is characterized as a chronic and recurring psychiatric condition that arises from prolonged drug use in susceptible individuals, leading to compulsive drug consumption despite harmful consequences. It is now acknowledged that changes in neuronal activity within the brain reward circuit, triggered by drug use, drive neuronal population-specific epigenetic and genetic responses shaping the lasting behavioral alterations characteristic of addictions [1, 2].
Addictive substances hijack the reward system by artificially increasing dopamine (DA) concentration in the mesolimbic system [3], which persistently modulates excitatory glutamate transmission within the reward system, especially in the ventral part of the striatum, the nucleus accumbens (NAc) [4, 5]. The NAc is regarded as a main target for drugs of abuse notably because it is at the intersection of converging glutamate inputs from limbic, thalamic, and cortical regions, which encode elements of drug-associated stimuli and environment, alongside DA signals that sense reward prediction error and incentive values [6].
DA and glutamate signal integration is mostly processed by medium-sized spiny neurons (MSNs) receiving glutamate axon terminals and DA inputs that converge onto their dendritic spines [7, 8]. MSNs are mainly divided into two distinct populations based on their expression of DA receptor (DAR) type 1 (D1R) or type 2 (D2R) receptors, which are G protein-coupled receptors (GPCRs) positively and negatively coupled to adenylyl cyclase through their respective coupling to the Gs/olf and Gi/o subtypes [9, 10], even though a subset of NAc MSNs expresses both receptors [11]. Although beyond the scope of this article, it is important to keep in mind that dopamine receptors (DARs) also signal through multiple effectors and downstream intracellular cascades in addition to G proteins as already reviewed elsewhere [12].
These two MSN sub-populations also display distinct projections within the cortico-basal ganglia network with D1R-MSN and D2R-MSN preferentially forming the direct pathway and indirect pathway, respectively. Cell-type-specific manipulations of MSN activity through optogenetic, chemogenetic, as well as MSN-specific ablation or inhibition of synaptic transmission, support that activation and inhibition of NAc D1R-MSN respectively promotes and dampens addictive-like behavior in preclinical models, while manipulating NAc D2R-MSN activity leads to opposite behavioral outcomes [13–21]. However, this dichotomic view of two MSN subpopulations forming parallel pathways whereby D1R-MSNs and D2R-MSNs respectively promote and reduce drug-evoked adaptation has been challenged. In fact, a subset of NAc D1R-MSN has been shown to join the indirect pathway and control specific components of drug response [22]. Furthermore, the two MSN subpopulations can modulate their activity through reciprocal inhibitory collaterals between D1R-MSN and D2R-MSN [23] and the modulation of D2R-MSN to D1R-MSN collaterals has been shown to contribute to behavioral sensitization to cocaine by promoting D1R-MSN activity [24].
Nevertheless, a widely accepted view is that the raise of DA caused by addictive substances stimulates D1R, which activates D1R-MSNs and promotes reinforcement. Meanwhile, the stimulation of D2R, which inhibits D2R-MSNs, counteracts aversion. This explains why a stimulus is highly rewarding when both D1R and D2R are concomitantly activated. Based on these findings, it has been therefore proposed that an imbalance in the activity of D1R-MSN and D2R-MSN triggered by drugs of abuse, whereby the activity of D1R-MSN prevails over D2R-MSN, may lead to compulsive drug use and addiction [2, 5, 25].
In this context, significant research has focused on the cellular and molecular mechanisms behind the imbalance in activity between the two MSN subpopulations evoked by addictive substances, as understanding these processes could lead to the development of novel therapeutic strategies in the field of addiction. In this respect, we found that cocaine elicits a local signaling pathway downstream from D1R that potentiates N-methyl-D-aspartate glutamate receptors (NMDARs) and triggers the activation of extracellular signal-regulated kinase 1/2 (ERK1/2) in D1R-MSN, which is known to play key roles in controlling epigenetic and genic responses and long-term behavioral alterations to this drug [26–29]. Even though the functional role of DA- and glutamate-dependent signaling crosstalk in drug-evoked adaptations is well-acknowledged, targeting the cognate receptors to alleviate symptoms is associated with a loss of efficacy over time and the appearance of severe sides effects, likely due to the involvement of these receptors in fundamental physiological functions [29–31]. As an alternative, a promising strategy may lie on the selective targeting of molecular mechanisms responsible for the deleterious dialogue between DA and glutamate that is triggered by drugs of abuse.
Activation of local intracellular signaling pathways downstream DAR is important to modulate the functions of multiple neighboring receptors, whether other GPCRs or ion channels. However, multiple evidence indicates that direct physical interactions between receptors serve as a powerful mechanism by which receptors can influence each other’s functions through allosteric modulations. These receptor oligomers can form either between identical receptors (homomers) or between different types of receptors (heteromers). Besides their unique ability to control the dynamic spatiotemporal modulation of partner receptors’ functions, receptor heteromers display properties that are distinct from component receptors, therefore making them prime targets for the development of more selective pharmacological treatments for a vast number of neurological and psychiatric diseases, including addiction [31–36]. DARs, particularly in heterologous systems, have been shown to form numerous heteromers with other GPCRs, ionotropic channels, and transmembrane proteins, leading to functional alterations in the partner receptors, changes in ligand binding affinity, and biased signaling. The diversity and biophysical properties of DAR heteromers have been extensively reviewed elsewhere [30, 37]. Furthermore, numerous studies have highlighted the critical role of the physical interaction between DARs and NMDARs in their mutual modulation [30, 38–41], underscoring their relevance to drug addiction.
This review will provide a brief overview of current methodologies for detecting receptor heteromers and then focus on DAR heteromers that have been shown to play a functional role in vivo in preclinical models of psychiatric disorders, particularly in models of addictions in which the roles of those receptor heteromers have been the most extensively investigated. We will also discuss the potential benefits of targeting specific DAR heteromers in the treatment of eating disorders. Finally, we will consider DAR heteromers that have been implicated in conditions beyond psychiatric disorders, highlighting how their targeting could also be advantageous in the treatment of addiction and other psychiatric diseases for future studies.
Current methodologies for detecting receptor heteromers
A consensus has emerged to define receptor heteromers as macromolecular complexes composed of at least two receptors possessing biochemical properties different from those of the monomers of which they are composed, and that are lost when the protomer interaction is disrupted [42–44]. It is classically considered that the maximal distance between protomers is around 10 nm to consider direct protein-protein interaction. Various techniques have been developed to demonstrate such interaction between receptors [45]. However, these approaches are mostly implementable in heterologous systems through ectopic expression of fusion proteins. This limitation raises the question of the genuine existence of receptors heteromers in vivo in physiological conditions. Herein we describe the main techniques used to characterize receptor heteromers, including those implemented on tissues as a proxy for interaction of endogenous receptors.
Co-immunoprecipitation
Co-immunoprecipitation has been routinely used as a primary approach to investigate potential receptor complexes. It consists of detecting the presence of a protein (a receptor) using an antibody in a lysed tissue/cell extract, immobilizing and purifying it before biochemical analysis of the proteins that have been precipitated with the protein of interest, i.e., the proteins linked to the antigen detected. This technique can be a first step in demonstrating an interaction between two receptors, but it has several limitations. First, the use of detergents to solubilize membrane proteins can induce artificial interactions or, on the contrary, disrupt existing interactions; second, it does not allow the resolution of the localization of the complexes in cellular subtypes and sub-compartments [45]; third, it does not demonstrate direct protein-protein interaction.
Bioluminescence resonance energy transfer or fluorescence/Förster resonance energy transfer techniques
Bioluminescence resonance energy transfer (BRET) or fluorescence/Förster resonance energy transfer (FRET) techniques (Figure 1A and 1B) have been extensively used to demonstrate heteromerization of receptors (ectopically) through expression of fusion proteins in heterologous systems. The principle is based on the transfer of energy between a donor and an acceptor, resulting in the emission of light by the acceptor and a decrease in emission by the donor [46]. Proteins of interest can be tagged with fluorophores, typically cyan fluorescent protein (CFP) and a derivative of green fluorescent protein (GFP) [e.g., yellow fluorescent protein (YFP)], referred to as FRET or with YFP and the luciferase enzyme (Rluc), referred to as BRET. When the tagged proteins are sufficiently close (< 10 nm), in the case of FRET, stimulation of the fluorophore with the shortest wavelength (the donor; and therefore, releasing the greatest energy, in this case CFP), will lead to an increase in its fluorescence intensity, enabling the acceptor (in this case YFP) to be stimulated. As a result, the fluorescence intensity of the acceptor increases, while that of the donor decreases. Measure of the YFP signal emitted in relation to that of the donor reveals the interaction between the two proteins of interest.
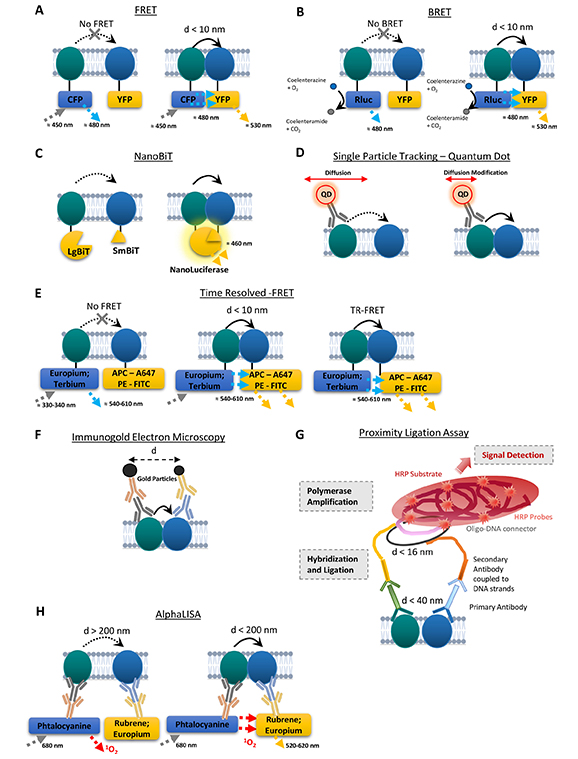
Schematic representations of the main techniques to assess the close proximity between receptors. (A) Fluorescence resonance energy transfer (FRET). Left: when the receptors are distant enough, no FRET signal occurs. Right: FRET signal is detected when the distance between the receptors is lower than 10 nm. (B) Bioluminescence resonance energy transfer (BRET). Luciferase degrades coelenterazine into coelenteramide resulting in 480 nm luminescence emission. Left: when the receptors are distant enough, no BRET signal occurs. Right: BRET signal is detected when the distance between the receptors is lower than 10 nm. (C) NanoLuciferase Binary Technology Assay (NanoBiT). Left: no luminescence emission occurs when the two receptors are distant. Right: when the protomers are close enough, the two subunits complement to form an active nanoluciferase that emits at 460 nm. (D) Quantum dot based single particle tracking. The initial diffusion of one receptor tagged with a QD (left) can be modified by its interaction with another receptor (right). (E) Time resolved FRET. Using europium or terbium as donors, combined to ad hoc acceptors (e.g., allophycocyanin, Alexa Fluor 647, phycoerythrin or fluorescein) (left) allows the detection of a FRET signal when the protomers are close enough (< 10 nm; middle) that lasts after the excitation of the rare-earth fluorescent lanthanides stops, allowing for a prolonged emission of a long-lived fluorescence and with a better excitation and emission spectrum compatibility both enhancing the signal-to-noise ratio (right). (F) Immunogold electron microscopy. The gold particles of different sizes, fused to specific antibodies allow the detection of receptors. The distance between gold particles can be measured to assess the proximity between the receptors. (G) Representation of brightfield in situ proximity ligation assay (PLA). The hybridization and ligation only occur when the receptors are close enough (16 nm at strand levels; 40 nm at epitope level). The oligonucleotides can also be labelled with a fluorophore. (H) Amplified luminescent proximity homogeneous assay (AlphaLisa). The ALPHA signal is detected only when the two protomers are in close proximity (< 200 nm). d: distance; HRP: horseradish peroxidase; CFP: cyan fluorescent protein; YFP: yellow fluorescent protein; Rluc: luciferase enzyme; O2: dioxygen; CO2: carbon dioxide; 1O2: singlet oxygen; APC: allophycocyanin; A647: Alexa Fluor-647; PE: phycoerythrin; FITC: fluorescein; LgBiT: large subunit of the nanoluciferase; SmBiT: small subunit of the Nanoluciferase; QD: quantum dot
For BRET, luciferase is its own energy source, and on the same principle, when the proximity/interaction conditions are met, the energy released will stimulate the emission of a signal from the acceptor fluorophore. The advantage of BRET lies in the bioluminescence of the luciferase, which avoids polluting the sample with the donor’s stimulating light beam, thus improving the signal by reducing background noise. Luminescence results from the catalytic degradation of coelenterazine by luciferase [45, 47–49].
Bimolecular fluorescence/luminescence complementation techniques
A large spectrum of bimolecular fluorescence/luminescence complementation techniques has been implemented over the last 20 years and is extensively reviewed in [50]. Of note, one of the latest techniques is the NanoLuciferase Binary Technology Assay (NanoBiT) (Figure 1C), used to detect several interactions between GPCRs in cultured cells. The proteins/receptors of interest are fused with two fragments composing the NanoLuciferase, a small subunit (1.3 kDa, Small BiT, SmBiT) and a larger subunit (18 kDa, Large BiT, LgBiT), that have low affinity for each other. When the two receptors are close enough, the two subunits complement and form an active NanoLuciferase that generates a luminescent signal [50]. This technique was used to detect the interaction between GPCR such as D2R-mAChR1 on HEK293T cells [51] or the antagonist-dependent modulation of D2R−D2R homodimers [52].
Sequential resonance energy transfer technique
Sequential resonance energy transfer (SRET) (sequential BRET-FRET technique): In order to detect higher orders receptor complexes, Navarro and colleagues [53] have combined a BRET and subsequent FRET approach to demonstrate the interaction between adenosine A2A receptor (A2AR), D2R, and cannabinoid receptor type 1 (CB1R) in heterologous system.
For all these resonance energy transfer (RET) approaches, there are several limitations regarding signal specificity. First, the binding of fusion proteins can potentially alter the conformation of the receptors within the complex, consequently impacting the RET signal. Second, tagging of the proteins could interfere with the ability of each protomer to interact with other proteins (including the other protomer in the complex), or with the functioning of the complex itself [44]. Third, the transient transfection classically used results in strong expression of the tagged receptors of interest, which can artificially promote interactions that might not occur with endogenous receptors. Finally, these approaches cannot be implemented for the detection of endogenous interaction on tissue ex vivo.
Time-resolved fluorescence energy transfer
More recent improvements in these techniques have increased the FRET/BRET signal while reducing noise, enabling more accurate detection of GPCR oligomers. Time-resolved FRET (TR-FRET) (Figure 1E) is based on the use of rare-earth fluorophore lanthanides, which emits a long fluorescence of the order of several milliseconds (e.g., europium, terbium). The fluorescence emitted persists after the light source has been switched off, enabling the FRET signal to be generated while avoiding the background noise created by the initial excitation beam. The improved spectral compatibility of these donor fluorophores with the acceptors also helps improving the signal-to-noise ratio [45, 54–57]. However, although this technique provides a better signal than FRET alone, it requires several controls and the implementation of corrections to obtain a specific TR-FRET signal (e.g., removal of several types of signals polluting the TR-FRET signal, such as sample autofluorescence, the signal due to acceptor excitation, or that arising from spectral overlap between donor and acceptor emission). Interestingly, by using specific receptor ligands respectively fused to donor and acceptor, this technique has been used to highlight the presence of endogenous oxytocin receptor homodimers in rat mammary glands (native tissue) [54] and D2R-A2AR heteromer in the striatum (striatal membrane preparations) [56]. However, this technique has so far failed to identify the same oligomers from human tissues, potentially due to conservation of samples [45, 54, 56]. Even though TR-FRET is a promising approach for the detection of endogenous heteromers, it remains challenging to implement, and lacks spatial resolution for the localization of receptor complexes in cell subtypes and sub-compartments.
This latter aspect requires detections of heteromers in situ on preserved tissue. Immunohistochemistry-based methods have been developed to investigate protein-protein interaction. The main caveat being the loss of resolution regarding direct protein-protein interaction, inherent to the use of antibodies to detect and label antigens.
Single particle tracking
Single particle tracking (SPT) relies on fluorescent microscopy techniques allowing spatial isolation and computational localization of single fluorophores. The fluorophore can be directly fused to the protein of interest, requiring ectopic expression, or through binding of antibodies against extracellular domains for detection of endogenous proteins [58]. This latter approach has been used to detect the close proximity between receptors on cell surface (Figure 1D), mostly using antibodies directed against extracellular domains. Such an approach has notably been used for detection of the interaction between D1R and the GluN1 subunit of NMDAR [41, 59], as well as the interaction between mGluR5 and the GluN1 subunit of NMDAR [60] or between EPHB2R and the GluN2A subunit of NMDAR [61]. Because of the inherent need for high-resolution microscopy and computational analyses, such an approach is still hard to implement in 3D models such as tissue sections and remains to date mostly limited to (primary) cell cultures.
Immunogold electron microscopy
Immunogold electron microscopy (IEM) (Figure 1F) uses gold particles coupled to secondary antibodies to visualize receptor expression at the subcellular level. Electron microscopy offers better spatial resolution, sensitivity, and ultrastructure than light microscopy. Gold particles coupled to secondary antibodies are of different sizes, allowing not only the localization of different proteins within a neuronal compartment, but also the measurement of the distance between two different particles, so as to identify potential oligomers [45, 56, 62]. IEM can be used on tissue sections, though it remains a technically challenging approach that does not allow relative quantification of heteromer density.
Proximity ligation assay
Proximity ligation assay (PLA) (Figure 1G) is a technique based on classical immunohistochemistry methods, developed in the early 2000s [63], which has since been used to demonstrate close proximity, suggestive of interaction, between two proteins. Since its development, PLA has been extensively used for in situ detection of receptor complexes [45, 56, 64–68]. Primary antibodies raised in different species recognize antigens of interest, and are then themselves recognized by secondary antibodies. The particularity lies in the coupling of secondary antibodies to small complementary single-stranded DNA sequences. When the antigens and therefore the DNA strands are in close proximity (16 nm at strand level), they can interact in the presence of oligonucleotide connectors. This interaction generates an amplifiable circular DNA strand, serving as a template for a local DNA amplification reaction by circular replication. The product of this amplification can then be detected by hybridization with complementary oligonucleotides labelled with a fluorophore or conjugated to HRP (horseradish peroxidase), and subsequently detected by the substrate DAB (3,3’-diaminobenzidine). This close proximity between receptors then appears as a dot, visualized by confocal microscopy (with fluorophore), or brightfield transmission microscopy (HRP coupled to DAB). Several studies have successfully implemented this method on human brain samples, notably to study the expression of receptor complexes density in comparison with mouse models (D2R-A2AR) or in the context of pathologies (e.g., addiction for D2R-NMDAR) [67, 69].
PLA is easily implementable and the signal obtained quantifiable with regular imaging techniques. Moreover, it offers the opportunity to localize receptor complexes in a subpopulation-specific and sub-compartment manner. The main limitation remains the maximal distance between antigens detected that, when used with both primary and secondary antibodies, largely exceeds 10 nm. Of note, the primary antibodies can be directly labelled with single-stranded DNA sequences, decreasing the maximal necessary distance between antigens to allow for the subsequent reactions. Moreover, the use of fixatives can impact (positively or negatively), the proximity of antigens.
A new detection method has recently emerged based on the PLA approach: MolBoolean. It uses proximity probes and rolling circle amplification as well, but in contrast to PLA, MolBoolean allows the detection of free and interacting protomers on the same sample, enabling the normalization of the detected interacting proteins to the quantity of free proteins [70]. It was recently used to detect the D2R-A2AR interaction in striatal MSN [71].
AlphaLisa approach
The AlphaLisa approach (Figure 1H) has been developed based on the AlphaScreen technology (amplified luminescent proximity homogeneous assay). This technique uses secondary antibodies coated with either donor or acceptor latex nanobeads. The donor bead contains a photosensitizer (phtalocyanine) and the acceptor bead contains a chromophore (ex: rubrene, europium). When both antigens are in close enough proximity (< 200 nm), the photostimulation of the donor with a 680 nm light induces the release of singlet oxygen that triggers emission of fluorescence from the acceptor bead (520–620 nm) (LOCI™, luminescent oxygen channeling immunoassay technology) [72, 73]. This approach has been validated through the detection of D2R-A2AR heteromers in WT mice, as well as in a model of Parkinson’s disease (PD) (6-OHDA lesioned rats) and in post-mortem brains from PD subjects and healthy controls [72]. This method has also been used to detect a decrease of D2R-A2AR interaction in the striatum in a mouse model of schizophrenia (PCP) or in the caudate of schizophrenic subjects [73] and to detect striatal D2R-mAChR1 interaction in a mouse model of movement disorder [51].
As mentioned, a main caveat of antibody-based approaches is the loss of spatial resolution due to the size and flexibility of antibodies that may enhance the probability of proximity. Moreover, in particular, for GPCRs, antibodies may lack of specificity. These approaches also do not allow to discriminate between transient versus stable protein-protein interaction, PLA and Immuno Gold allowing offering a “snapshot” of protein complexes at the time of fixation. Emerging evidence suggest that heteromers formed by class A GPCRs are transient (at maximum 100 of ms), a temporal resolution that can only be reached through SPT. Because of the respective limitations of the aforementioned approaches (Table 1), combination of these techniques has been performed to demonstrate heteromerization between two or more receptors [56, 62]. Nonetheless, final validation of interaction specificity can be reached through disruption of the interaction between the receptors that should result in loss of receptor proximity or of heteromer-specific signaling. Some examples will be described below, focusing on heteromers formed by D1R or D2R.
Main techniques to assess receptor heteromerization
| Method | Proximity/resolution | Sample type |
|---|---|---|
| Co-IP | N.A./sampled tissue | Lysed tissue |
| In situ hybridization | > 500 nm–1 µm | Heterologous system/native tissue |
| Immunogold | 20–30 nm | Native tissue |
| PLA | 16 nm (strand level)–40 nm (epitope level) | Native tissue/heterologous system |
| BRET | < 10 nm | Heterologous system |
| FRET | < 10 nm | Heterologous system |
| TR-FRET | < 10 nm | Heterologous system/native tissue/membrane preparations |
| SRET | < 10 nm | Heterologous system |
| AlphaLisa | < 200 nm | Membrane preparations |
| NanoBiT | N.D./low molecular weight of NanoLuc (19 kDa) | Heterologous system |
| SPT | 100–300 nmMovement detection < 25 nm | Heterologous system/culture of neurons |
N.A.: not applicable; N.D.: not determined; Co-IP: co-immunoprecipitation; PLA: proximity ligation assay; BRET: bioluminescence resonance energy transfer; FRET: fluorescence/Förster resonance energy transfer; TR-FRET: time-resolved FRET; SRET: sequential resonance energy transfer; SPT: single particle tracking
DA and glutamate NMDAR heteromerization in addiction
D1R-NMDAR heteromers
NMDARs are ligand and voltage-gated cation channels with high calcium (Ca2+) permeability that are critical in regulating synaptic transmission in the CNS. Many neurological and psychiatric disorders, including addiction, have thus been linked to alterations of NMDAR functioning [74]. NMDARs are formed through the heterotetrameric assembly of 2 mandatory GluN1 subunits with 2 modulatory GluN2(A–D) and/or GluN3(A–B) subunits, which mostly differ in their C-terminal domains (CTD) [75]. Subunit compositions shape kinetics of NMDAR-mediated currents and Ca2+ influx, as well as post-translational modifications of these subunits. For instance, phosphorylation of GluN2B on the Ser1303 residue by Ca2+/calmodulin-dependent protein kinase II (CaMKII) [76] and protein kinase C (PKC) [77], or the phosphorylation of Y1472-GluN2B by the non-receptor tyrosine of the Src kinase family (SFK), have been shown to enhance NMDAR functions [27, 77, 78], notably by favoring surface expression of the receptor [79].
The first description of a direct interactions between D1R and NMDAR has been reported by the group of Lee et al. and Pei et al. [38, 39] in cultured cells overexpressing both receptors and hippocampal tissues. Using blot overlay assays, the existence of two direct interaction sites between D1R and NMDAR subunits has been established. The C-terminus t3 domain of D1R [D1R-t3 (S417-T446)] binds to GluN2A subunits, while the D1R-t2 (L387-L416) region interacts with the C1 cassette (D864-T900) of GluN-1a subunits of NMDAR (Figure 2A). The functional roles of the interaction between D1R and GluN2A subunits of NMDAR have been less studied and remain elusive compared to the roles of D1R binding to GluN1 subunits, which have been more deeply investigated in different models. In the next paragraphs, this later D1R-GluN1 interaction will therefore be referred to as the D1R-NMDAR heteromer for simplicity. D1R-NMDAR interaction has been described in vivo in the mouse striatum [68], as well as in human post-mortem striatal samples [67]. Mass spectrometry experiments demonstrated that the association between the D1R-t2 fragment and the C1 cassette of GluN1 is driven by electrostatic interactions between an arginine-rich epitope of GluN1 and acidic residues within the D1R-t2 [80]. Interestingly, such molecular mechanism has already also been shown to underly the heteromerization between D2R and A2AR [81]; (see section D2R and A2AR heteromerization in addiction). Given their remarkable similarity and high degree of conservation across species, the epitopes involved in D2R-A2AR and D1R-NMDAR heteromerization may constitute a general mechanism underlying receptor-receptor interactions, which has been likely preserved through evolution as a result of their involvement in physiological functions. Consistent with the implication of acidic residues in D1R-NMDAR interactions, Woods and colleagues showed [80] that phosphorylation of the D1R-S397 residue within the D1R-t2 fragment strengthens its interaction with C1 cassette of GluN1. This was further confirmed by using FRET on COS-7 cells and high-resolution microscopy on cultured hippocampal neurons, which shows that an increased D1R-NMDAR interaction occurs with a phosphomimic version of D1R (i.e., D1R-S397D) when compared to the wild type form of D1R. It was also established that D1R-S397 phosphorylation relies on a mGluR5-dependent activation of casein kinase 1 (CK1), therefore supporting that mGluR5 signaling is an important regulator of D1R-NMDAR heteromerization in hippocampal neurons [59].
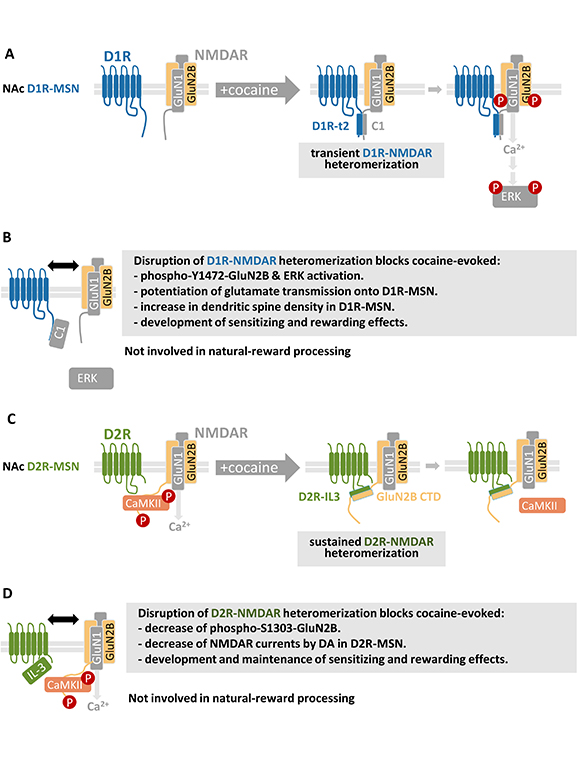
Modulation and functions of DAR-NMDAR heteromerization in response to cocaine. Schematic representation of cocaine-evoked (A) D1R-NMDAR and (C) D2R-NMDAR heteromerization and associated downstream signaling cascades. Impact of D1R-NMDAR (B) and D2R-NMDAR (D) disruption on molecular, cellular, and behavioral adaptations induced by cocaine. See main text for details. NAc: nucleus accumbens; D1R: DA receptor type 1; MSN: medium sized-spiny neurons; NMDAR: N-methyl-D-aspartate glutamate receptor; ERK: extracellular signal-regulated kinase
Besides post-translational modifications, studies on the impact of D1R and NMDAR agonists and antagonists on heteromer formation yielded contradictory results. Some authors reported a D1R agonist-dependent decrease of D1R-NMDAR heteromerization in cultured hippocampal neurons [38], while experiments in cell lines overexpressing both receptors demonstrated no effect of D1R or NMDAR agonists and antagonists [82]. Finally, it was later shown that a co-stimulation protocol of D1R and NMDAR with a low dose of glutamate combined with a D1R agonist induces an increase in endogenous D1R-NMDAR heteromers in cultured striatal neurons [68]. These contradictory results suggest that molecular mechanisms shaping D1R-NMDAR heteromers formation differ depending on the cell-type studied and/or on the strategies used to stimulate or antagonize partner receptors. The discrepancies observed on the impact of receptor agonists on heteromer formation may also depend on the cellular localization of these heteromers, which as yet unclear. Fiorentini and coworkers [82] demonstrated the presence of D1R-NMDAR heteromers in the striatal post-synaptic density (PSD), suggesting their presence at, or at close proximity to, synapses. Accordingly, application of NMDA to organotypic striatal cultures has been shown to recruit D1R at dendritic spine through a “synapse trapping” mechanism by which activated NMDAR act as a scaffold to recruit laterally diffusing D1R via their heteromerization within dendritic spines [83]. This supports that D1R-NMDAR heteromerization may preferentially occur at synapses. However, Zhang and colleagues [84] showed that the CTD of GluN1 subunits of NMDAR and D1R bind to the synaptic scaffold protein PSD-95 through domain partly overlapping with D1R-t2, therefore positioning PSD-95 as an inhibitor of D1R-NMDAR heteromerization that would limit D1R-NMDAR interactions at synapses. In agreement with this hypothesis, Ladepeche and colleagues [41] gathered evidence showing that D1R-NMDAR heteromers were enriched at peri-synaptic sites. The precise localization of D1R-NMDAR with regard to synapses is therefore not yet fully elucidated. This is an important issue since it has been previously shown that modulatory role DA on NMDAR functions differs depending on the localization of NMDAR at synaptic or extrasynaptic sites [85].
Based on these results a general consensus would therefore be that D1R-NMDAR heteromers are mainly located in the peri-synaptic and synaptic areas. This is in agreement with studies on their functional roles on synaptic plasticity showing a facilitating effect of D1R stimulation on NMDAR functions [86, 87] but see [38]. Indeed, the use cell-penetrating peptide disrupting D1R-NMDAR heteromerization in combination with electrophysiological recordings showed that D1R-NMDAR heteromers were key in controlling the facilitatory effect of D1R on NMDA currents and NMDAR-mediated long-term potentiation (LTP) in both the hippocampus [88] and in NAc D1R-MSN [68]. By contrast, the heteromerization of D1R with GluN2A subunits of NMDAR is not involved in the modulation NMDAR functions by DA in NAc D1R-MSN [68]. However, an intriguing question remains as the interaction between D1R and GluN1, which is the obligatory subunit virtually present in all NMDAR, selectively potentiates calcium influx and post-synaptic currents mediated by GluN2B-containing NMDAR [68]. This shows that D1R-NMDAR interactions are critical for the facilitation of NMDAR by DA but this crosstalk is bidirectional. In fact, it was established that D1R-NMDAR heteromer formation also allows NMDAR to increase both D1R cell surface expression and D1R-mediated cAMP accumulation [39], therefore showing that D1R-NMDAR heteromerization allows a mutual enhancement of both partner receptor functions. As multiple psychiatric diseases, and in particular, addiction, have been associated with changes in glutamate and DA transmission in the NAc leading to an increased activity of D1R-MSN over D2R-MSN, targeting D1R-NMDAR interactions may be of therapeutic relevance.
D2R-NMDAR heteromers
Liu and colleagues [89] were the first to demonstrate a direct physical interaction between D2R and GluN2B subunits of NMDAR within PSD-enriched fractions of the rat hippocampus, prefrontal cortex, and striatum. They identified the specific protein domain involved in the D2R-NMDAR interaction, with GST pull-down experiments revealing that the residues R220-A234 within the third intracellular loop of D2R (D2R-IL3) were essential for the binding of D2R to the CTD of GluN2B subunits of NMDAR. Given that alterations in DA and glutamate signaling integration in the striatum are crucial for regulating adaptations induced by psychostimulants [2], this study investigated how a single injection of a high-dose of cocaine modulates D2R-NMDAR interactions in the striatum. They observed that an acute cocaine injection triggers an increase of D2R-NMDAR heteromerization, paralleled by a decreased of phosphorylation of GluN2B on the S1303 residue, a post-translational modification known to enhance Glu2B-containing NMDAR functions [77, 78]. This was dependent on D2R signaling since the D2R antagonist Eticlopride blocked both cocaine-mediated inhibition of phospho-GluN2B-Ser1303 and D2R-NMDAR interaction, while a D2R agonist had opposite effects [89]. By using a penetrating peptide corresponding the R220-A234 fragment of D2R-IL3, which disrupts D2R-NMDAR heteromerization, it was shown those heteromers are required for the D2R-mediated inhibition of NMDAR currents. This inhibition of NMDAR relies on a competition mechanism whereby D2R-NMDAR heteromerization induced by cocaine disrupts the binding of CaMKII to GluN2B subunits of NMDAR and reduces CaMKII activity (Figure 2C). However, it should be mentioned that these results contradict more recent work supporting that the stimulation of D2R does not modulate NMDAR functions [90]. Nevertheless, at the behavioral level, disruption of D2R-NMDAR interactions inhibits the immediate hyperlocomotion and stereotypy responses induced by a single high-dose of cocaine [89]. However, this study did not explore the impact of D2R-NMDAR heteromerization on long-term alterations caused by the drugs of abuse, which is important with regard to the chronic nature of addiction. Nonetheless, this pioneering study was the first to establish that D2R-NMDAR heteromerization is required for the inhibition of NMDAR function by DA in D2R-MSNs, suggesting that these heteromers could play a role in addictive-like behavior.
Roles of DAR-NMDAR heteromerization in drug-evoked long-term adaptations
As previously described, DAR-NMDAR heteromers serve as cell-type specific integrators of DA and glutamate signals across both NAc MSN subpopulations, with D1R-NMDAR interactions enhancing NMDAR currents in D1R-MSN [68], while D2R-NMDAR heteromerization inhibits NMDAR in D2R-MSN [89], in response to DA. Considering the significance of a prevailing activity of D1R-MSN over D2R-MSN in promoting compulsive drug use [25], we investigated whether DAR-NMDAR heteromerization might contribute to the cellular, molecular, and behavioral responses to cocaine.
The effect of cocaine on DAR-NMDAR heteromerization in vivo was investigated using the locomotor sensitization paradigm, where mice receive five daily injections of a fixed dose of cocaine. This paradigm induces a progressive locomotor sensitization, which persists even after several weeks of abstinence and serves as a widely used model for studying lasting adaptations caused by drugs of abuse [2, 91]. Detection of heteromers through PLA showed that behavioral sensitization is associated with an increase in D1R-NMDAR and D2R-NMDAR heteromerization in the core and shell subdivisions of the NAc, 24 hours after the last cocaine injection compared to the control group. This mechanism was DAR-dependent, as administering D1R or D2R antagonists before cocaine blocked D1R-NMDAR and D2R-NMDAR heteromerization, respectively. Noteworthy, the interaction kinetics differed, with D1R-NMDAR heteromerization being transient and returning to baseline after 7 days of abstinence, while D2R-NMDAR in the NAc core subregion outlasts this abstinence period (Figure 2A) [67].
The role of DAR-NMDAR heteromerization in regulating different phases of cocaine-induced responses in vivo has been investigated by mean of adeno-associated virus (AAV)-based strategy developed to disrupt either heteromer subtype in a temporally-controlled manner. Disruption of D1R-NMDAR interactions was achieved by using an AAV that enables a doxycycline-inducible expression of a peptide corresponding to the C1 cassette (D864-T900) of GluN1, which binds to D1R [38], and blocks D1R-NMDAR interaction while retaining the independent functions of D1R and NMDAR outside of their heteromerization [68]. A similar approach was employed to interfere with D2R-NMDAR heteromerization, using a virus driving an inducible expression of a peptide corresponding to a small fragment (T225-A234) located within the 3rd intracellular loop of D2R, which efficiently disrupts heteromerization in vivo [67]. Using these viral tools, it was shown that disruption of D1R-NMDAR interaction in the NAc prior to and during single cocaine injection prevents the D1R-mediated phosphorylation of Y1472-GluN2B subunits of NMDAR, which enhances NMDAR functions [92] and downstream calcium-dependent activation of ERK in D1R-MSN (Figure 2A and 2B) [67, 68]. The inhibition of D1R-NMDAR in the NAc prior to and during repeated cocaine exposure also blocked the development of locomotor sensitization, as well as cocaine-evoked potentiation of glutamate transmission onto D1R-MSN. However, in mice that have been sensitized to cocaine, D1R-NMDAR disruption during a 7-day withdrawal from cocaine followed by a challenge injection of cocaine, did not affect the maintenance of established behavioral sensitization. This supports that cocaine-induced D1R-NMDAR heteromerization in the NAc selectively controls early phases of cocaine responses. In contrast, disrupting D2R-NMDAR interactions blocked both the development and maintenance of these responses [67]. This differential role of both heteromer subtypes was also evidenced in the protocol of conditioned place preference (CPP), which is pavlovian conditioning based on the association of the hedonic properties of cocaine with a specific environment. Here again, the inhibition of D1R-NMDAR interactions before and during the conditioning phase the CPP paradigm blocked the development of cocaine-induced CPP, as well as the associated increase in dendritic spine density in D1R-MSN. However, in animals that have developed a CPP, subsequent inhibition of D1R-NMDAR heteromerization did not alter the kinetics of CPP extinction nor its reinstatement induced by a challenge injection of cocaine. Conversely, blocking D2R-NMDAR altered both the development and the reinstatement of CPP. These effects appear specific to drugs as interrupting either heteromer subtype preserved palatable food-evoked CPP (Figure 2B and 2D) [67]. These observations suggest that targeting D2R-NMDAR shows promise for the development of innovative strategies to treat addiction, which is supported the by the increased D2R-NMDAR heteromerization observed in post-mortem tissues from subjects with a history of psychostimulant misuse compared to matched controls [67]. This finding raises question as to whether the role of this heteromer can be generalized to other classes of drugs than psychostimulants, an issue that we are currently actively pursuing. A recent study has investigated the role of D2R-NMDAR interactions in response to morphine. Co-immunoprecipitation experiments showed an enhanced D2R-NMDAR heteromerization in the striatum of mice exposed to morphine and the use of a cell-penetrating peptide, which slightly dampens D2R-NMDAR interactions in cultured neurons, was able to alter morphine-induced CPP [93]. However, these observations must be interpreted with caution since the interfering peptide used, although altering D2R-NMDAR interactions in vitro, primarily targets the arginine-rich domain of D2R-IL3 that mediates D2R interaction with A2AR [81] (see section D2R and A2AR heteromerization in addiction), rather than D2R-NMDAR interactions, which questions the specificity of these findings.
D2R and A2AR heteromerization in addiction
The D1R has been shown to interact with adenosine type 1 receptor (A1R) but the roles of these complexes have been mostly studied in the context of motor behavior, including L-DOPA-induced dyskinesia in preclinical models of PD or the restless legs syndrome [94, 95], which is beyond the scope of this review. This paragraph will therefore focus on the heteromerization of D2R with A2AR, which has been deeply investigated, including in preclinical models of addiction.
The A2AR is a purinergic GPCR expressed in striatal D2R-MSN [96, 97], which binds adenosine, an endogenous nucleoside with multiple roles in the CNS, notably in controlling neural excitability, synaptic plasticity, and neuronal degeneration [98]. The therapeutic use of A2AR antagonists may represent a promising strategy in neuropsychiatric diseases [98], since caffeine consumption, which antagonizes A2AR, alleviates depressive-like symptoms in preclinical models of the disease, including chronic unpredictable stress-induced social aversion, anhedonia, and helpless behavior observed in forced-swim test [99]. These observations therefore highlight the value of studying A2AR signaling in psychiatric disorders. The purinergic system plays an antagonistic role on DA signaling and indirect evidence for an involvement of D2R-A2AR heteromers in this antagonistic crosstalk originally comes from the observation that caffeine was also able to increase the antiparkinson actions of Levodopa and DAR agonists in rat models of PD [100]. Furthermore, it was later on showed that a A2AR agonist was able to decrease the affinity of D2R for its agonist apomorphine in plasma membranes purified from striatal samples [101], supporting allosteric modulations between D2R and A2AR possibly occurring as a result of D2R-A2AR heteromer formation. Consistently, multiple approaches, including imaging, BRET and co-immunoprecipitation, have been used to demonstrate the existence of D2R-A2AR heteromers in living cells and helped elucidating the molecular mechanisms behind their interaction and functional implications [94]. Several in vitro studies indeed revealed the existence of A2AR-D2R heteromerization by showing substantial colocalization of recombinant of A2AR and D2R in primary cultures of striatal neurons and immortalized cell lines [102, 103] and the use of FRET and BRET techniques supported the existence of D2R-A2AR complexes [103–106]. More recently, D2R-A2AR heteromers were detected by PLA in situ in the striatum of mice [66, 107], rat [108], and sheep [109], but also in human brain samples [69, 110]. Furthermore, detection of D2R interaction with A2AR using the “MolBoolean” imaging technique allowed to assess the proportion of individual A2AR or D2R receptors and D2R-A2AR complexes in vitro in heterologous cells and in primary striatal neurons as well as in vivo in sections from rat and primate PD models. This study revealed that a major fraction of D2R interacts with A2AR in MSN, while a significant proportion of A2AR are not involved in with D2R-A2AR complexes [71].
Functionally A2AR and D2R, which are respectively coupled to Gs/olf and Gi/o, form a heterotetrameric structure capable of binding with adenyl cyclase type 5 (AC5) [111, 112]. Electrostatic interactions between transmembrane domain (TM) 6 form the homodimer interface, while heteromeric interactions involve TM 4 and 5 of one receptor within each homodimer [111] and D2R interaction with A2AR relies on an arginine-rich domain within the IL3 domain of D2R, which is critical for it binding to A2AR through electrostatic interactions [81].
D2R-A2R heteromerization induces reciprocal allosteric interactions between both receptors since A2AR ligands reduce the efficacy and the binding of D2R agonists [37, 102, 109], while D2R stimulation inhibits A2AR-dependent activation of AC5 and reduces downstream cAMP levels in vitro [111] and in D2R-MSN in vivo in mice [113]. Moreover, patch-clamp recording of identified D2R-MSN showed that D2R-A2AR interactions are required for the D2R-mediated inhibition of NMDAR currents [114], therefore supporting that D2R-A2AR heteromerization should be considered as potential target in the field of addiction as it could contribute to the deleterious imbalance between D1R- and D2R-MSN activities induced by drugs of abuse.
Accordingly, D2R-A2AR heteromerization has been shown to increase in NAc shell of rats trained to self-administer cocaine [108]. From a functional stand point, the use of peptides corresponding to the A2AR TM5, which disrupts D2R-A2AR interactions, infused into the rat NAc shell inhibits the A2AR agonist-induced decrease of cocaine self-administration in rats [115]. Even though further characterization of the implication of cocaine-induced D2R-A2AR heteromerization in the NAc on cocaine-self administration would be of interest, these data strongly support that targeting D2R-A2AR interactions is a promising strategy to preserve D2R signaling, which is reduced in response to drugs of abuse [116, 117].
Noteworthy, the presence of D2R-A2AR complexes has also been reported on the plasma membrane of striatal astrocytes. Indeed, confocal imaging revealed that D2R and A2AR receptors were co-expressed on a subpopulation of striatal astrocytes in adult rats (40% of GFAP-positive astrocytes), particularly on vesicular glutamate transporter VGLUT1-positive astrocytes [118, 119]. Furthermore, co-immunoprecipitation experiments demonstrated the interaction between the A2AR and the D2R on purified rat striatal astrocytes, which was also confirmed by in situ detection of D2R-A2AR proximity through PLA in rat striatum slices [120]. Using an A2AR agonist, or a synthetic interfering peptide corresponding to the IL3 fragment of D2R that binds to A2AR [81], it was shown that these receptor complexes functionally interact at the astrocytic level to control glutamate release. Disruption of D2R-A2AR interaction indeed prevents the D2R-induced decreased of glutamate release induced by the stimulation of A2AR [118, 119, 121]. Based on the above-mentioned studies showing that targeting D2R-A2AR heteromerization is a promising strategy, it would be of interest to unravel to characterize the specific roles of D2R-A2AR heteromerization in astrocytes in drugs of abuse-evoked adaptations.
D2R and D1R heteromerization
The use of BAC transgenic mice expressing fluorescent reporter proteins in D1R- and D2R-MSN [11], or cell-type-specific viral tagging [122], revealed that 2–5% of MSN in the NAc core subdivision express both D1R and D2R, while this proportion can reach up to 10–15% in the shell subdivision [122]. Accordingly, D1R and D2R mRNA were shown to be co-expressed in a subset of MSN within the NAc and dorsal striatum in the mouse, rat, and rhesus monkey [123, 124]. The co-expression of D1R and D2R was also reported at the protein level [125–128] and D1R-D2R complexes were observed through PLA in the striatum of rodents and non-human primates [129–131]. However, the existence of such D1R-D2R heteromers in vivo has been challenged by Frederick and colleagues [128] who demonstrated that D1R-D2R complexes were not detected by PLA in MSN co-expressing D1R and D2R.
From a functional stand point, using receptor overexpression in heterologous systems, the group of Perreault et al. [127] made the original observation that D1R-D2R interactions result in a shift towards a non-canonical pathway whereby D1R-D2R heteromers signal through Gq, therefore eliciting a phospholipase C-mediated Ca2+ release from intracellular stores [127, 132]. Through PLA, the same group showed that cocaine exposure was able to promote D1R-D2R heteromerization in the NAc and dorsal striatum [123]. With the aim of evaluating the role D1R-D2R heteromerization in cocaine-evoked responses, a cell-penetrating interfering peptide corresponding to the t2 fragment of D1R CTD that binds to 2 adjacent arginine residues within the D2R-IL3 was used [133]. This approach indeed blocked D1R-D2R interactions, as well as the coupling of D1R-D2R complexes to Gq in vitro [132]. In vivo, the use of the cell-penetrating D1R-t2 peptide resulted in an amplification of cocaine-induced gene expression, as well as the sensitizing and rewarding effects of this drug [129] but decreased despair behavior in an acute model of depression [131]. However, it is worth mentioning that the D1R-t2 peptide, although disrupting D1R-D2R interactions, also interferes with D1R-NMDAR heteromerization [39, 41, 88] and has been shown to, by itself, activate the ERK pathway in the NAc and potentiate glutamate synapses in the hippocampus [41, 68], which could also explain its amplifying effects on cocaine-evoked responses. This supports the need of developing more selective strategies to investigate the role of D1R-D2R interactions in vivo, which is particularly relevant with regard to recent findings showing that deleting D1R selectively in D2R-MSN has major consequences on the fine-tuning of D1R-MSN and D2R-MSN activities, the modulation of basal locomotion activity, and several responses to cocaine [124]. Further work is therefore required to determine whether D1R-D2R heteromerization is involved in these important functions achieved by MSN expressing both receptors.
Towards new heteromers in drug and food addiction?
While the blockade of monoaminergic transporters by cocaine is undoubtedly a main mechanism responsible for the behavioral adaptations induced by cocaine, several evidence support that cocaine’s effects are mediated by its direct binding at the Sigma-1 receptor [134]. The functions of this atypical transmembrane receptor remain fuzzy, though disruption of Sigma-1 pathway has been implicated in a variety of disorders such as neurodegenerative diseases, in addition to psychostimulant addiction [135]. Interestingly, Sigma-1 receptor may interact with a variety of hormone and neurotransmitter receptors. In particular, heteromerization with the D2R—but not D3R or D4R—was evidenced through BRET in heterologous system [136]. Different Sigma-1 receptor agonists such as cocaine induce changes in the D2R-Sigma-1 heteromer through modifications of the D2R-D2R homomer. Cocaine also affects D2R signaling (detected through cAMP assays and ERK1/2 phosphorylation) in a Sigma-1 receptor-dependent manner. D2R and Sigma-1 receptor interaction was detected through coimmunoprecipitation on mouse striatal tissue and PLA on striatal slices. A similar negative cross-talk between D2R and Sigma-1 receptor regarding D2R-dependent ERK1/2 phosphorylation was detected on striatal slices from WT mice, while this effect was absent in Sigma-1 receptor KO mice [136].
The Sigma-1 receptor has also been shown to interact with the D1R, detected through BRET in heterologous system. Cocaine induces modifications of the subcellular distribution of Sigma-1, as well as of the quaternary structure of D1R homomers composing the D1R-Sigma-1 heteromer. Moreover, the binding of cocaine to the Sigma-1 receptor alters D1R-dependent pathways such as cAMP accumulation and ERK1/2 phosphorylation. This effect on D1R-dependent ERK1/2 signaling was recapitulated in mouse striatal slices by using WT vs Sigma-1 receptor KO mice [137]. D1R and Sigma-1 receptor may also form higher-order oligomers notably with the ghrelin receptor growth hormone secretagogue receptor 1a (GHS-R1a) as shown through BiFC (bimolecular fluorescence complementation) and SRET in heterologous system and indirectly with PLA in primary cultures of striatal neurons [138]. Interestingly, cocaine can inhibit GHS-R1 signaling through interaction of GHS-R1 with Sigma-1 receptor [139], which could participate in the anorexigenic effect of cocaine, ghrelin being classically considered as the “hunger” hormone.
Even though highly speculative, alterations of such heteromer-mediated crosstalk could be at the cornerstone of common pathogenic mechanisms between addiction and eating disorders that share dysfunction of the reward system. In the same vein, both D1R and D2R have been proposed to form heteromers with GHS-R1, leading to the potentiation of cAMP- and phospholipase C-dependent signaling, respectively [140]. Further identification and characterization of such receptor complexes might pave the way for the identification of discrete pathogenic mechanisms, but also identification of targetable factors with higher specificity and lower side effects.
Conclusions
Literature searches corresponding to the terms “receptor heteromer” indicate an increasing number of articles published in this field over the last 20 years. This surely illustrates the interest, both fundamental and potentially therapeutic, aroused by this mode of modulation of receptor function and downstream signaling. However, the number of studies demonstrating the existence of those heteromers and their specific roles in vivo, at least in the brain, remains limited. This can be explained, at least in part, from a technical point of view. Even though recent development in high-resolution microscopy helped to gain an unprecedent access to the dynamics of association and dissociation of these receptor complexes in living cultured neurons [59], there are only a few methods available for visualizing these interactions in vivo in the brain, and each method has a number of limitations (see Table 1). The field is also often facing a lack of precise information about the residues that are mandatory to form receptor complexes. Furthermore, partly overlapping domains of the same receptor may be involved in its heteromerization with different receptor partners, which often precludes the development of strategies targeting a receptor heteromer of choice without off-target effects. A better knowledge of the 3D structure of TM and cytoplasmic domains involved in receptor heteromerization is therefore required. This could be achieved through crystallographic structure or 3D in silico molecular modeling of the receptor heteromer interface, as already done for D2R-A2AR interactions [141]. This would help refining the receptor interaction interface through the identification of critical amino acids within binding pockets and developing small druggable compounds selectively targeting receptor heteromerization in vivo, therefore increasing the specificity of current interfering peptide-based strategies.
Herein, we focused on the description of receptor heteromers for which modulation and functions have been established in vivo in psychiatric disorders, mostly in preclinical models of addictions. However, it is worth mentioning that other receptor heteromers, which have not been studied in the field of addiction may be of importance in the field. Among them, the heteromerization of D3R with bf2 subunit of acetycholine receptor is of particular interest as it has been shown to shape VTA DA neurons [142, 143] or D1R-D3R interactions [144]. As illustrated in this review, much effort has been made to unravel the role of DAR heteromerization in the NAc in preclinical models of addiction. Because dysfunction of the mesolimbic system and DA signaling are a common feature of multiple psychiatric disorders, it would be important to expand our knowledge on the modulation and roles of DAR heteromers in preclinical models of other psychiatric disorders, as common alterations of DAR heteromer formation and function could, at least in part, participate to the high prevalence of comorbidities between addiction and other psychiatric disorders.
Abbreviations
| A2AR: | adenosine A2A receptor |
| AAV: | adeno-associated virus |
| AC5: | adenyl cyclase type 5 |
| BRET: | bioluminescence resonance energy transfer |
| CaMKII: | calcium calmodulin-dependent protein kinase II |
| CFP: | cyan fluorescent protein |
| CPP: | conditioned place preference |
| CTD: | C-terminal domain |
| DA: | dopamine |
| D1R: | dopamine receptor type 1 |
| DAB: | 3,3’-diaminobenzidine |
| DAR: | dopamine receptor |
| ERK1/2: | extracellular signal-regulated kinase 1/2 |
| FRET: | fluorescence resonance energy transfer |
| HRP: | horseradish peroxidase |
| IEM: | immunogold electron microscopy |
| MSNs: | medium-sized spiny neurons |
| NAc: | nucleus accumbens |
| NMDAR: | N-methyl-D-aspartate glutamate receptor |
| PD: | parkinson’s disease |
| PLA: | proximity ligation assay |
| PSD: | post synaptic density |
| RET: | resonance energy transfer |
| SPT: | single particle tracking |
| SRET: | sequential resonance energy transfer |
| TM: | transmembrane domain |
| TR-FRET: | time-resolved fluorescent energy transfer |
| YFP: | yellow fluorescent protein |
Declarations
Author contributions
AV, AP, and MCA: Writing—original draft. PT and PV: Conceptualization, Writing—review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Our research work was supported by the “Centre de la Recherche Scientifique” (CNRS), “l’institut National de la Santé et de la Recherche Médicale” (INSERM), “l’institut National de la Recherche pour l’agriculture et l’environnement” (INRAE), Sorbonne University and University of Bordeaux; University of Bordeaux’s IdEx “Investments for the future” program/[GPR BRAIN_2030] (PT), the “Agence Nationale pour le Recherche” (ANR) ANR-FrontoFat ([ANR-20-CE14-0020] to PT), ANR-Dropstress ([ANR-18-CE37-003] to PV), ANR-Striacode ([ANR-23-CE37-0006] to PV), Institut de Recherche en Santé publique (IReSP) Aviesan APP-addiction 2019 ([20II146-00] to PV and PT), IReSP Aviesan APP-addiction 2023 ([23IIsSP089] to PV and PT). AP is a recipient of a PhD fellowship from the “Ecole Universitaire de Recherche” (EUR Neuro, Bordeaux Neurocampus). AV is the recipient of a PhD fellowship from the French ministry of research, MCA is the recipient of 4th year PhD fellowship from the “Fondation pour la Recherche Médicale” (FRM). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2025.
Publisher’s note
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.