
Abstract
Stroke is the third leading cause of death and disability in industrialized countries. The estimated costs of stroke to the healthcare system are $85 billion in the United States and $40 billion in the European Union. Despite the extensive research over the past decades, only therapies aimed at restoring blood flow to the affected area have been successful. However, the high risk of causing intracranial hemorrhage limits the application of this type of therapy to a small number of patients. Several studies have shown that, in addition to its well-known regulatory function in erythropoiesis, erythropoietin (EPO) is a potent neuroprotective agent against ischemic stroke. However, the use of EPO to treat stroke requires long-term protocols, high doses, and multiple administrations, which may cause thromboembolic complications due to increased hematocrit and blood viscosity, making EPO treatment unsuitable. To mitigate these adverse effects, various EPO analogues with neuroprotective properties but lacking erythropoietic activity have been investigated. This review aims to provide an overview of the protective mechanisms of EPO and its derivatives in the treatment of stroke.
Keywords
Acute ischemic stroke, excitotoxicity, oxidative stress, erythropoietin analogues, neuroprotectionIntroduction
After heart disease, stroke is the second leading cause of death and disability worldwide [1]. According to the World Health Organization, stroke affects a total of 17 million people globally each year, resulting in 6.5 million deaths [1]. Since age is a major risk factor for stroke, the global ageing population will lead to an increasing number of stroke cases in the near future. Between 15% to 30% of stroke survivors often live with significant functional limitations [2] making stroke a major public health and economical issue. In 2019, the direct and indirect costs associated with stroke amounted to $85 billion in the United States and $40 billion in the European Union [3]. However, despite extensive research conducted over the past decade, the limited success of therapeutic strategies makes developing an effective stroke therapy one of the primary challenges in clinical neuroscience in the 21st century.
A stroke is a sudden loss of brain function caused by the alteration of the blood flow to the central nervous system (CNS). The neuropathological outcome depends on multiple factors, such as the duration and severity of ischemia, the presence of collateral vasculature, systemic blood pressure, etiology, and location in the brain [4]. Stroke may be classified into two main types: ischemic and hemorrhagic. Ischemic stroke occurs when there is a sudden decrease in blood flow to a specific brain area, leading to a loss of function. In contrast, hemorrhagic stroke results from a ruptured artery in the brain, causing bleeding in the region supplied by the affected artery. Approximately 88% of all strokes are acute ischemic strokes (AIS), 10% are intracerebral hemorrhages, and 2% are subarachnoid hemorrhages due to trauma or aneurysm rupture [5–7]. Based on tissue responses and biological events following the onset of stroke symptoms, three phases can be identified: (1) acute phase (the first 48 h), (2) the subacute phase (between 48 h and 6 weeks), and (3) the chronic phase (beginning 2 months after the stroke). The severity of these phases varies among individuals and depends on several factors, primarily the location and size of the affected area, the extent of collateral circulation, the metabolic state of brain tissue, patient’s age and comorbidities [8, 9].
Cerebral ischemia can be permanent if blood flow is not restored, or transient if blood flow is reestablished. The reduction of blood flow can affect a specific region of the brain (focal ischemia) or result in a complete shutdown of cerebral circulation (global ischemia), as seen in cardiac arrest [9, 10]. Brain damage depends on both the intensity and duration of the reduced blood flow. Blood flow in the infarcted area is not uniform, as there is a flow gradient influenced by collateral circulation, which plays a crucial role in stroke progression. AIS-related damage is determined by a complex sequence of factors acting over time and space [9–11].
In humans, normal cerebral blood flow (CBF) is approximately 50–60 mL of blood per 100 g of brain tissue per minute, ensuring a continuous supply of oxygen and glucose necessary for ATP synthesis. In neurons, ATP is primarily used to maintain the membrane potential, as well as for the synthesis and release of neurotransmitters. Since neurons lack the ability to store oxygen and glucose, they require a continuous blood supply. As a result, a severe reduction in blood flow, even for a shortperiod, leads to a drastic drop in cellular ATP and the loss of membrane potential, ultimately ceasing neuronal activity and causing cell death if oxygen is not rapidly restored [12].
In AIS, the decrease in blood flow is not uniform, and blood flow in the infarcted area is characterized by the loss of neuronal activity. The result is a central zone where blood flow is less than 15% of its normal physiological value, known as the “core”, and a surrounding area where blood flow is reduced but remains above 15%, referred to as the ischemic penumbra [13]. Beyond the penumbra, there is the region called oligemia, where blood flow is reduced but does not cause functional or metabolic alterations, and no apparent damage is observed [14]. In the ischemic core, the reduction in blood flow below 15% fails to supply sufficient oxygen and glucose, preventing mitochondrial oxidative phosphorylation from generating enough ATP to sustain neural viability [15]. As a result, neurons rapidly undergo necrosis, leading to irreversible brain tissue damage [16].
In the penumbra, blood flow is less severely reduced, typically receiving between 15% and 30% of its baseline level [17]. Neurons lose functional activity while maintaining their structural integrity because they receive enough oxygen to sustain metabolic activity, allowing the cells to remain viable, although their physiological functions are significantly diminished. Within the penumbra, neurons experience a decrease in electrical excitability but without irreversibe disruption of their ion gradients [18]. If CBF is not restored, neuronal death occurs through the activation of programmed cell death, primarily via mitochondrial apoptosis [19], which, unlike necrosis, requires energy. If CBF is not restored within the first 6 hours, penumbral tissue may progress toward cell death, contributing to the centrifugal expansion of the infarct [20–22]. The death of the neurons converts the penumbra into an ischemic core, further exacerbating neurological deterioration, which can ultimately lead to patient death [12]. Since the penumbra receives blood from collateral arteries, neurons can remain viable for several hours after stroke onset. Restoring blood flow in the penumbra, either by reopening the occluded vessel or by increasing collateral circulation, can salvage the penumbral tissue, allowing it to revert to a non-ischemic state and improving the patient’s neurological function [23].
While a CBF reduction above 15% induces changes in neurons that compromise their viability and functionality, it also triggers a hypoxic response by activating mechanisms that promote neuronal survival for hours, and even days, after a stroke occurs. For this reason, the period during which the penumbra remains viable presents a therapeutic window of opportunity, primarily depending on the severity of ischemia, CBF from adjacent vessels, the selective vulnerability of the affected neuronal tissue, and various systemic factors such as glycemia, systemic blood pressure and body temperature [16].
The aim of this review is to highlight the neuroprotective effects of NeuroEPO in AIS, an erythropoietin (EPO) analogue that retains neuroprotective potential while avoiding the negative side effects associated with the excessive increases in hematocrit and blood viscosity due to EPO’s erythropoietic activity. To achieve this, we first review the pathophysiological events occurring during AIS, including excitotoxicity, the oxidative and nitrosative stress, mitochondrial dysfunction leading to neuronal death by apoptosis, and inflammation. We then discuss the limited success of therapies aimed at restoring blood flow in the infarcted area, as well as therapies focused on activating mechanisms that protect neurons after infarction. Additionally, we examine the structure and function of EPO and its receptors along with its clinical applications in stroke treatment. Finally, we introduce the different EPO derivatives currently identified that lack erythropoietic activity and explore the mechanisms and potential uses of NeuroEPO for treating AIS.
Pathophysiology of AIS
Over the past 30 years, experimental and clinical findings have contributed to characterizing the pathophysiological basis of stroke [9, 10, 16, 22, 23]. The reduction in blood flow disrupts cellular homeostasis, triggering the ischemic cascade, which involves multiple processes, including physiological, biochemical, molecular, and genetic mechanisms that lead to the breakdown of cellular integrity. The key events of the ischemic cascade include ionic imbalance, glutamate-mediated excitotoxicity, Ca2+ overload, oxidative stress, mitochondrial dysfunction, blood-brain barrier (BBB) disruption, brain inflammation, microglial activation, and endothelial injury, ultimately damaging neurons, glial cells and endothelial cells [9, 10, 24–26].
Bioenergetic failure
The brain is highly vulnerable to ischemia in part due to its high metabolic demands. Although it constitutes only about 2% of total body mass, the brain consumes approximately 20% of the body’s oxygen and 25% of its basal glucose intake [27]. These high levels of oxygen and glucose are essential for generating the ATP required to maintain and restore membrane potential and to synthesize neurotransmitters [28]. It is estimated that glutaminergic synapsis accounts for most (~80%) of the brain’s energy expenditure, highlighting the close relationship between brain activity, glutamatergic neurotransmission, and energy requirements [29]. The brain lacks the ability to store oxygen and has only limited reserves of carbohydrate substrates and high-energy phosphate compounds. Since neurons have high energy demands, relying almost exclusively from ATP generated through mitochondrial oxidative phosphorylation, they require a continuous oxygen supply, making the brain particularly susceptible to ischemia [12].
The first effect of ischemia following its onset is a severe decrease of oxygen and glucose supply to neurons, leading to insufficient ATP synthesis by the electron transport chain (ETC) to sustain Na+/K+-ATPase normal activity. Within minutes of AIS onset, neurons affected by ischemia lose the ability to maintain Na+ and K+ gradients across the cell membrane. This disruption activates voltage-sensitive Ca2+ channels, causing excessive Ca2+ influx into neurons and triggering the excessive release of neurotransmitters, particularly glutamate, into the extracellular space [30–34]. Simultaneously, neurotransmitter reuptake from the extracellular space is reduced [35, 36], further contributing to their accumulation in the extra-neuronal space (Figure 1).
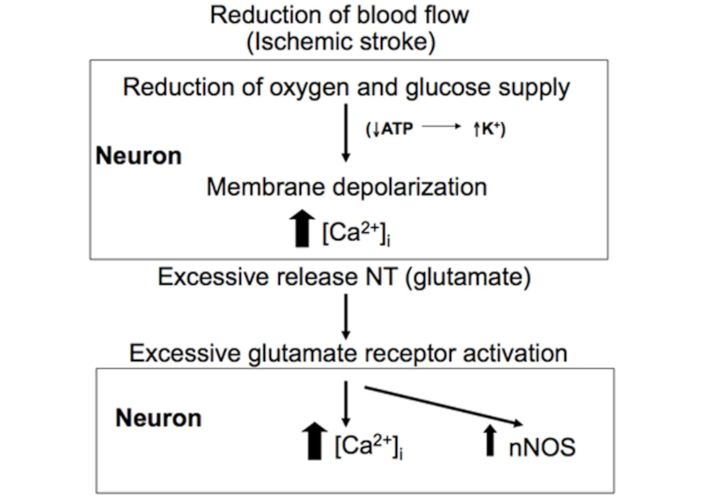
Ischemic stroke leads to excessive glutamate release. During ischemic stroke, the reduction in blood flow to the brain in an insufficient supply of oxygen and glucose, leading to inadequate ATP production required to maintain ionic gradients. This disruption causes excessive Ca2+ influx and an over release of glutamate. Reprinted from [37]. © 2012 The Author(s). CC BY 3.0
Excitotoxicity
In 1969, the term excitotoxicity was coined to describe the condition in which excessive glutamate acts on excitatory receptor, leading to cell death [37]. Excitotoxicity refers to neuronal death mediated by the excessive activation of ion channel-linked glutamate receptors, particularly N-methyl-D-aspartate (NMDA) receptors, resulting in cellular Ca2+ overload [32, 38–41], although astrocytes may also suffer damage by excessive glutamate levels [42]. Within the complexity and heterogeneity of mechanisms leading to neuronal death, glutamate excitotoxity is the central process underlying delayed neuronal death in AIS [39, 43–45]. Excitotoxicity disrupts cellular Ca2+ homeostasis, promotes free radical generation, induces mitochondrial dysfunction and triggers apoptosis [40]. Although each of these mechanisms can independently cause neuronal death, they act synergistically.
In the brain, glutamate is the most abundant neurotransmitter, playing essential roles in presynaptic and postsynaptic neurons as well as glial cells. It exhibits a Janus effect: While appropriate glutamate release is crucial for physiological functions and neuronal survival, excessive glutamate release contributes to pathological changes, including cell death [32, 34, 39, 46, 47]. Glutamate concentration at excitatory synapses is exquisitely fine-tuned regulated, as disruption of its homeostasis can lead to brain pathology, as observed in several neurodegenerative diseases. In the resting state, the extracellular glutamate concentration in neurons is mantained below 1 μM, while its cytoplasmic concentration is significantly higher (approximately 2 mM) and its concentration within synaptic vesicles reaches 100 mM. Upon the arrival of an action potential at presynaptic terminals, glutamate is released into the synaptic cleft, transiently increasing its concentration to approximately 1 mM [48]. However, this elevated concentration lasts only a few milliseconds, as glutamate is rapidly removed through enzymatic degradation and reuptake by high-affinity glutamate transporters (excitatory amino acids transporters, EAAT) located in pre- or post-synaptic neurons, but primarily on astrocytes, from which it is recycled into pre-synaptic neurons by the glutamate-glutamine cycle [49, 50]. Since astrocytes are responsible for the majority of extracellular glutamate uptake, whereas only a small fraction is recovered by neuronal uptake [46], they play a central role in the glutamate cycle in the brain. In astrocytes, glutamate is converted into glutamine by the enzyme glutamine synthetase, which is exclusively expressed in these cells [51]. The synthesized glutamine is then released by astrocytes and taken up by presynaptic neurons via glutamine transporters located on the neuronal membrane. Inside the neuron, glutamine is deaminated into glutamate, which then accumulates in synaptic vesicles, ready to be released into the synaptic cleft during neurotransmission [49, 50].
Overactivation of glutamate receptors and Ca2+ overload
After an ischemic stroke, within a few minutes, neurons release high levels of glutamate, which can exceed the capacity of the glutamate-glutamine cycle to maintain homeostatic glutamate levels. The presence of elevated glutamate in the extracellular medium leads to the activation of both synaptic and extra-synaptic receptors in neighboring neurons.
In the brain, the neurons have two kinds of the glutamate receptors: a) ionotropic glutamate receptors (iGluRs), which include NMDA, AMPA, and kainite receptors, are tetramers forming a central pore allowing ion flow upon activation; b) metabotropic glutamate receptors (mGluR), which are coupled with a G-protein [52–54]. Although all glutamate receptors contribute to the excitotoxic cascade [53], the NMDA glutamate receptor (NMDAR), located in the synaptic and extra-synaptic neuron membranes of the CNS [55, 56], is considered the primary mediator of excitotoxic damage in AIS [54, 57, 58]. NMDAR is a tetrameric protein complex composed of two essential GluN1 subunits and two GluN2 and/or GluN3 subunits. More than a dozen NMDAR subtypes have been characterized and this diversity is critical for its role in cell survival and death [58, 59]. Unlike other receptors, such as AMPA and kainite, NMDAR is a voltage-dependent channel that remains blocked by Mg2+ under resting conditions. AMPA receptor (AMPAR) activation by glutamate leads to membrane depolarization, which removes the Mg2+ block from NMDAR. This allows glutamate binding to NMDAR, opening the channel and permitting Ca2+ influx into the postsynaptic neuron due to its high permeability to Ca2+ and other cations [54].
Depending on the type of subunit that forms the tetramer, NMDAR exhibits different functions [60]. The properties of NMDAR depend on the type of GluN2 subunit that makes up the GluN1/Glun2 complex [61, 62]. It has been proposed that NMDARs containing GluN2B are excitotoxic and are predominantly located at the extra synaptic sites, while NMDARs containing GluN2A are neuroprotective and are primarily localized at the synaptic sites [55, 63]. In AIS, specific GluN2 subtypes (A–D) appear to play a pivotal role [64]. In neurons, NMDARs are part of a structure known as postsynaptic density (PSD), which appears electron-dense under an electron microscope [65, 66]. The PSD includes members of the membrane-associated guanylate kinase family (MAGUKs), which are essential structural proteins that anchor and organize NMDARs, governing the overall molecular organization of PSD. These MAGUK proteins consist of three PSD 95/large/zonula occludens-1 (PDZ) domains and one SH3-GK supermodule [66]. The PDZ domains of MAGUK proteins bind to the GluN2 subunits of the NMDARs [67–69]. Increased Ca2+ input induces binding of neuronal nitric oxide synthase (nNOS) to NMDAR via NMDAR-PSD95-nNOS complex [70].
In ischemic brain, excessive activation of the NMDAR-PSD-nNOS complex leads to excessive Ca2+ influx and activation of nNOS, resulting in the overproduction of nitric oxide (NO•) [65]. This leads to an accumulation of Ca2+ and NO• in the neuronal cytosol [71–74]. Excess of NO•, together with hydrogen peroxide (H2O2), which is produced from superoxide radical (O2•–) by the enzyme superoxide dismutase (SOD), triggers the formation of peroxynitrite (ONOO–), a highly reactive free radical that promotes cellular damage and ultimately neuron death [71, 72]. In AIS, Ca2+ entry through NMDARs activates cell death pathways more effectively than Ca2+ influx through other channels [75]. However, the efficiency of Ca2+ in triggering the excitotoxic signaling cascade via NMDAR can be reduced by disrupting the interaction between components of the NMDAR-PSD-95 or nNOS-PSD-95 complexes. In experimental animals, altering the interaction of NMDAR-PSD-95 or nNOS-PSD-95 complexes using small peptides has been shown to reduce the excitotoxic effects of Ca2+ and increase neuronal resistance to focal cerebral ischemia [76, 77]. The importance of the NMDAR/PSD-95 complex interaction in cerebral ischemia is further supported by findings showing that inhibiting PSD-95 binding to the NMDAR decreased brain damage without affecting NMDAR function [77]. In neuronal cultures, the binding of low-molecular-weight peptides to PDZ domains in PSD-95 induces structural changes in the PSD-95 complex, decreases NO• production, and reduces excitotoxicity [65].
It is well established that when excessive Ca2+ input surpasses the ability of mitochondria and the endoplasmic reticulum to uptake Ca2+ and regulate Ca2+ homeostasis in the cytosol, the resulting Ca2+ overload activates a cascade of mechanisms leading to neuronal death in the AIS [9, 73, 78]. There is strong evidence that loss cellular Ca2+ homeostasis is a key factor in neuronal death following an AIS [75, 78, 79]. As in other organs, Ca2+ is essential for proper brain function, so maintaining Ca2+ homeostasis in neurons is strictly regulated and any disruption can have severe consequences for the brain. Neurons possess specialized homeostatic mechanisms to control cytosolic Ca2+ concentration [78].
In the resting state, there is a significant difference in free Ca2+ levels between the extracellular medium, where Ca2+ concentration is estimated at 1–2 mM, and the cytosol, where Ca2+ concentration is approximately 100 nM. The maintenance of homeostatic Ca2+ levels in the cytosol results from a complex balance between Ca2+ influx, buffering, internal storage, and efflux [73, 80]. An overload of Ca2+ in the cytosol can saturate the buffering capacity of mitochondria and the endoplasmic reticulum, leading to mitochondrial damage and, ultimately, neuronal death [81, 82]. This Ca2+ overload triggers the activation of various enzymes, including lipases (phospholipase A2), proteases (calpain), phosphatases (calcineurin), Ca2+-dependent endonucleases and nNOS [38, 75, 78, 83–85]. Together with excessive free radical formation, these enzymatic activities cause mitochondrial dysfunctioin, cell membrane rupture and, ultimately, neuronal death [9, 10, 78, 86] (Figure 2).
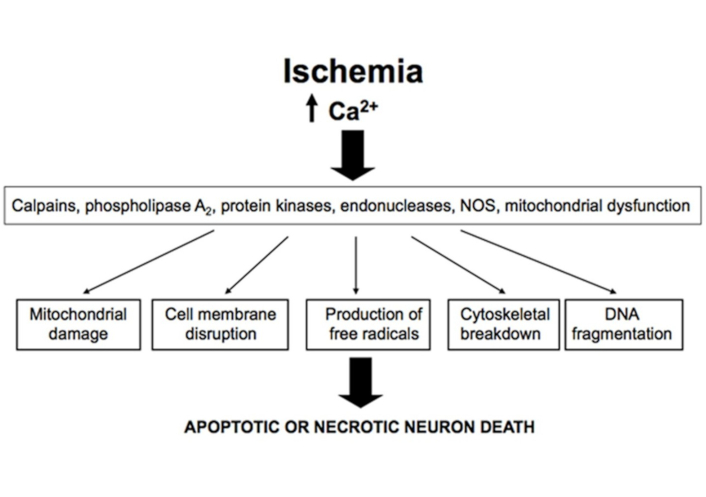
Effects of excessive Ca2+ accumulation in neurons after ischemia. Excessive cytoplasmic Ca2+ accumulation in neurons following ischemia induces activation of lipases, proteases, and endonucleases, which can lead to mitochondrial dysfunction, increased free radical production and DNA fragmentation, ultimately resulting in neuronal death. Reprinted from [37]. © 2012 The Author(s). CC BY 3.0
Other pathways independent of glutamate may contribute to the increase in intracellular Ca2+ levels. It has been reported that transient receptor potential (TRP) membrane channels, such as TRPM7 and TRPM2, are activated during ischemia contributing to elevated neuronal Ca2+ levels, as TRP channel blockage reduces Ca2+ accumulation in neurons exposed to excitotoxicity [87, 88]. Additionally, voltage-gated Ca2+ channels and Ca2+-permeable acid-sensing ion channels also contribute to increased cytosolic Ca2+ levels in ischemic neurons [89]. These findings indicate that Ca2+ accumulation in neurons within the infarcted area disrupts Ca2+ homeostasis, which is the primary cause of ischemic damage, rather than the specific pathway of Ca2+ entry into neurons. This may explain why drugs that reduce Ca2+ inflow through NMDARs have not been successful in clinical AIS treatments. However, since that NMDARs are the primary route for Ca2+ accumulation in ischemic neurons, reducing Ca2+ entry via NMDA receptor results in lower Ca2+ buildup in neurons and a subsequent reduction in brain damage.
In AIS, excessive NMDAR stimulation leads to excessive Ca2+ influx into the cytosol, activating nNOS and increasing NO• levels, as well as promoting Ca2+ accumulation in mitochondria. Excessive mitochondrial Ca2+ disrupts oxidative phosphorylation, leading to increased reactive oxygen species (ROS) production, heightened oxidative stress and mitochondrial dysfunction, ultimately resulting in increased apoptosis cell death in the ischemic area [90–95]. Mitochondrial dysfunction, oxidative/nitrosative stress, calpain activation and inflammation are among the key events triggered by Ca2+ overload due to excessive NMDAR activation in AIS neurons (Figure 3).
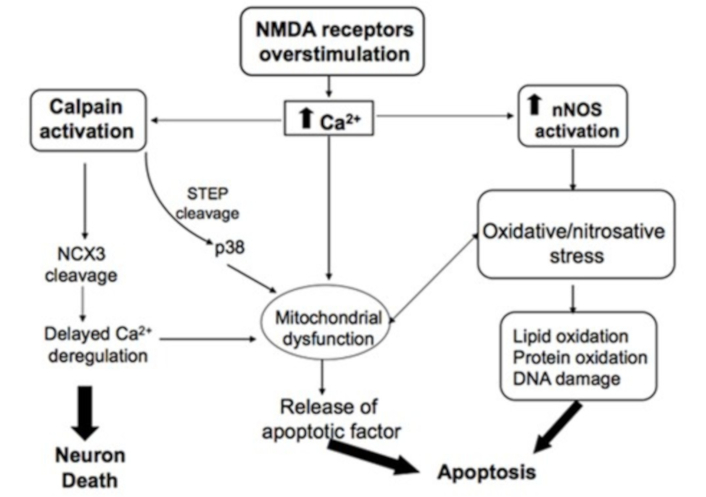
Main excitotoxic events induced by NMDAR activation in neurons. Excessive NMDAR stimulation by glutamate leads to elevated cytosolic Ca2+ levels. Ca2+ overload in the cytosol primarily results in the activation of calpains and nNOS, as well as mitochondrial dysfunction. Calpain activation leads to the inactivation of the Na+/Ca2+ exchanger (NCX3), a key regulator of intracellular Ca2+ homeostasis. Activation of nNOS increases nitric oxide levels, contributing to nitrosative stress, which can promote neuronal death either directly, through the oxidation of lipids, proteins and DNA, or indirectly by amplifying the mitochondrial apoptotic pathway. Reprinted from [37]. © 2012 The Author(s). CC BY 3.0
Oxidative and nitrosative stress in AIS
One mechanism involved in the excitotoxic cascade in AIS is oxidative stress, a term used to describe a state in which “an imbalance between prooxidants and antioxidants in favor of the oxidants, that leads to a disruption of cellular redox homeostasis” [96]. Redox processes play a fundamental role in nearly all biological functions, from bioenergetics and metabolism to vital cellular activities [97]. Cell physiology is closely linked to maintaining a delicate balance between oxidant production and elimination. Redox homeostasis results from the equilibrium between oxidant systems and an antioxidant system, and the normal functioning of cells depends on their ability to maintain this redox state. Neurons continuously produce superoxide radicals, mainly via the ETC during mitochondrial oxidative phosphorylation. While the production of radicals was once thought to pose a threat to cellular survival and contribute to aging, it is now understood that oxidizing species, particularly hydrogen peroxide, act as second messengers involved in essential cellular functions, primarily redox signaling. Under physiological conditions, a sophisticated antioxidant system maintains oxidative activity within homeostatic levels [98]. The antioxidant system include: i) low-molecular-weight molecules, such as ascorbic acid, glutathione, thiols, α-tocopherol, carotenoids, and polyphenols; ii) enzymes that directly neutralize oxidants, such as SOD, catalase, and glutathione peroxidase (GPx); iii) enzymes that remove peroxides, such as glutaredoxins, thioredoxins, and peroxyredoxins; iv) enzymes that repair oxidative damage, such as methionine sulfoxide reductase, disulfide reductases/isomerases, and sulfiredoxins; v) systems responsible for removing damaged material, such as proteasomes, lysosomes, proteases, phospholipases, and DNA repair enzymes [99].
The normal functioning of cells depends on a delicate balance between the production and elimination of oxidants. However, when antioxidant systems fail to maintain redox homeostasis, either due to excessive oxidant production and/or reduced antioxidant production, a state of oxidative stress occurs. The most common oxidants induced by excitotoxicity are derived from oxygen and nitric oxide, known as ROS and reactive nitrogen species (RNS), respectively. ROS can be classified into two main groups; free radicals and non-radical molecules. In mammals, the primary free radical ROS are the superoxide radical (O2•–) and hydroxyl radical (OH•), while the main non-radical ROS is hydrogen peroxide (H2O2). The principal RNS are the nitric oxide (NO•) and peroxinitrite (ONOO–) [100].
In mammals, ROS and RNS are continuously generated by various biochemical processes involved in cellular metabolism, both under physiological as pathological conditions. Under homeostatic conditions, hydrogen peroxide and nitric oxide are essential for normal neuronal function due to their roles in cell signaling processes [100]. However, under oxidative stress conditions, their harmful effects on lipids [101] and proteins [102] in cell membranes, as well as on DNA [103], make them key contributors to cellular damage, ultimately leading to cell death. The recognition of the significance of ROS in the physiology of living organisms has led to the emergence of a new discipline in Biology, Redox Biology, which studies the oxidation and reduction processes associated with life [97].
Several factors make neurons particularly vulnerable to oxidative stress, exposing the CNS to oxidative damage [104]. High ROS production results from a high metabolic activity and energy demand of neurons, which is primarily met through mitochondrial oxidative phosphorylation. When neuronal membranes contain high levels of polyunsaturated fatty acids and have reduced levels of endogenous antioxidant enzymes, particularly catalases, lipid oxidation of the neuronal membrane occurs [105].
In neurons, there are two main physiological sources of ROS: NADPH oxidases (NOX) and mitochondria. NOX family enzymes are transmembrane proteins that transfer electrons across biological membranes, using oxygen as the electron acceptor and producing the superoxide radical as a reaction product [106]. NOX was initially discovered in phagocytic cells, where it was identified as the first system to generate ROS as its primary function. In phagocytic cells, NOX is responsible for the “respiratory burst”, a mechanism by which NOX activation leads to the production of large amounts of ROS in the phagolysosome, oxidizing lipids and proteins and ultimately destroying the engulfed bacterium. Later, it was found that NOX is expressed in nearly all tissues, where its main function is ROS generation [106]. In neurons, NOX activity plays a crucial role in altering cell fate and modulating neuronal activity. NOX enzymes are involved in synaptic potentiation and long-term learning, although the cognitive impairment observed in NOX-deficient mice is relatively mild, suggesting that NOX has a modulatory rather than essential role. ROS generation by NOX enzymes has been implicated in various CNS diseases. In stroke, increased NOX expression in rats has been associated with greater brain injury [107], whereas in NOX-deficient mice exhibit a significant reduction in ischemic damage size [108].
ROS production derived from mitochondria is particularly important in high-energy demanding tissues. In humans, the brain has significantly higher metabolic activity compared to other organs. It consumes nearly ten times more of oxygen and glucose. Despite comprising only 2% of body weight, the brain accounts for 20% of the body’s oxygen consumption and 25% of its glucose consumption [27]. More than 90% of this oxygen is used by the mitochondrial ETC, where oxygen serves as the final electron acceptor. Under quiescent conditions, not all electrons travel through the ETC efficiently; some and are prematurely reduced by oxygen. Between 0.2% and 2% of the electrons supplied by NADH and FADH2 to the ETC leak from complexes I and III, directly reacting with oxygen to form the superoxide radical O2•– [109, 110]. The continuous requirement for an ATP production via oxidative phosphorylation makes mitochondria a significant source of O2•–. The lifespan of O2•– is 1 nanosecond, and it can rapidly undergo dismutation either spontaneously or enzymatically, primarily through SOD, which converts O2•– into O2 and H2O2 [111, 112]. Depending on the redox state, H2O2 can participate in cellular redox signaling, serve as a substrate in the Fenton reaction (in the presence of ferrous ion, Fe2+) to produce hydroxyl radicals (OH•), or to be reduced to H2O by glutathione [113–115]. The production of mitochondrial ROS is influenced by factors such as the mitochondrial membrane potential (Δψm), matrix pH, and the oxygen tension [116]. Mitochondria can also generate ROS through enzymes of the tricarboxylic acid (TCA) cycle, including aconitase, pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, as well as through outer mitochondrial membrane (OMM) enzymes, such as cytochrome P450 and enzyme monoamine oxidase [117]. In ischemic regions, elevated ROS levels play a critical role in the pathogenesis of AIS [118].
As a key component of oxidative stress, RNS, including NO• and ONOO–, are involved in brain damage in AIS. In neurons, nNOS is part of the NMDAR complex via PDZ domains and its activation by Ca2+ leads to the production of NO• through the catalytic conversion of arginine to citrulline. In AIS, excessive Ca2+ influx through NMDAR triggers the activation of both neuronal and inducible NOS, resulting in excessive NO• production [72, 119]. Accumulating evidence suggests that excessive NO• production from nNOS and iNOS significantly contributes to brain damage in AIS [72, 110, 120]. High levels of O2•– and NO• rapidly react to form OH• and ONOO–, both highly toxic species due to their strong reactivity with biomolecules such as lipids and proteins. These reactive species can also activate pathways that disrupt the BBB and exacerbate brain damage [72, 121]. In AIS, increasing ROS/RNS levels in the brain [118, 122, 123] indicate that oxidative and nitrosative stress play a crucial role in neurological damage following AIS [118, 124–126].
Mitochondria and NOX family are the main sources of ROS in AIS. NOX contributes to oxidative stress and ischemic brain damage by oxidizing NADPH to NADP+ and reducing O2 to O2•– [106, 127]. NO• and ONOO– have been shown to inhibit mitochondrial respiratory complexes, particularly complex IV, leading to increased O2•– production [90]. High levels of RNS can lead to ATP depletion through a mechanism initiated by DNA oxidation caused by RNS. Damaged DNA molecules activate poly(ADP-ribose) polymerase (PARP) leading to the depletion of its substrate, NAD+, which is a crucial electron donor for the mitochondrial ETC [128]. ROS and RNS can: a) directly cause the neuronal death by oxidazing lipids and proteins, irreversibly altering the architecture of essential cellular structures; b) indirectly activate pathological processes such as mitochondrial dysfunction [122, 129], the caspase cascade, and ultimately, neuronal apoptosis [109]. Excessive ROS production is a key factor in ETC impairment within mitochondria, leading to decreased ATP synthesis. This, in turn, exacerbates O2•– formation, further increasing mitochondrial dysfunction, which can trigger the intrinsic mitochondrial apoptotic pathway [93, 130] and reduce antioxidant activity [131].
The role of calpains in excitotoxicity
Another key mechanism induced by excitotoxicity, mediated by the accumulation of Ca2+ in neurons within infarcted areas, is the activation of calpains, a family of Ca2+-dependent cysteine proteases abundant in the CNS, with essential roles in synapses and memory [91]. Under physiological conditions, Ca2+ regulates the activity of a limited number of calpain molecules. However, when cytosolic Ca2+ levels rise to the micromolar range, calpains become overactivated, contributing to excitotoxic damage [91]. This occurs, in part, due to their ability to impair the activity of Na+/Ca2+ exchangers, which are essential for maintaining Ca2+ homeostasis in neurons [132]. The activation of calpain by increased cytosolic Ca2+ levels may also trigger the intrinsic mitochondrial apoptosis pathway. Calpains promote the cleavage of the pro-apoptotic component BH3 (Bid) to its truncated active form (tBid), which subsequently activates Bax. Bax then translocates to the OMM, where it induces pore formation, facilitating the release of cytochrome c (cyt c) into the cytosol, where it binds to the apoptosis-inducing factor (AIF), leading to increased caspase cascade activation and neuronal death [74]. In animal models of AIS, the use of calpain inhibitors has demonstrated neuroprotective effect [133].
Mitochondrial dysfunction and neuronal death by apoptosis in AIS
Mitochondria, especially in neurons, serve as the center of various biological processes, including proliferation, differentiation, Ca2+ signaling regulation, redox homeostasis and cell death control [134]. Under certain conditions, such as increased Ca2+ levels and oxidative stress, mitochondria lose their ability to produce sufficient ATP to sustain essential functions. This condition, known as mitochondrial dysfunction, is a key factor in many neuropathologies, including AIS [94, 135].
For mitochondria to perform their normal functions, the creation of an electrochemical gradient between the mitochondrial intermembrane space (IMS) and the inner mitochondrial membrane (IMM) is essential. This gradient generates a force known as the protonmotive force (PMF), which drives the protons to flow through complex V (F1F0-ATP synthase) for ATP synthesis. This process that requires the re-entry of protons into the matrix via complex V, leading to a decrease of the Δψm [136]. Uncoupling proteins (UCPs) located in the IMM regulate the proton gradient by facilitating proton flow into the mitochondrial matrix [137]. Alterations in Δψm occur when changes in mitochondrial membrane permeability take place and often represent the decisive event that determines neuronal survival or death. The permeability of the mitochondrial membrane is primarily regulated by the opening and closing of the mitochondrial permeability transition pore complex (mPTP) [130]. This complex is a non-selective, solute-permeable channel (< 1.5 kDa) present in the IMM. Although its exact molecular composition remains uncertain, F1F0-ATP synthase, adenine nucleotide translocase (ANT), and matrix cyclophylin D (CypD) are known components of the mPTP. Mitochondrial Ca2+ concentrations and redox balance play crucial roles in maintaining Δψm by regulating mPTP opening and closing. Under physiological conditions, transient mPTP opening helps regulate mitochondrial bioenergetics, cellular metabolism and ROS production. However, excessive Ca2+ accumulation in the mitochondrial matrix and oxidative stress lead to prolonged mPTP opening [138, 139], resulting in the leakage of protons, pyridine nucleotides (NAD+, NADP) and TCA cycle substrates from the mitochondrial matrix. This renders both the TCA cycle and the ETC inoperable, ultimately activating pathways that cause neuronal death [92, 117]. Overall, mPTP opening in the IMM results Δψm loss, increased ROS production and the excessive opening of voltage-dependent anion channels (VDAC) in the OMM. This allows substrates up to 5 kDA to pass through VDAC [140], further contributing to Δψm dissipation and severe mitochondrial dysfunction.
Loss of control over mitochondrial membrane permeability is a critical event in cell death. In AIS, Ca2+ overload, combined with oxidative and nitrosative stress in the infarcted area leads to the opening of the mPTP, resulting in the loss of Δψm and the activation of pathways that ultimately lead to neuronal death [92, 117, 141, 142]. The regulation of mitochondrial outer membrane permeabilization (MOMP) is essential for proper mitochondrial activity [143]. Sustained mPTP opening causes mitochondrial swelling and rupture of the OMM [144], triggering pathways that lead to neuronal death [92, 117]. In most cases, prolonged mPTP results in a state where the mitochondria rapidly hydrolyze both cytosolic and mitochondrial ATP. This ATP depletion causes the death by necrosis. However, if ATP synthesis is maintained, transient mPTP opening leads to apoptosis, a highly regulated and controlled process of programmed cell death [74, 145].
The maintenance of MOMP depends on the relationship between the levels of antagonist (Bcl-2 and Bck-xl) and agonists (Bax, Bad, and Bid) apoptotic proteins of the B-cell lymphoma 2 (Bcl-2) family. Activation of the intrinsic mitochondrial apoptotic pathway begins with the activation of the proapoptotic BH3-only protein and cleavage of Bid into its truncated form (tBid) by caspase-8. This process leads to the activation of inactive Bax in the cytosol, converting it to its active form. Once activated, Bax translocates to the OMM, where it forms pores that facilitate the release of cyt c into the cytosol. In the cytosol, cyt c binds to apoptosis protease-activating factor-1 (Apaf-1), leading to the formation of the apoptosome. This structure promotes the activation of procaspase-9, which in turn triggers the caspase cascade ultimately resulting in the activation of caspase-3. Caspase-3 then promotes the cleavage of enzymes such as PARP, which activates deoxyribonuclease, which leads to DNA fragmentation and neuronal death by apoptosis [146, 147].
Mitochondrial dysfunction can contribute to brain damage after a stroke through a mechanism other than apoptosis or necrosis, involving iron metabolism and oxidative stress. Ferroptosis is a form of caspase-independent programmed cell death caused by excessive lipid peroxidation of cell membranes, driven by iron and leading to oxidative stress through the Fenton reaction. This process ultimately results in cellular disruption and neuronal death [113–115].
Therapeutic treatment of AIS
Current stroke management prioritizes rapid diagnosis, timely intervention, and comprehensive rehabilitation. Clinically, the primary goal is to minimize brain damage. The following sections outline the current clinical approaches, including pharmacological and surgical treatments, highlighting their strengths and limitations.
Thrombolysis and thrombectomy for AIS treatment
Because the AIS is caused by a decrease in blood flow, the primary goal for treatment is the rapid restoration of blood flow to the affected brain area. For this reason, research has primarily focused on identifying suitable thrombolytics. For years, the only treatment authorized by the U.S. Food and Drug Administration (US-FDA) for AIS has been the administration of the tissue plasminogen activator (tPA), a thrombolytic drug that dissolves the clot. Treatment with tPA has proven effective, with significant improvement largely depending on the time elapsed between stroke onset and initiation of tPA therapy. Administering tPA as early as possible remains the most effective strategy for achieving a favorable outcome after stroke. tPA treatment should begin within 4.5 h of stroke symptom onset. Treatment outside this therapeutic window can result in intracerebral hemorrhage, where the risks outweigh the benefits [148, 149].
Several factors limit the use of tPA, including the need for an accurate diagnosis of stroke type (which requires imaging techniques that are not always available), knowledge of the stroke onset, and the risk of intracerebral hemorrhage. As a result, only 1–3% of stroke patients receive this treatment, with a success rate of less than 35% and full recovery in only about half of treated patients [150]. Despite significant advances in acute-phase stroke therapy over the past five years, including ongoing developments in thrombolytic strategies [150, 151] and mechanical thrombectomy [152, 153], most patients remain untreated due to limited therapeutic windows and strict selection criteria.
Similarly, endovascular thrombectomy, another US-FDA-approved procedure that mechanically restores blood flow, is restricted to a subset of patients [153]. Mechanical clot retrieval must be performed within 6–24 h of stroke symptom onset. Although thrombectomy extends the therapeutic window, the number of eligible ischemic stroke patients remains limited due to the risk of intracranial hemorrhage [154].
Molecular and cellular neuroprotection in AIS
In response to the large number of AIS patients who are not eligible for thrombolytic and/or endovascular thrombectomy therapy, significant efforts have been directed toward developing a drug capable of protecting the brain from the damage caused by AIS over time [155]. While several compounds have shown success in treating ischemic stroke in animal models, most have failed in clinical trials [156]. Several factors may explain this failure, including differences in the characteristics of AIS models used in animals versus those used in clinical trials, the heterogeneity of AIS in humans compared to the relative homogeneity in animal models, and the fact that preclinical studies are conducted on healthy young animals, whereas clinical trial participant are typically elderly individuals [157, 158].
EPO in AIS therapy
Biological functions of EPO
EPO is a sialoglycoprotein that plays a crucial role in regulating erythropoiesis. It is synthesized and secreted by type I peritubular interstitial fibroblasts near the corticomedullary border in the kidney and released into the bloodstream [158–161]. EPO controls red blood cell production in the bone marrow by binding to its receptor on the cell membrane of red blood cell precursor cells. This binding activates a signaling pathway initiated by the phosphorylation of Janus tyrosine kinase 2 (JAK2), which prevents apoptosis of colony-forming unit-erythroid (CFU-E) cells and promotes their proliferation and differentiation into mature erythrocytes [162]. In the human body, EPO regulates the production of approximately 200 billion new red blood cells every day.
Human EPO was purified from the urine of patients with aplastic anemia in 1977 [163] and its gene was cloned in 1985 [164, 165]. In 1986, treatment with recombinant human EPO (rHuEPO), produced using recombinant DNA technology from Chinese hamster ovary cells, was administered to 12 patients diagnosed with end-stage renal disease and anemia, resulting in a significant increase in hemoglobin levels in 9 of the patients [166]. Since its approval by US-FDA in 1989, EPO has been used clinically to treat anemia caused by insufficient EPO production [167]. Shortly after, it was approved in Europe and other parts of the world. Since then, more than 1 million people have benefited from the use of rHuEPO [168].
For a time, the only clinical application of EPO was in treating various types of anemia, particularly those associated with chronic kidney disease. However, studies beginning in the late 20th century revealed that, in adult humans, EPO is also expressed in other tissues, including the brain [169, 170], where it does not serve an erythropoietic function [171–175]. The first animal studies on EPO’s effects in stroke demonstrated its potent neuroprotective properties [176–182]. Subsequent studies [179–181] confirmed EPO’s neuroprotective effects in ischemic stroke treatment. Alongside these neuroprotective effects, the extensive clinical use of EPO in thousands of patients with due to chronic kidney disease [159] has paved the way for exploring EPO as a potential treatment for AIS in humans [182–187].
Structure and expression of EPO
EPO is a glycoprotein with a molecular mass ranging from 30 to 34 kDa, depending on the carbohydrate content. The peptide core of mature EPO consists of a chain of 165 amino acids, folded into four α helices that are linked by two disulfide bridges: one between cysteines 6 and 161, and another between cysteines 29 and 33 [188]. EPO is composed of approximately 60% protein and 40% carbohydrate, with four oligosaccharide chains terminated by sialic acids, three N-glycosylation sites at asparagine 24, 38 and 83, and one O-glycosylation site at serine 126 [189]. The sialic acid content in the EPO molecule plays a critical role in its plasma half-life, which under normal conditions ranges from 4 to 6 hours [190]. AsialoEPO is rapidly degraded by hepatocytes [191], whereas increasing glycosylation is clinically used to enhance erythropoietic activity by prolonging EPO’s half-life. The strength of EPO’s interaction with its receptor and its biological activity depends on the degree of glycosylation [192].
In addition, the interaction of EPO with its receptors and the modulation of its biological activity are controlled by the distribution of glycosylated chains within the EPO molecule [192]. Like other asialo-glycoproteins, asialo-EPO is rapidly removed via hepatocytes galactose receptors [191], as galactose is the preterminal sugar of the glycans. Completely desialylated EPO has a plasma half-life of approximately 1.4 minutes. Conversely, the introduction of additional N-glycans into recombinant EPO through site-directed mutagenesis results in prolonged in vivo survival of the molecules [192].
The human EPO gene is located on the long arm of chromosome 7 (q11–q22) [193]. It consists of five exons, which encode a prohormone of 193 amino acids, and four introns [165]. The active form of the hormone results from the excision of a 27-residue peptide, which is cleaved prior to secretion. The expression and secretion of EPO by kidney cells are primarily regulated by hypoxia and occur in response to anemic and hypoxic conditions [168, 194]. Tissue O2 pressure (pO2) depends on hemoglobin concentration, hemoglobin’s O2 affinity and blood flow rate [195]. Cells sense oxygen concentration decreases through hypoxia-inducible factor 1α (HIF-1α) stabilization [196–198]. HIF-1α is a subunit that, under hypoxia conditions, binds to the HIF-1β subunit to form HIF-1, a member of the HIF family. Under normoxic conditions, prolyl hydroxylase (PHD) and factor inhibiting hypoxia (FIH) use oxygen to hydroxilate the HIF-1α. This hydroxilated form of HIF-1α binds to von Hippel-Lindau ubiquitin E3 ligase (pVHL), leading to ubiquitination and proteasomal degradation. Under hypoxia, PHD and FIH are inhibited, preventing pVHL from binding to HIF-1α, which is then stabilized and able to associate with HIF-1β. HIF-1 translocates to the nucleus, where it binds to the p300 coactivator. This complex then binds to the hypoxia responsive element (HRE) in the EPO gene promoter, inducing EPO expression [198]. This transcriptional mechanism activates 100–200 genes involved in erythropoiesis, angiogenesis, autophagy, and energy metabolism [199]. Beyond hypoxia, EPO production can also be induced by several other stimuli, including mechanical damage, infection, metabolic stress, elevated temperature, intense neural activity, and ischemic stress [174].
Structure and expression of EPO receptors
The fact that EPO exerts different functions in different tissues implies the existence of different molecules acting as EPO receptors (EPOR) [168, 179, 183]. In CFU-E cells, EPO binds to a receptor formed by the homodimerization of two identical 66 kDa monomers expressed on the cell membrane. The binding of EPO to EPOR leads to the formation of EPOR-EPOR (EPOR2) complex, known as the canonical isoform. The interaction induces the phosphorylation of the JAK2, which is constitutively bound to the EPOR, thereby triggering the signaling pathway that promotes the expression of anti-apoptotic proteins, facilitating precursor cells in their transition to erythrocytes. Different isoforms of EPOR have been identified in the CNS. In neurons, along with EPOR2, the beta common receptor (βcR), also known as CD131, has been identified [200]. The binding of EPOR monomer to βcR leads to the formation of the EPOR/βcR heterodimer, whose activation initiate the same JAK2 signaling pathway as EPOR2 [201]. In the CNS, neurons, astrocytes, microglia, and endothelial cells express EPOR/βcR [202]. EPO derivatives, including neuro-EPO, carbamylated erythropoietin (CEPO), and asialo-EPO, preferentially bind to EPOR/βcR [202].
The functions of endogenous EPO in the CNS
Different studies confirm that the brain synthesizes EPO, which can carry out non-erythroid biological functions [171–173, 203]. In the brain, EPO has various effects, acting both as an inhibitor (of inflammation, apoptosis, and oxidative stress) and as a stimulator (promoting neuroprotection, tissue protection and repair, neuronal plasticity, memory enhancement, cell proliferation, and neurogenesis) [175, 179, 183, 204, 205].
The diversity of functions exerted by EPO in the CNS may, in part, be due to the different isoforms of EPOR and the various intracellular signaling pathways activated by the phosphorylation of JAK2 [202]. Phosphorylation of JAK2, in turn, leads to phosphorylation of phosphatidylinositol 3-kynase (PI3K), Ras-mitogen-activated protein kinase (MAPK), and signal transducer and activator of transcription (STAT5), which modulates the expression of various genes involved in proliferation, differentiation and cell survival [205, 206]. EPO may protect neurons through a combination of mechanisms, including limiting the production of ROS, enhancing the expression of anti-apoptotic proteins, inducing anti-inflammatory cytokines and promoting neurogenesis [207].
EPO therapy in preclinical studies of AIS
Abundant evidence confirms the potential of EPO for neuroprotection in the treatment of stroke. The neuroprotective efficacy of rHuEPO has been tested in several animal species (mouse, rat, gerbil, and rabbit) using models of focal and global cerebral ischemia, primarily by inducing stroke through permanent or transient occlusion of the middle cerebral artery (MCAO). This artery supplies blood to a large portion of the cerebral hemispheres and is responsible for approximately 80% of strokes. The results indicate that EPO promotes a reduction in neuronal death, improves outcomes, and significantly decreases brain infarct volumes [208–212].
Since 1998, exogenous EPO has been shown to exert potent neuroprotective effects in experimental models of focal and global cerebral ischemia [177–179, 182, 186, 208, 213, 214], yielding encouraging results in the treatment of ischemic stroke, particularly in preventing ischemic neuronal injury [176–179, 186, 215]. In mice, intracerebroventricular injection of EPO 24 hours prior to MCAO significantly reduced infarct volume [177]. The survival rate after 14 days in adult male C57 BL/6 mice treated with EPO (5,000 IU/kg) increased to 80%, compared to only 50% in untreated mice. EPO also improved neurobehavioral outcomes on days 3 and 7 post-MCAO and attenuated axonal injury caused by AIS [212]. Similarly, in a gerbil model of global ischemia, infusion of EPO into the lateral ventricles prevented hippocampal CA1 neuron death and increased synapse density in the same region [176]. In rats subjected to MCAO, treatment with EPO (5,000 IU/kg) reduced stroke size by 75% and suppressed apoptosis in ischemic penumbra [178]. Intravenous administration of a single dose of 1,000 IU/kg rHuEPO before the onset of a 60-minute transient MCAO protected the brain from ischemic damage [211]. A meta-analysis of sixteen studies exploring the efficacy of EPO in experimental focal cerebral ischemia concluded that administration of EPO AIS reduced stroke size by 32% and significantly improved neurobehavioral deficits [182]. Administration of EPO within 6 hours of stroke onset was more effective was more effective than administration after 6 hours [180]. Additionally, in mice undergoing transient cerebral ischemia, increased EPO expression was associated with a reduction in infarct size [216]. Other studies have demonstrated that, beyond reducing infarct volume, EPO also improves learning capacity in gerbils and prevents navigation disability in rats [176].
These results indicate that EPO is a potent neuroprotector agent capable of mitigating the various mechanisms that contribute to brain injury in stroke. Preclinical research demonstrates that EPO attenuates excitotoxicity, promotes intracellular Ca2+ homeostasis, reduces oxidative stress and inflammation, supports Δψm maintenance, and plays a crucial role in preventing mitochondrial dysfunction and the activation of the apoptotic cascade [177, 179, 183]. Additionally, EPO increases the levels of the anti-apoptotic protein Bcl-2 and facilitates the formation of tBid-Bad channels, which release cyt c from mitochondria, promoting neuronal recovery in ischemic penumbra [146, 177, 217].
Different doses and routes of EPO treatment in AIS
Since the beginning of its therapeutic use, rHuEPO has been administered via subcutaneous, intraperitoneal, intravenous and intranasal at high doses to ensure that it crosses the BBB and reaches sufficient levels to activate brain tissue cells expressing EPOR [218]. However, there is no convincing evidence demonstrating that, under conditions of BBB structural integrity, EPO can reach the brain parenchyma. Only molecules that are highly lipid-soluble and have a molecular mass of less than 400 Da, including amino acids, can pass through the BBB [219]. In contrast, EPO is a large (34 kDa), highly glycosylated and negatively charged protein, making it poorly suited to cross the BBB. Normally, proteins and peptides require a receptor-mediated translocation system to traverse the BBB. In the case of EPO, it has been suggested that it may cross via a receptor-mediated transcytosis mechanism [220]. The presence of EPOR in cerebral capillaries, along with findings that injection of labeled EPO is blocked by the administration of unlabeled EPO, led to the hypothesis that EPO accesses the brain through the BBB [180]. However, studies using radiolabeled EPO in rodents and primates showed that its entry into the brain is very slow and occurs via a non-specific mechanism [221].
In early studies on the efficacy of EPO in the brain, EPO was administered via intraventricular or intracerebral routes in animal models [101, 102, 222]. However, these invasive administration methods require surgical expertise and can cause brain damage and infections, making them unsuitable for clinical use. More recent studies in several species, including humans, have confirmed that when EPO is administered in high doses, both intravenously and intraperitoneally, a sufficient amount crosses the BBB to mediate neuroprotection against AIS [180, 182, 221].
Since only 1% of the administered EPO reaches the brain parenchyma [180, 221], effective AIS treatment would require the injection of a high dose of EPO. Although a relatively small fraction crosses the BBB and exerts its neuroprotective effects, the majority remains in systemic circulation, where it binds to EPOR in CFU-E cells, promoting their proliferation and differentiation into mature erythrocytes [162]. This leads to an increase in hematocrit levels, raising the risk of cerebral thrombosis and hypertension [223], which ultimately limits the clinical use of exogenous EPO in AIS treatment.
There is a great deal of variability in the dose of EPO to be used for the treatment of AIS. In rats, intravenous doses of rHuEPO between 500 to 5,000 IU/kg injected at 6, 24, and 48 h after initiation of AIS caused a significant reduction in infarct volume and neurologic deficit after 24 days [181]. Treatment with intra-arterial doses of 800 IU/kg of rHuEPO at the onset of AIS attenuated infarction volume and improved neurobehavioral outcomes at 2 h and 24 h after MCAO [224]. In humans, intravenous injection of a bolus of 16,000 IU of EPO, followed by additional injections of 8,000 IU every 12 h up to a total dose of 56,000 IU of EPO for 3 days, showed an improvement in neurological damage caused by AIS after 14- and 18-day evaluation [225]. In humans, subcutaneous administration of 5,000 IU 48 h and 72 h after stroke significantly recovered from neurological damage after 90-day evaluation [186], while in other studies the concentration supplied was between 3,500 to 10,500 IU of EPO per day [223, 226].
In an attempt to minimize the side effects associated with high-dose EPO administration, the intranasal route has been explored. This approach allows to bypass the BBB while reducing systemic exposure. The intranasal delivery of rHuEPO is an effective, safe and non-invasive method for introducing rHuEPO into the brain while avoiding side effects [227–229]. In rats with MCAO-induced AIS, intranasal administration of rHuEPO at doses of 4, 8, 12, and 24 IU, 10 minutes after occlusion, has been shown to effectively reduce infarct volume and cell damage in the ischemic hemisphere while improving behavioral dysfunction 24 hours after cerebral ischemia [230]. Comparatively, the intranasal route is 10 times faster, requires smaller doses of rHuEPO than the intraperitoneal route, and presents a lower risk of collateral damage [231, 232]. Thus, intranasal administration represents a promising method for efficiently delivering EPO and its derivatives to the brain.
The mechanism of EPO in AIS
The mechanism by which EPO exerts its function in the brain is similar to the one by which it induces erythropoiesis [206]. The difference lies in the EPOR isoform: while hematopoietic cells express the homodimer (EPOR2), non-hematopoietic organs present a heterodimeric receptor [205, 206]. In both cases, the binding EPO to its receptor triggers the phosphorylation of JAK2, which subsequently activates three main signaling pathways: phosphatidylinositol 3-kinase (PI-3K)/AKT (protein kinase B), MAPK, and signal transducer and activator of transcription 5 or 3 (STAT5 or STAT3) [206, 216]. STAT5 induces the expression of anti-apoptotic proteins (e.g., Bcl-xl), while PI-3K and MAPK inhibit pro-apoptotic proteins (Bax, Bad) [168]. The action of EPO concludes with the dephosphorylation of the EPOR complex and the internalization of the EPO/EPOR complex. After internalization, approximately 40% is degraded by the proteasome, while 60% of the EPO is recycled into the extracellular medium [233].
The clinical use of EPO as a neuroprotective agent against AIS
Results from experimental studies on the neuroprotective effects of EPO prompted the initiation of clinical trials to evaluate rHuEPO in patients with AIS. The first clinical study on the effects of EPO treatment in AIS was conducted by the Göttingen EPO Stroke Study, which demonstrated that rHuEPO is safe and has beneficial outcomes in AIS patients [215]. Safety was analyzed in 13 patients who received 33,000 IU of rHuEPO once daily for the first 3 days after the stroke. Efficacy was evaluated in 40 patients who received the same dose of rHuEPO within 5 hours of stroke onset. Treatment with rHuEPO did not raise safety concerns, as treated patients rHuEPO exhibited cerebrospinal fluid (CSF) levels of rHuEPO from 60 to 100 times higher than untreated, along with a reduction in infarct size. The study concluded that high doses of NeuroEPO were well tolerated by AIS patients and that, after 1 month, clinical outcomes had improved [215]. However, a subsequent trial conducted by the same group failed to demonstrate any beneficial effects of EPO in AIS [234]. The multicenter phase II/III clinical trial involved 460 AIS patients who received rHuEPO or a placebo within six hours of symptom onset. The results did not support a beneficial effect of rHuEPO treatment. Moreover, the mortality rate in the rHuEPO-treated group was 1.8 times higher than the placebo group. These negative findings dismissed the generated by earlier clinical trials. However, a thorough analyses of the methodology revealed significant differences between the phase I and phase II/III clinical trials. Surprisingly, the same protocol was not followed in both trials: in phases II and III, 63% of patients had previously been treated with tPA, whereas tPA was not used in phase I. A subsequent study analysing a subgroup of phase II/III patients who had not received tPA confirmed the results observed in phase I [184]. Further human studies have reaffirmed the neuroprotective effects of EPO, concluding that long-term EPO treatment significantly reduces the adverse neurological effects of AIS [186, 215, 225].
Side effects of EPO use on the brain
Along with the beneficial effects of EPO in stroke treatment, its therapeutic use is limited by the difficulty of crossing BBB, which requires the administration of high doses of rHuEPO. Three hours after an intravenous injection of 5,000 IU, less than 2% of rHuEPO crosses BBB [174]. While rHuEPO exerts the neuroprotective effect, its presence rHuEPO in the bloodstream leads to increased hematocrit, blood viscosity, and blood pressure, all of which significantly raise the risk of thrombus formation and secondary stroke [224, 235]. These undesirable effects make rHuEPO unsuitable as a treatment for AIS. Recognizing both the beneficial and adverse effects of EPO in stroke treatment, research has focused on developing EPO derivatives with neuroprotective activity but without erythropoietic effects.
EPO derivatives without erythropoietic activity
Several EPO-derived molecules with reduced or no erythropoietic activity but with neuroprotective effects have been tested for the treatment of AIS [205, 206]. These analogues include asialo-EPO [236, 237], CEPO [238, 239] and rHuEPO with low sialic acid content (NeuroEPO) [204].
Preclinical EPO derivatives in AIS treatment
CEPO
CEPO is a modified form of EPO in which the carbamylation of lysine residues alters its conformation and function. This medication allows CEPO to retain its neuroprotective activity while losing its hematopoietic activity [240]. The structural alteration prevents CEPO from binding to EPOR2 homodimer while preserving its ability to bind to the EPOR-βcR heteroreceptor. As a result, CEPO specifically targets cells expressing the EPOR-βcR heterodimer. In vitro and in vivo experiments have shown that, due to its anti-apoptotic and anti-inflammatory properties, CEPO prevents or reverses neuronal damage but lacks the ability to activate signaling pathways leading to cell proliferation [238–240].
Asialo-EPO
Asialo-EPO is a derivative of rHuEPO obtained by removing sialic acid from glycosylated chains using the enzyme sialidase. This modification accelerates the degradation of asialo-EPO by shortening its half-life without altering its affinity for EPOR. Consequently, asialo-EPO retains its neuroprotective capacity while significantly reducing hematopoietic activity, preventing an increase in hemoglobin concentration [236].
Effect of NeuroEPO in AIS
NeuroEPO is a modified form of rHuEPO obtained from Chinese hamster ovary cells and produced by the Centre of Molecular Immunology in La Havana (Cuba). Unlike other EPO derivatives (asialo-EPO and CEPO), NeuroEPO does not undergo any chemical modification. It differs from kidney-synthesized EPO only in its low sialic acid content, which eliminates its erythropoietic activity while preserving its neuroprotective properties [241]. Due to its low sialic acid content (4–7 mmol/mmol protein in NeuroEPO compared to 12–14 mmol/mmol protein in rHuEPO), intravenously administered NeuroEPO is rapidly degraded by the liver and has a very short half-life. To overcome this limitation, NeuroEPO formulations have been developed for intranasal administration, providing a direct and efficient route to the brain [209, 212, 241]. Furthermore, nasal administration, tested in various rodent and primate stroke models, has demonstrated superior therapeutic efficacy compared to conventional rHuEPO, without inducing its adverse effects [209, 230, 242, 243].
Preclinical studies of NeuroEPO in AIS
Preclinical studies of NeuroEPO in an in vivo model of stroke
The neuroprotective effect of NeuroEPO in AIS has been analysed in animal models. In Mongolian gerbils with stroke induced by MCAO, an intranasal dose of NeuroEPO (249 UI/10 μL) administered immediately after ligation improved neurological status and cognitive function, promoted recovery of the penumbra area, and reduced damage in specific brain regions such as the hippocampus, temporal cortex and thalamus [209]. In a similar study, using the same intranasal dose administered every 8 hours for 4 days, animals showed a 25% higher survival rate, as well as improved neurological scores and behavior 7 days after the ischemia event [230]. Another study, with a longer follow-up of five weeks, demonstrated reduced mortality, significant improvements in sensory and motor functions and a delayed neuronal death in the brain [188].
Preclinical studies of NeuroEPO in an in vitro model of stroke
Since excitotoxicity is the primary mechanism involved in neuronal damage after stroke [40], the effect of NeuroEPO in stroke was analyzed using a primary culture model of cortical neurons exposed to high glutamate levels. Neuron viability, morphological changes and the cellular and molecular mechanisms mediating its action, such as oxidative stress, mitochondrial dysfunction and apoptosis, were assessed [242, 243]. To determine the direct effect of NeuroEPO on neurons, all cultures were treated with cytosine arabinoside type 2 to eliminate astrocytes and other glial cells. While the removal of these cells prevents neuron-glia interactions, it allows for a more direct evaluation of NeuroEPO’s effect on neuronal excitotoxicity.
After 9 days, untreated neurons displayed thick and abundant cellular processes connected to neighboring neurons (Figure 4-IA). However, 24 hours after exposure to high doses of glutamate for 15 minutes to induce excitotoxicity, neuronal morphology showed evident deterioration. The neurons became smaller, their cell bodies retracted, dendritic loss was significant and they lost contact with neighboring neurons (Figure 4-IB). These excitotoxic effects were attenuated when neurons were treated with NeuroEPO (Figure 4-IC). The protective effect of NeuroEPO was evidenced by the tendency to preserve the characteristics of untreated neurons (Figure 4-IA). Although some neurons exhibited retracted shapes with shorter dendrites, many retained their size and dendritic connections with neighboring neurons, resembling those in the untreated group.
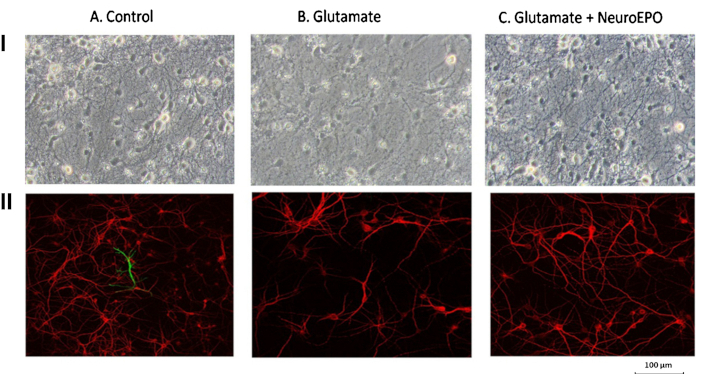
Protective effect of NeuroEPO on glutamate-induced changes in neuronal morphology and viability. Neurons were obtained from the cerebral hemispheres of Wistar rat embryos at 17 days of gestation. Panel I. After 9 days in culture, phase-contrast microphotography analysis revealed an abundant number of neurons with numerous filaments establishing contact with neighboring cells (IA). Excitotoxic treatment (IB) resulted in smaller cells with fragmented thin processes and a shrunken cell body, losing contact with neighboring cells. The excitotoxic effect was attenuated in the presence of NeuroEPO (IC), as neurons retained the morphology observed in untreated neurons and maintained connections with neighboring cells. Panel II. Immunocytochemistry images using microtubule-associated protein 2 (MAP2) (red) and glial fibrillary acidic protein (GFAP) (green) as specific markers for neurons and glial cells, respectively. In untreated cells (IIA: Control), neurons are present, with only a few glial cells in the culture. Glutamate exposure induced significant neuronal death (IIB). However, the presence of NeuroEPO (IIC) reduced glutamate-induced mortality (compared to Figure IIB), resulting in neuronal morphology and density similar to those of the untreated control group (Figure IIA). Modified from [243] with permission from IOS Press (the publication is available at IOS Pres through http://dx.doi.org/10.3233/JAD-180668). © 2018– IOS Press and the authors.
Immunocytochemical analysis, using the MAP2 antibody as a neuronal marker and the GFAP antibody as a glial cell identifier, confirmed that prior treatment with cytosine arabinoside successfully eliminated glial cells (Figure 4-IIA). Additionally, glutamate led to significant neuronal loss (Figure 4-IIB), whereas subsequent NeuroEPO mitigated excitotoxic damage (Figure 4-IIC), further confirming the neuroprotective effect of NeuroEPO.
Using DAPI for DNA staining, it was observed that, compared to neurons not treated with glutamate, excitotoxicity increased the number of pyknotic nuclei and reduced their size. These morphological changes and DNA alterations are indicators of apoptosis [244, 245]. The increase in neuronal death and apoptosis in primary cortical neuron cultures induced by excitotoxicity was mitigated by NeuroEPO treatment. NeuroEPO reduced the number of nuclei with chromatin condensation, indicating a protective effect (Figure 5).
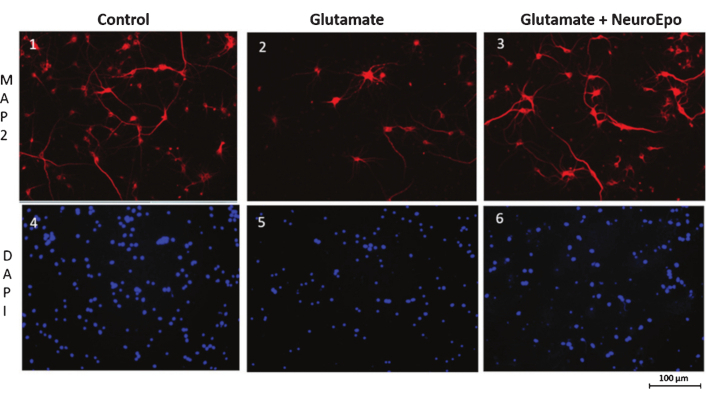
Staining of neurons and nuclei in cells exposed to glutamate followed by treatment with NeuroEPO. Cells images 1, 2, and 3 were stained with anti-MAP2 and examined using fluorescence microscopy, while images 4, 5, and 6 were stained with DAPI and analyzed with confocal microscopy. Compared to cells not treated with glutamate (1), glutamate induced neuronal cell death (2). Treatment with NeuroEPO (3), administered after glutamate exposure, attenuated glutamate-induced neuronal death. Compared to the control (4), glutamate treatment (5) increased the number of pyknotic nuclei, reduced their size and caused DNA condensation. NeuroEPO (6) mitigated these effects. Modified from [243] with permission from IOS Press (the publication is available at IOS Pres through http://dx.doi.org/10.3233/JAD-180668). © 2018– IOS Press and the authors.
Oxidative stress induced by excitotoxicity is one of the main mechanisms leading to neuronal death in AIS [109]. Excitotoxicity increases the production of oxidants while decreasing components of the antioxidant system, resulting in oxidative stress in the infarcted area. Treatment with NeuroEPO reduces excitotoxicity-induced oxidative stress by enhancing cellular antioxidant activity and decreasing oxidative activity [242, 243]. The neuroprotective effect of NeuroEPO appears to be associated with its ability to restore the neuron’s redox balance. These effects of NeuroEPO are similar to those observed with rHuEPO [246].
Treatment with NeuroEPO significantly attenuated apoptosis in neurons exposed to glutamate. NeuroEPO increases Bcl-2 expression and reduces Bax expression, maintaining the Bcl2/Bax ratio at levels similar to those found in neurons not treated with glutamate. Compared to glutamate-treated neurons, NeuroEPO reduces the expression of key indicators of the mitochondrial apoptotic pathway, including Bax, cyt c, and caspase-3, while increasing Bcl-2 expression. The reduction in pro-apoptotic indicator expression does not necessarily require a direct action of NeuroEPO on each component of the apoptotic pathway; it may result from the overall increase in Bcl-2 expression. Notably, the upregulation of Bcl-2 induced by NeuroEPO in glutamate-treated neurons is also observed in untreated neurons. Bcl-2 plays a critical role in maintaining the structural integrity of the OMM [143, 247] by inhibiting the activation of pro-apoptotic proteins, primarily Bax and Bad, and preventing the release of cyt c into the cytosol. Preserving mitochondria integrity helps maintain the redox balance, preventing mitochondria from becoming the primary source of oxidants responsible for oxidative stress. This supports the hypothesis that the neuroprotective effect of NeuroEPO against excitotoxicity is largely due to its ability to upregulate Bcl-2 expression. By maintaining mitochondria integrity, Bcl-2 neutralizes the main source of glutamate-induced oxidants.
These findings make NeuroEPO a highly promising candidate for AIS treatment. While the use of cell cultures allows for precise control of experimental conditions compared to whole-organism studies, the results should be interpreted with caution. The neurons used in this study were derived from embryonic tissue, i.e., young neurons, whereas infarcted neurons in human stroke patients are typically aged.
Preliminary studies on the effect of NeuroEPO on AIS in humans
To date, NeuroEPO has not been used in any clinical trials for the treatment of AIS, as pharmaceutical companies have shown little interest. Only a study in phase I has been conducted in healthy volunteers (Trial registration: Cuban Public Registry of Clinical Trials RPCEC00000157, June 10, 2013). In this study, intranasal NeuroEPO was administered to 25 healthy volunteers, 14 women, with a mean age of 27 years. NeuroEPO quickly reached the CSF and the brain. The results showed that NeuroEPO is a safe, well-tolerated product that does not stimulate erythropoiesis in healthy volunteers [241]. NeuroEPO was also been tested in patients with Parkinson’s disease (stages 1 and 2 on the Hoehn and Yahr scale). Nasal administration of NeuroEPO was found to be safe and well tolerated [248]. Currently, NeuroEPO is undergoing phase II-III clinical trials for Alzheimer’s and Parkinson’s diseases, with very promising results [249].
Conclusions
Stroke remains a leading cause of mortality, claiming 5.5 million lives annually, and leaves approximately 5 million survivors with disabilities requiring long-term medical care. The search for an effective stroke treatment remains one of the greatest challenges in clinical neuroscience. Despite promising results in preclinical models, most neuroprotective agents have failed to translate into successful stroke therapies in humans.
Since the late 20th century, EPO has been recognized not only as a key hormone for anemia treatment but also as a potent neuroprotectant due to its antioxidant and anti-apoptotic properties. However, its erythropoietic activity leads to increased hematocrit and blood viscosity, raising the risk of thrombosis and secondary stroke, which ultimately disqualifies it as a viable treatment for AIS. This limitation prompted the development of EPO derivatives that preserve neuroprotective properties while eliminating erythropoietic activity. Among these, CEPO, asialo-EPO, and Neuro-EPO have shown promise.
This review highlights NeuroEPO as a particularly promising candidate for treating AIS due to its potent neuroprotective effects, safety profile and ability to bypass the BBB via nasal administration. NeuroEPO protects neurons by attenuating excitotoxicity, enhancing antioxidant defenses and maintaining mitochondrial membrane integrity through upregulation of anti-apoptotic Bcl-2 family proteins. Its non-erythropoietic nature and efficient brain delivery without rapid hepatic degradation make it a strong candidate for clinical translation.
Given these advantages, NeuroEPO warrants further investigation in clinical trials to establish its efficacy as a therapeutic option for AIS. If confirmed, it could represent a significant advancement in stroke treatment, offering a safe and effective neuroprotective strategy.
Abbreviations
| AIS: | acute ischemic stroke |
| BBB: | blood-brain barrier |
| CBF: | cerebral blood flow |
| CEPO: | carbamylated erythropoietin |
| CFU-E: | colony-forming unit-erythroid |
| CNS: | central nervous system |
| CSF: | cerebrospinal fluid |
| Cyt c: | cytochrome c |
| EPO: | erythropoietin |
| EPOR: | erythropoietin receptor |
| ETC: | electron transport chain |
| FIH: | factor inhibiting hypoxia |
| HIF-1α: | hypoxia-inducible factor 1α |
| IMM: | inner mitochondrial membrane |
| JAK2: | Janus tyrosine kinase 2 |
| MAGUK: | membrane-associated guanylate kinase |
| MAPK: | mitogen-activated protein kinase |
| MCAO: | middle cerebral artery |
| MOMP: | mitochondrial outer membrane permeabilization |
| mPTP: | mitochondrial permeability transition pore complex |
| NMDA: | N-methyl-D-aspartate |
| NMDAR: | N-methyl-D-aspartate receptor |
| nNOS: | neuron nitric oxide synthase |
| NOS: | nitric oxide synthase |
| NOX: | NADPH oxidase |
| OMM: | outer mitochondrial membrane |
| PARP: | poly(ADP-ribose) polymerase |
| PDZ: | postsynaptic density 95/disc-large/zona occludens |
| PHD: | prolyl hydroxylase |
| PSD: | postsynaptic density |
| rHuEPO: | recombinant human erythropoietin |
| RNS: | reactive nitrogen species |
| ROS: | reactive oxygen species |
| SOD: | superoxide dismutase |
| TCA: | tricarboxylic acid |
| tPA: | tissue plasminogen activator |
| TRP: | transient receptor potential |
| VDAC: | voltage-dependent anion channel |
| βcR: | beta common receptor |
| Δψm: | mitochondrial electron potential |
Declarations
Author contributions
RR: Conceptualization, Writing—original draft. JRT: Writing—review & editing, Validation, Supervision. Both authors read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2025.
Publisher’s note
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.