 Systematic Review
Systematic Review

Affiliation:
1Laboratório de Neurociência, Instituto de Química da Universidade de São Paulo, São Paulo 05508–000, Brazil
ORCID: https://orcid.org/0000-0002-3350-4569
Affiliation:
1Laboratório de Neurociência, Instituto de Química da Universidade de São Paulo, São Paulo 05508–000, Brazil
ORCID: https://orcid.org/0000-0002-2555-7652
Affiliation:
1Laboratório de Neurociência, Instituto de Química da Universidade de São Paulo, São Paulo 05508–000, Brazil
ORCID: https://orcid.org/0000-0001-9317-9713
Affiliation:
1Laboratório de Neurociência, Instituto de Química da Universidade de São Paulo, São Paulo 05508–000, Brazil
ORCID: https://orcid.org/0000-0002-4962-0302
Affiliation:
1Laboratório de Neurociência, Instituto de Química da Universidade de São Paulo, São Paulo 05508–000, Brazil
ORCID: https://orcid.org/0000-0002-9416-3083
Affiliation:
1Laboratório de Neurociência, Instituto de Química da Universidade de São Paulo, São Paulo 05508–000, Brazil
ORCID: https://orcid.org/0000-0002-4258-2649
Affiliation:
1Laboratório de Neurociência, Instituto de Química da Universidade de São Paulo, São Paulo 05508–000, Brazil
ORCID: https://orcid.org/0000-0002-8276-8149
Affiliation:
1Laboratório de Neurociência, Instituto de Química da Universidade de São Paulo, São Paulo 05508–000, Brazil
ORCID: https://orcid.org/0000-0002-3850-4867
Affiliation:
1Laboratório de Neurociência, Instituto de Química da Universidade de São Paulo, São Paulo 05508–000, Brazil
2Chengdu University of Traditional Chinese Medicine, 37 Shi-er Qiao Road, Chengdu 610075, Sichuan, China
ORCID: https://orcid.org/0000-0001-6971-2506
Affiliation:
2Chengdu University of Traditional Chinese Medicine, 37 Shi-er Qiao Road, Chengdu 610075, Sichuan, China
Affiliation:
1Laboratório de Neurociência, Instituto de Química da Universidade de São Paulo, São Paulo 05508–000, Brazil
Email: henning@iq.usp.br
ORCID: https://orcid.org/0000-0002-2114-3815
Explor Neuroprot Ther. 2022;2:36–64 DOI: https://doi.org/10.37349/ent.2022.00017
Received: August 30, 2021 Accepted: December 08, 2021 Published: February 23, 2022
Academic Editor: Thomas Müller, St. Joseph Hospital Berlin-Weissensee, Germany
Aim: Excitotoxicity results from unusually increased activation of excitatory amino acid receptors leading to neuronal death. Since glutamate is the main excitatory neurotransmitter in the central nervous system, it is also the most common excitotoxicity trigger. This uncontrolled neuronal response participates in various neurodegenerative diseases, such as ischemia, hypoglycemia, Huntington’s, Parkinson’s and Alzheimer’s disease. Thus, the investigation in the field expanded a lot in the past decade, leading to in vitro modelling adaptations. However, much performed work on glutamate-induced excitotoxicity is methodologically inconsistent in the literature. The field lacks reproducibility, which is one of the main fundaments of empirical science. In this regard, the literature was summarized and the main methodological features were critically evaluated, aiming to guide the researchers that are starting in the field.
Methods: Published data since 1985 from PUBMED were collected and analyzed to observe which in vitro experimental conditions of excitotoxicity were reproducible. The suggested methods were based on the characteristics of excitotoxicity, such as abnormal intracellular calcium mediated signaling, mitochondria impairment, reactive oxygen species accumulation and cell death. Various conditions and comparative controls were used to design the standard investigation of excitotoxicity, such as culture medium content (presence of glutamate and aspartate), time interval of induction and the concentration of the inductor, based on the most reproducible published ones.
Results: Our results and critical analysis point to some experimental conditions to consider, such as primary cultured neurons are more sensitive to glutamate and the response obtained is more robust than in other models; excitotoxicity mediated effects are better observed one hour following the stimulus; the culture medium should contain low levels of glutamate or aspartate or glycine. Online available phosphoproteomic data on excitotoxicity using the primary cortical neurons in vitro model supported the same conditions proposed by us.
Conclusions: This manuscript will facilitate the design of any research for excitotoxic or neuroprotective compounds in physiological and pathophysiological conditions by standardizing and improving experimental conditions.
Excitotoxicity is the result of an unusual increase in activation of excitatory amino acid receptors. Glutamate is most commonly the cause of such an event because it is the main excitatory neurotransmitter in the central nervous system [1–5], although excitotoxicity can also be caused by glycine, aspartate or quinolinic acid (QA) [6].
The harmful effects of glutamate were first described by Hayashi [7] in 1954, and since then it has attracted intense attention, due to its elevated concentration compared to other amino acids in the brain, and to its ability to induce convulsion and necrosis [8].
Glutamate has three ligand gated ion channel receptor subtypes named after its exogenous agonists, the N-methyl-D-aspartate (NMDA), kainic acid (KA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA). The abnormal excessive activation of the receptors sensitive to these agonists, mainly of NMDA subtypes, increases Ca2+ flux into the cell. Thereby Ca2+ overload increases the activation and production of enzymes, such as endonucleases, phospholipases, caspases and proteases leading to necrosis and apoptosis [1].
Glutamate receptor agonists trigger intracellular Ca2+ concentration ([Ca2+]i) increase through ionotropic and metabotropic glutamate receptor activation. The activation of NMDA receptor (NMDAR) by NMDA, glutamate or QA and the co-activator glycine increases the plasma membrane permeability to Ca2+ ions. Alternatively, glutamate also induces transient [Ca2+]i increase through activation of G protein-coupled metabotropic receptors activating phospholipase C-β to cleave phosphatidylinositol biphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). In turn, IP3 triggers the release of endoplasmic reticulum (ER) intracellular Ca2+ ions through the IP3 receptor. The Ca2+ signaling is an important second messenger and disruption of its homeostasis is related to the excitotoxicity process in excitatory neurons. Following [Ca2+]i increase, mitochondrial Ca2+ uniporter (MCU) transports Ca2+ ions to the mitochondrial matrix (Ca2+ buffering) in a gradient favorable manner, which in turn disrupts mitochondrial membrane potential interrupting ATP production and initiating cell death mechanism. Moreover, the NMDAR interacts with neuronal nitric oxide synthetase (nNOS) in a postsynaptic density protein-95 (PSD-95)-dependent manner. Thus, overstimulation of NMDAR induces the formation of nitric oxide, reactive oxygen species (ROS) and peroxynitrites that also contributes to cell death induction [9].
Aspartate, another endogenous amino acid present in the brain, activates NMDAR. Although aspartate has a weaker affinity than glutamate to these receptors, high levels of L-aspartate can trigger excitotoxicity, because high levels of this amino acid can prevent glutamate uptake, increasing the glutamate concentrations in the synaptic cleft and activating glutamate receptors. It is worth mentioning that even though L- and D-aspartate bind to NMDAR, only L-aspartate is metabolized [10].
Metabotropic glutamate receptors (mGluRs) also have a critical role in excitotoxicity. They are also divided into three subclasses, group I (mGluR1 and 5), group II (mGluR2 and 3) and group III (mGluR4, 6, 7, and 8), that are classified according to their pharmacology and amino acid homology [11]. These receptors have been described to modulate the passage of Ca2+ and K+ through ionotropic glutamate receptor channels. Therefore, the activation of group II receptors can inhibit the NMDAR-induced Ca2+ response and have neuroprotective properties [11].
ATP receptors are also involved in excitotoxicity. P2X receptors are ionotropic Ca2+ permeable receptors, and the activation of these receptors can induce the same Ca2+ dependent deleterious behavior in cells, as described for glutamate-induced excitotoxicity. In reported cases of ischemia and multiple sclerosis, ATP is released from dead or damaged neurons, therefore inducing the toxic cascade of events [12].
The field of science investigating excitotoxicity has provided incredible achievements in the past few decades, especially for the understanding of mechanisms of neurodegeneration and psychiatric disorders. Despite extensive research of excitotoxicity roles in many different diseases and disorders, the field still lacks standards and key features to characterize the detection and mensuration of the excitotoxicity effects, especially in vitro. In this way, many recent findings on in vitro models were placed in a table format, highlighting methodological conditions used for quantifying excitotoxicity and its underlying mechanisms features. In addition, the application of search engine tools of PUBMED for published papers was used in order to compare the protocols.
The highly demand of standardization on the field comes with the fast growth of investigations of excitotoxicity in various diseases. Here, some studies rapidly advanced in the past years are summarized.
In cases of ischemia, it has been observed that excitotoxicity is the primary neuron death mechanism, and this occurs due to energy failure. To prevent the extracellular glutamate from reaching high concentrations, the cell produces glutamate transporters that bind to the amino acid and transport it back into the interior of the cell, but this mechanism depends on ATP. Thus, during nutrient starvation situations, such as an interruption of blood flow, the neurons are not able of upregulating inward glutamate transport and subsequently NMDAR is over activated, leading to excitotoxicity [13, 14].
Huntington’s disease (HD) is a genetic, neurodegenerative disease that affects mostly the striatum. The mutation in the huntingtin (HTT) gene, responsible for the disorder, expands the polyglutamine repetitions in the HTT protein, healthy individuals present 19 to 29 repetitions HTT, whereas individuals with the altered gene present 36 or more of these polyglutamine repetitions [mutant HTT (mHTT)]. The disease has a deleterious effect specifically in medium sized γ-aminobutyric acid (GABA)-ergic spiny neurons, and although the exact reason for this specificity is not known, many hypotheses were raised, with one of them being the excitotoxic hypothesis [15].
Although every function of HTT has not been fully explained, this protein most likely has a role in axonal transport [16], and when mutated, mHTT impedes mitochondrial trafficking along neurons. Thus, the neurons die as a result of energy failure. Moreover, mHTT forms insoluble aggregates that sequester other proteins and dysregulate not only the proteasome system but also vesicle trafficking and gene transcription.
The suggestion that excitotoxicity is one of the mechanisms involved in HD comes from studies indicating that the injection of QA induces a similar neurodegeneration to that observed in the HD and studies demonstrating an increase in excitotoxicity in the HD model YAC128. A possible explanation for these events comes from the interaction between mHTT and the PSD-95. This scaffold protein is responsible for the concentration and organization of neurotransmitter receptors, thus glutamate signaling could be impaired, leading to excitotoxicity [17, 18].
Alzheimer’s disease (AD) is a neurodegenerative disease responsible for the majority of cases of dementia, usually identified by impairment in memory, judgment, decision making, orientation to physical surroundings, language and neuronal loss in the cortex and hippocampus. The cause of the neurodegeneration is associated with increased deposit of the peptide amyloid-β (Aβ) that is produced by proteolytic processing of the transmembrane amyloid precursor protein, or APP, that is cleaved by α-secretase, or if this process fails, it can be cleaved by β and γ-secretases. Additionally, a small fraction of this peptide may be produced in the ER and the Golgi apparatus [19].
One interesting characteristic of neurons in patients with AD is that the dendrites contain an increased amount of vacuolar structures, indicating that the hyper-stimulation of the lysosomal system is a part of the pathology of the disease. Some recent studies elucidated how this stimulation can be toxic for the cell and that is because autophagic vacuoles are enriched with beta-carboxyl terminal fragment (βCTF, another precursor of Aβ) and γ-secretases [20], and both of these proteins can produce Aβ, accelerating the progression of the disease.
Excitotoxicity was theorized to be a part of AD due to a number of similarities that both conditions share; one as well as the other presents an increase in ROS [1], AD patients treated with a NMDAR inhibitor drug called memantine demonstrated a better preservation of memory. Thus, the cell loss observed in the disease is usually restricted to glutamatergic neurons [21]. This evidence was not considered coincidence, and further research demonstrated a link between both conditions, as follows. For example in immunoprecipitation assays [22], Aβ oligomers were co-immunoprecipitated with the NR1 subunit of the NMDAR suggesting that Aβ binds in proximity or directly to subunits of this receptor. Furthermore, studies indicate that the amount of glutamate or kainate needed to induce excitotoxic damage in neurons was approximately 50% less when the cells had been exposed to Aβ. In view of that, neurons in AD are more susceptible to excitotoxic damage [22].
Hypoglycemic brain damage occurs only in extreme cases of low sugar content in the blood, such as in conditions of insulin-secreting tumors, insulin overdoses and suicide tentatives. This specific type of brain damage was first described during clinical trials, as consequence of the methodical use of hypoglycemia to treat schizophrenia, in which patients would be injected with increasing doses of insulin, varying from 15 to 150 units until the individual would enter the state of shock. This procedure would be repeated 3 to 6 times a week, depending on the expected result [23]. Clinical trials reported behavioral changes, and that one could not survive a long-lasting hypoglycemic coma, and that low levels of glucose in the organism interfered with the central nervous system, causing deleterious effects.
During the hypoglycemic shock, concentrations of pyruvate and acetyl coenzyme A (CoA) are reduced, since the production of both depends on glucose’s metabolism. Thus, in order to maintain energy production, the cell switches from glucose to fatty acids and phospholipids as energetic substrates. Although the neurons are capable of maintaining their energy metabolism, the decrease in glycolysis generates an increase in oxaloacetate levels, thus altering the balance of the glutamic oxaloacetic transaminase reaction, favoring aspartate production. As previously mentioned, aspartate is an NMDAR agonist, and during hypoglycemic shock, this amino acid leaks across the cell membrane and excessively activates its receptors, inducing excitotoxic effects [24].
Parkinson’s disease (PD) is a neurological disorder that affects approximately one percent of the population over 65 years of age. This neurodegenerative disorder is characterized by neuronal loss in the substantia nigra, dopamine deficiency in parts of the brain and the presence of Lewy bodies, which are filamentous aggregates of α-synuclein. Most patients present the cardinal symptoms of bradykinesia, resting tremor, rigidity, and postural instability [25]. Actual treatments include the use of levodopa, a dopamine agonist that attempts to compensate for the loss of dopamine, and monoamine oxidase-B inhibitors, as expression levels of this enzyme is augmented in PD [26]. As observed in many studies, when the ubiquitin-proteasome system is inhibited in rats, these animals start to develop symptoms seen in PD such as parkinsonism and selective neurodegeneration [27]. These results suggest that the system responsible for degradation of misfolded, old and defective proteins is impaired in PD, and a failure in this crucial proteolytic process in cells could lead to the production of protein aggregates, as seen in Lewy bodies, and other cellular dysfunctions. In addition to that, brains of individuals with PD present overproduction of ROS. Even though the cause of this phenomenon is unclear, it is most likely that there is a defect in complex I of the respiratory chain in mitochondria.
Interestingly, the neurotransmitter dopamine has a critical role in intracellular Ca2+ homeostasis. Some studies indicate that not only dopamine can induce an increase in Ca2+ concentration, but that the application of glutamate followed by the addition of dopamine to neurons stimulates a smaller rise in cytosolic [Ca2+]i compared to neurons treated solely with glutamate. Thus, a neurological disease that impairs dopaminergic neurons, such as PD, creates the environment for a dysregulation of Ca2+ homeostasis, leading to excitotoxicity [10]. Moreover, the group I mGluR5 metabotropic receptor was overexpressed in both PD and AD brains, and it has been described that the antagonists of this specific receptor improve motor function in rats affected by the disease [28].
This search was performed with PUBMED searching tools with its indexed papers, following the request example “((excitotoxicity) AND (in vitro) AND (Glutamate)) NOT (review[Publication Type])”, “((excitotoxicity) AND (in vitro) AND (NMDA)) NOT (review[Publication Type])”, “((excitotoxicity) AND (in vitro) AND (Kainate)) NOT (review[Publication Type])”, “((excitotoxicity) AND (in vitro) AND (Quinolinic acid)) NOT (review[Publication Type])”, “((excitotoxicity) AND (primary neuron)) NOT (review[Publication Type])”, “((excitotoxicity) AND (cortical neuron)) NOT (review[Publication Type])”, “((excitotoxicity) AND (stem cell)) NOT (review[Publication Type])”, “((excitotoxicity) AND (PC12)) NOT (review[Publication Type])”, “((excitotoxicity) AND (SH-SY5Y)) NOT (review[Publication Type])”, “((excitotoxicity) AND (HT-22)) NOT (review[Publication Type])”. The obtained papers are cited in a table, highlighting the different methodological conditions, including Model (cell line), Species, Medium, Inductor, Concentration, Treatment time, Time after induction, Methods for validation, and so on.
Distribution frequencies of the methodological conditions were obtained with the PUBMED literature bank about the work published in the field since 1985. Ninety-two articles were analyzed in relation to the percentage distribution of the use of agonist concentration, cell culture medium, pulse duration and the defined time used to quantify the excitotoxic effects after the application of stimuli. The GraphPad Prism5 software was used for this purpose.
A phosphoproteomic data deposited into the ProteomeXchange Consortium via the PRIDE repository with the dataset identifier PXD008353 was analyzed. In this data set, mouse primary cortical neurons were overstimulated with the neurotoxic concentration of glutamate (100 μmol/L). After certain insult time intervals (between 5 and 240 min), the sample was prepared and submitted to quantitative proteomic analysis approaches to identify neuronal proteins and their phosphorylation dynamic. Nine hundred and seventy-two combinations of phosphopeptide groups were identified, derived from 383 different neuronal proteins [29]. Mouse primary cortical neurons treated with glutamate revealed a statistically significant change in phosphorylation levels of over 100 proteins (P < 0.05 and log2 fold change greater than |±0.5|). These proteins were separated into two groups—those with log2 fold change greater than 0.5 and those with less than –0.5. Each group was submitted to Gene Ontology enrichment analysis on the Panther platform with the entire mouse genome as the background list [30].
Aiming to find conditions that are more reproducible in the field of in vitro excitotoxicity, search engine tools of PUBMED for published papers since the first appearance of the term in 1985 were used in order to compare the most reproducible protocols (Figures 1 and 2A). Some articles have their details summarized in Table 1.
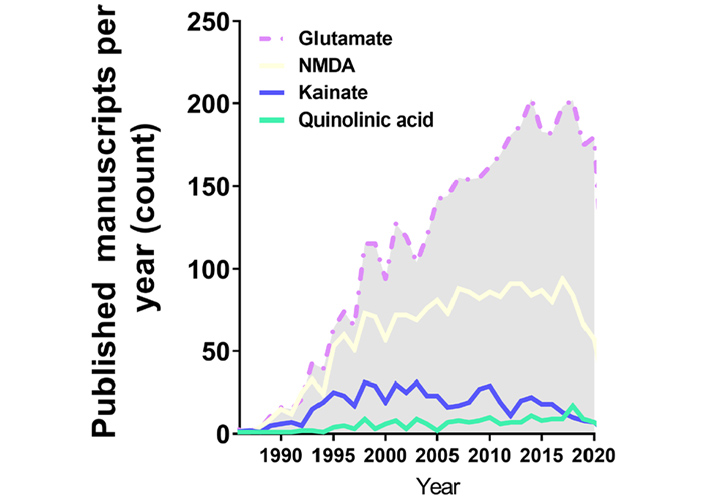
Annual publications in the excitotoxicity field regarding the application of excitotoxicity inducer in in vitro cell models over the years. This search was performed with PUBMED searching tools with its indexed papers, following the request example “((excitotoxicity) AND (in vitro) AND (Glutamate)) NOT (review[Publication Type])”, “((excitotoxicity) AND (in vitro) AND (NMDA)) NOT (review[Publication Type])”, “((excitotoxicity) AND (in vitro) AND (Kainate)) NOT (review[Publication Type])”, “((excitotoxicity) AND (in vitro) AND (Quinolinic acid)) NOT (review[Publication Type])”
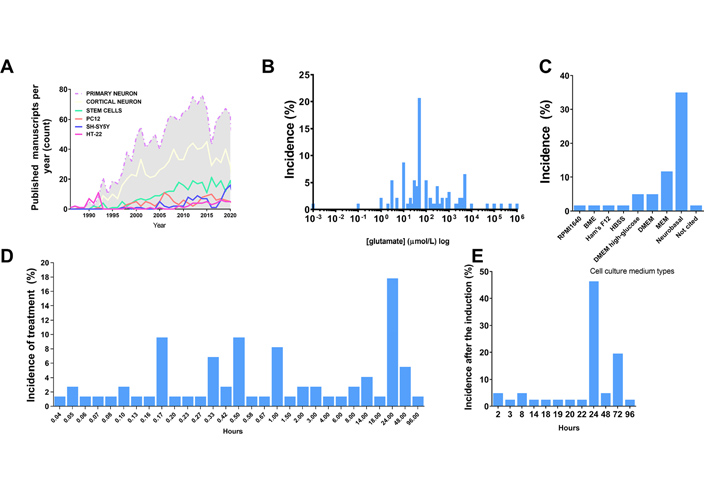
Excitotoxicity induction. A. Distribution of the amount of published studies over the years using different agonists in the excitotoxicity field. Data acquired through PUBMED search engine of indexed papers, following the request example “((excitotoxicity) AND (primary neuron)) NOT (review[Publication Type])”, “((excitotoxicity) AND (cortical neuron)) NOT (review[Publication Type])”, “((excitotoxicity) AND (stem cell)) NOT (review[Publication Type])”, “((excitotoxicity) AND (PC12)) NOT (review[Publication Type])”, “((excitotoxicity) AND (SH-SY5Y)) NOT (review[Publication Type])”, “((excitotoxicity) AND (HT-22)) NOT (review[Publication Type])”; B. distribution of agonist concentration incidence in published manuscripts regarding in vitro models (% of published studies that tested that concentration among the total retrieved by search); C. distribution of cell culture medium type incidence in published manuscripts regarding in vitro models (% of published studies that tested that type of medium among the total retrieved by search); D. incidence distribution of the duration of the pulse induced by the agonist in published manuscripts regarding in vitro models (% of published studies that tested that condition among the total retrieved by search); E. distribution of the time set used for the quantification of excitotoxic effects after application of stimuli in published manuscripts regarding in vitro models (% of published studies that tested that condition among the total retrieved by the search)
Experimental aspects used in published excitotoxicity papers
| Ref | Model (cell line) | Species | Medium | Inductor | Concentration | Treatment time | Time after induction | Methods for validation | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| [31] | Primary cortical neurons | Rat | DMEM high glucose, Neurobasal | Glutamate | 10, 50 and 100 μmol/L | 20 min | 24 h | Morphology analysis and ELISA | Decreased cell survival rate |
| [32] | Primary cortical neurons | Rat | DMEM high glucose, Neurobasal | Glutamate | 50 and 100 μmol/L | 24 h | 24 h | TUNEL, CCK-8 and [Ca2+]i measurement | Decreased cell survival rate, augmented [Ca2+]i and increased number of TUNEL cells |
| [33] | Primary cortical neurons | Mouse | Neurobasal | NMDA | 50 and 100 μmol/L | 10 min | 24 h | Trypan blue and LDH assay | Decreased cell survival rate, increased LDH release |
| [34] | Primary cortical neurons | Rat | Neurobasal | NMDA | 50 μmol/L | 20 min | 20 h | LDH assay, Hoechst-33342 staining, patch-clamp and WB | Increased LDH release; fragmented chromatin blockade of the NMDA-induced currents and cleaved caspase-3 reactivity |
| [35] | Primary hippocampal neurons | Rat | Neurobasal | NMDA | 100 μmol/L | 24 h | 24 h | LDH and MTT assays, Hoechst-33342 and PI staining, Ca2+ imaging and patch-clamp | Decreased MTT metabolization; increased LDH release, increased [Ca2+]i, fragmented chromatin and inward current |
| [36] | Primary cortical neurons | Rat | Neurobasal | NMDA | 15, 20, 30, 40 and 50 μmol/L | 30 min | 20 h | WB, LDH assay, DAPI staining and Ca2+ imaging | Increased [Ca2+]i, higher LDH release; cleaved caspase 3 reactivity, and increased condensed chromatin staining |
| [37] | PC12 | Rat | RPMI | Glutamate | 2, 5 and 10 mmol/L | 48 h | 48 h | MTT assay | Reduced MTT metabolization |
| [38] | HT22 | Mouse | DMEM high glucose | Glutamate | 5 mmol/L | 14 h | 14 h | WST-1 assay, viable cell counting, ROS analysis and WB | Increased ROS production; higher Bax and cleaved caspase-3 expression and decreased Bcl-1 expression and decreased cell survival |
| [39] | Organotypic hippocampal slice | Rat | Not informed | Kainate | 5 μmol/L | 24 h | 24 h | PI staining and WB | Increased percentage of PI+ cells, and higher expression of BAX and PARP proteins |
| [40] | Primary cortical neurons | Mouse | Neurobasal | Kainate | 100, 200, 400, and 800 μmol/L | 1, 2, 4 and 8 h | 8 h | CCK-8, oxygen consumption, mitochondrial superoxide measurement, membrane potential and ATP production | Augmented mitochondrial superoxide production, decreased mitochondrial membrane potential, reduced ATP production and decreased survival rate |
| [41] | Primary cortical neurons | Rat | Neurobasal | Glutamate | 50, 100, 200, 500 and 1,000 mmol/L | 30 min | 24 h | LDH assay, morphology analysis and viable cell counting | Decreased cell survival rate and higher LDH release |
| [42] | Primary cerebellar granule neurons | Rat | BASAL MEDIUM EAGLE | Glutamate | 1, 10, 20, 100, 250 and 500 μmol/L | 24 h | 24 h | MTT assay | Decreased MTT metabolization |
| [43] | Primary hippocampal neurons | Rat | Not informed | Glutamate | 100 μmol/L | 24 h | 24 h | TUNEL assay and mitochondrial activity measurement | Increased TUNEL+ cells and collapsed mitochondrial activity |
| [44] | Primary cortical neurons and co-culture with primary astrocytes | Rat | DMEM high glucose | Glutamate | 10, 25, 50, 100, 250, 500, and 1,000 μmol/L | 10 min | 72 h | Immunostaining | Increased glutamate transporter expression and decreased MAP2+ neurons |
| [45] | Primary cortical neurons | Rat | Neurobasal | Glutamate | 30 μmol/L | 3 h | 3 h | MTT assay, immunostaining and Ca2+ imaging | Diminished MAP2+ cells, higher Ca2+ influx |
| [46] | Mesencephalic cells | Rat | MEM and Ham’s F-12 | Glutamate | 150 μmol/L | 24 h | 24 h | Immunostaining | Diminished number of PHRF-1+ and TH+ cells |
| [47] | Dopaminergic differentiated SH-SY5Y | Human | HBSS | Glutamate, NMDA, kainate, AMPA and (1S, 3R)- ACPD | Glutamate: 0.1, 0.5, 1, 1.5, 2, 3 and 4 mmol/L; NMDA, kainate, AMPA and (1S, 3R)- ACPD: 0.1, 0.5, 0.75 and 1 mmol/L | 96 h | 96 h | MTT assay and trypan blue test | Augmented trypan blue stained cells with and decreased MTT metabolization |
| [48] | HT22 | Mouse | DMEM | Glutamate | 3 and 5 mmol/L | 2 h and 18 h | 2 h and 18 h | Morphology analysis, MTT assay, total ATP measurement and annexin-V/PI staining | Mitochondrial fission and fragmentation, mitochondrial depolarization; decreased ATP levels, decreased MTT metabolization and higher number of annexin+ cells |
| [49] | Primary embryonic hippocampal neurons | Rat | DMEM | Synthetic Aβ 1–42 | 0.1, 1, 10, 50, and 100 μg/mL | 24 h | 24 h | Cell viability based on morphology and photo negative projection | Decreased cell viability |
| [50] | HEK-293 cells-huntington model | Human | MEM | NMDA and glutamate | NMDA: 3, 10, 30, 100 and 1,000 μmol/L; Glutamate: 100 μmol/L | 6 h | N/A | β-galactosidase assay, Hoechst 33258 staining | Decreased cell survival rate, increased apoptotic nuclear morphology |
| [51] | Primary cortical neurons | Mouse | Neurobasal | NMDA and glutamate | NMDA: 100 μmol/L and Glutamate: 100 μmol/L | 350 s, 6 min and 1 h | N/A | Ca2+ imaging, oxygen consumption and ATP measurement | Increased [Ca2+]i and ATP and oxygen consumption |
| [52] | Primary striatal neurons | Mouse | Neurobasal | NMDA | 500 μmol/L | 10 min | 24 h | TUNEL assay | Increased percentage of TUNEL+ cells |
| [53] | Striatal neuronal progenitor cell lines | Mouse | DMEM | NMDA | 500 and 50 μmol/L | 30 min | 24 h | Hoechst 33258 staining | Decreased cell survival rate |
| [54] | Fetal primary neurons | Human | Not informed | Glutamate | 10 mmol/L | 24 h | 24 h | DAPI staining | Decreased cell survival rate |
| [55] | Primary cortical neurons | Rat | Neurobasal | Glutamate | 100 μmol/L | 20 min | N/A | Ca2+ imaging | Increased [Ca2+]i and mitochondrial depolarization |
| [56] | Primary hippocampal and striatal neurons | Rat | Not informed | NMDA | 100 μmol/L | 1 h | 24 h | ApoAlert MitoSensor Kit and whole-cell patch clamp | Augmented number of apoptotic cells and inhibition of the NMDA currents |
| [57] | Primary hippocampal, cortical and midbrain neurons and glial cells | Rat | MEM | Glutamate | 5 and 100 μmol/L | 2.5, 3.5, 14, 15, 16, 20, 25 and 40 min | 24 h | Ca2+ imaging, PI and Hoechst 33258 staining | Increased [Ca2+]i, decreased cell survival rate |
| [58] | Primary cerebellar neurons | Rat | Neurobasal | Glutamate | 1, 2 and 5 mmol/L | 24 h | 24 h | Immunostaining and WB | Downregulated expression of neuronal and anti-apoptotic markers, enhanced expression of inflammatory and senescence markers and mitochondrial damage |
| [59] | Embryonic stem cell 8-week derived neurons | Human | MEM | Glutamate | 200 μmol/L | 24 h | 24 h | Ca2+ imaging, whole-cell patch clamp and CellTiter-Glo Luminescent Cell Viability Assay | Higher peak Ca2+ current, and decreased cell survival rate |
| [60] | Primary striatal neurons | Mouse | Neurobasal | NMDA | 500 and 3,000 μmol/L | 10 and 30 min | 3 and 24 h | TUNEL assay, Ca2+ imaging, caspase activity, whole-cell patch clamp and mitochondrial membrane potential measurement | Increased [Ca2+]i, increased number of TUNEL+ cells, mitochondrial membrane depolarization increased NMDAR-evoked current higher activity of caspase 9 and 3 |
| [61] | Primary co-culture of cortical or hippocampal neurons and astrocytes | Rat | Neurobasal | Glutamate | 100 μmol/L | 3, 6, 8, 10, 12, 16, 20, 25, and 90 min | N/A | Mitochondrial membrane potential measurement, Ca2+ imaging, PI and Hoechst 33258 staining | Mitochondrial depolarization, increased [Ca2+]i, and decreased cell survival rate |
| [62] | Primary hippocampal neurons | Rat | Neurobasal | Glutamate | 100 μmol/L | 24 h | N/A | PI and Hoechst 33342 staining, LDH assay, MTT assay, Ca2+ imaging, WB | Decreased cell survival rate, mitochondrial dysfunction, increased [Ca2+]i, increased expression of procaspase-3 and reduced of Bcl-2 |
| [63] | Primary hippocampal and cortical neurons | Rat | MEM | (S)-(−)- Bay K 8644, FPL 64176, NMDA and glutamate | (S)-(−)-Bay K 8644: 1 μmol/L; FPL 64176: 5 μmol/L; NMDA: 100 μmol/L; Glutamate: 200 μmol/L | 5 and 30 min | 20 and 24 h | PI staining, immunostainin, whole-cell recording patch clamp, electron microscopy and Ca2+ imaging | Increased [Ca2+]i and mitochondrial dysfunction |
| [64] | Primary hippocampal neurons and glial cells | Rat | Neurobasal | Glutamate | Glutamate: 100 μmol/L; glycine: 10 μmol/L | 3, 4, 10, 12, 14, 16, 25, 30, 35, 40 min and 24 h | 24 h | Ca2+ imaging, PI and Hoechst 33342 staining | Mitochondrial depolarization and decreased cell survival rate |
| [65] | Primary neuron- astrocyte co-culture | Mouse | Neurobasal | NMDA | 100 μmol/L | 25 min and 24 h | N/A | Ca2+ imaging, immunostaining, WB and Hoechst 33258 staining | High superoxide production, increased [Ca2+]i, DNA damage and decreased cell survival rate |
| [66] | Primary hippocampal neurons | Mouse | Neurobasal | NMDA | 20 μmol/L | 10 min | 18 and 20 h | Hoechst 33258 staining, Ca2+ imaging and whole-cell patch clamp | Decreased cell survival rate, enhanced Ca2+ signals and mitochondria membrane depolarization |
DMEM: Dulbecco’s Modified Eagle’s medium; ELISA: enzyme linked immunosorbent assay; TUNEL: transferase dUTP nick end labelling; CCK-8: cell counting kit-8; LDH: lactate dehydrogenase; WB: western blotting; MTT: methyl thiazolyl tetrazolium; PI: propidium iodide; DAPI: 4’,6-diamidino-2-phenylindole; PC12: pheochromacytoma 12; RPMI: risk of permanent medical impairment; WST-1: water-soluble tetrazolium salt-1; BAX: B-cell lymphoma-2-associated X; Bcl-1: B-cell lymphoma 1; PARP: poly(ADP-ribose) polymerase; MEM: minimal essential medium; ACPD: aminocyclopentane-1,3-dicarboxylic acid; MAP2+ cells: Microtubule-associated protein 2-positive cells; PHRF-1+ cells: PHD and RING finger domain-containing protein 1-positive cells; TH+ cells: tyrosine hydroxylase-positive cells; HBSS: Hanks’ balanced salt solution
Surprisingly, methodological approaches for simulating excitotoxicity are extremely heterogeneous, varying from the employed stimuli (drug type, concentrations and incubation time) to the detection methods used for comparisons. Since the field is extremely important to elucidate the disease mechanisms and for drug/therapy design, in this article the methods and some common mistakes are discussed, followed by suggestions of some standard methods, pointing out the best research practices during excitotoxicity investigation experiments.
Through the years, several alternatives for in vitro cell modeling were developed, such as primary cultures, commercial cell lines, differentiated embryonic cells and induced pluripotent stem cells (iPSCs). Our in-depth comparison revealed advantages and drawbacks for every in vitro cell model.
Despite the scientific advance in stem cell research regarding differentiation and the development of purified commercial cell lines, our literature search identified the majority of the papers in the field of excitotoxicity using primary cortical neurons as the cellular model (Table 1 and Figure 1). Primary cortical neurons provide a simplified environment reflecting in vivo morphology and expression patterns of molecular markers, characteristic of the given tissue [67, 68]. However, for primary cell culture isolation and maintenance, the personnel need specific training regarding animal handling and might require institutional animal care and ethical committee approval in accordance with each country’s laws. Primary cultures are limited in their lifespan, as they become senescent, and are subject to cell heterogeneity and animal background interference [67, 69], which may be the cause of the diverse experimental conditions obtained in the field (Figure 2A and B). In addition, the study of alternative methods to primary cultures has been implemented in order to make research more appropriate to the animal user’s guiding principles regarding the 3 R’s—Replace, Reduce, and Refine [70].
Commercial cell lines are homogeneous and less costly alternatives for in vitro modeling. As a main advantage, cell lines provide uniform results after drug treatment, while they can be easily expanded and used a wide variety of conditions, avoiding the issues of ethical committee’s approval and generating high-speed data. However, these cell lines do not fully exhibit the in vivo phenotype or primary cell culture phenotype in a given cell population through culture passages, in which cells accumulate genetics and epigenetics modifications that restrict their use [68, 71]. Moreover, it is recommended to acquire cell lines from certified suppliers for avoiding drawbacks with misidentified and cross-contaminated cells [72]. The most frequently used cell lines in our research about excitotoxicity were PC12, HT-22 and SH-SY5Y cells, and the amount of yearly published manuscripts in excitotoxicity’s field using them have not changed much since the early 2000’s (Figure 1). These cell cultures need to be supplemented with animal serum, which can desensitize some receptors to be investigated. The most used serum, fetal bovine serum, has around 1 mmol/L of glutamate [73], and NMDAR desensitization is already high at 2 mmol/L concentration of the agonist and can be pronounced in chronic exposure, since calcium-calmodulin dependent kinase can induce the receptor desensitization [74].
The last available source of neuronal cells commonly identified here are stem cell-derived neurons, which have been increasingly used in this field of research, however still much less than primary cultured neurons (Figure 1). A brief overview of the completely historical aspects and milestones of neural stem cells (NSC) research were reviewed by Breunig et al. in 2011 [75] and Gage and Temple in 2013 [76]. NSC differentiation modelling is a promising field to overcome the limitations of both tumor derived/viral immortalized cell lines and primary neuron cell cultures. NSC self-renew and are multipotent, which means that they proliferate and can differentiate into every neuronal cell type [77]. Their self-renewable characteristics allow the use of a minimal number of animals as a source for primary cell culture. These cultures can be obtained by isolation of NSC from the animal fetal brain, or by differentiation of pluripotent stem cells (embryonic stem cells or iPSCs) [78]. However, studies indicate that neuronal differentiation generates changes in cellular susceptibility to excitotoxicity [79]. In view of that, the comparison of results between differentiating cell cultures of diverse origins is impaired and the protocols have to be adapted to each cell type in order to generate the excitotoxic condition that leads to neuronal death. These conditions are discussed in the next topic.
As previously explained, excitotoxicity can be triggered by various stimuli. Glutamate and NMDA were extensively present in the work developed in the past decades (Figure 2A and Table 1), meaning that NMDAR hyperactivation is the most elucidated mechanism of excitotoxicity in science. Recently, around 200 published studies per year are based on glutamate application, however, without consistency regarding concentration values and time of induction. This serious problem invalidates comparisons between works.
The mechanism of NMDA induced excitotoxicity depends on the activation level of NMDAR, which consists of the number of activated receptors and how long they are kept in active state. Upon binding to the ligand, the receptor undergoes conformational changes that trigger downstream signaling cascades. In the case of the NMDAR, a pore is open after binding of glutamate and glycine. The pore can only open when the cell membrane depolarizes, and removes Mg2+ from the channel. After that, the pore closes and enters a desensitization state, in which the ligand is still bound [74, 80]. This prevents excessive stimulation and influx of Ca2+ to the cytosol. Under conditions of excitotoxicity, the period of desensitization is impaired and depends on diverse mechanisms, such as higher affinity to glycine, membrane potential, pH, chemical reduction and oxidation, etc [80, 81]. These conditions can be easily reversed by decreasing the agonist concentration. PSD-95 anchorage to the NMDAR blocks its internalization, even after prolonged stimulation. In order to better simulate the process in vitro, the researcher must account for the fact that astrocytes and microglia control the availability of glutamate and NMDA in the synaptic cleft [82], which is impaired in pathological conditions [74, 80, 81, 83].
The literature investigation (Table 1), revealed that in most of the work in the field saturating concentrations of glutamate (30–100 μmol/L) were administered to neuronal primary cultures (Figure 2). The saturation measurement was based on Ca2+ influx and cell death measurements (Table 1). In addition, most studies were performed with Neurobasal or MEM, the composition of which lacks glutamate or aspartate, preventing pre-desensitization of NMDAR. Many other studies did not consider the possible desensitization of NMDAR and cultured the cells in aspartate- and glutamate-supplemented media, such as Ham’s F12 based systems (Table 1, Figure 2). Other studies did not consider the duration of the treatment, and therefore had to apply excessive high concentrations of glutamate for observing NMDA-induced excitotoxicity during longer periods. Published manuscripts with most successful data on excitotoxicity had a mean pulse duration of 30–60 min (Table 1, Figure 2). Besides that, most of the studies analyzed cell death and other parameters related to excitotoxicity after 24 h of the glutamate pulse (Figure 2). Some commercial cell lines are cultured in DMEM or F12 medium, and in this case, they demand much higher concentrations of agonist to induce cell death (Table 1) and pulses of at least 24 h duration.
The research in the field of glutamate receptor overstimulation in primary cultured neurons pointed to changes in the expression and phosphorylation of more than one hundred proteins, including neuronal survival regulators such as mitogen-activated protein kinases Erk1/2, glycogen synthase kinase 3 (GSK3) and microtubule-associated protein (Tau) [29]. These proteins are known to be related to AD by regulating cell death and consequent progressive brain atrophy [84]. In addition, phosphorylated tau (p-Tau) is a specific marker for AD, and p-Tau levels are related to the clinical severity of the disease [85].
Biological processes have been enriched through the lists of differentially phosphorylated proteins (DPs) after 15, 30 and 60 min of overstimulation by glutamate and plotted in Figure 3.
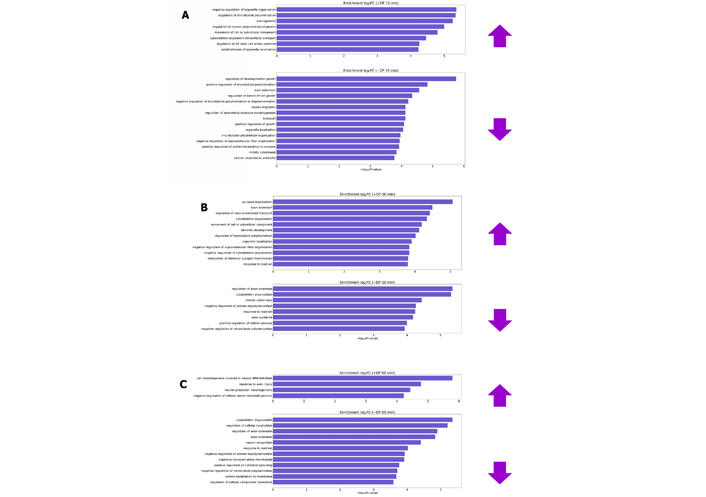
Biological processes enriched by differentially DP after glutamate treatment for 15 (A), 30 (B) and 60 min (C). Prefixes (+) and (–) are representatives of proteins that have obtained normalized fold change by the transformation of log2 (increasing and decreasing phosphorylation levels, respectively). log2FC: log2 fold changes; AV: atrioventricular
In the first 15 and 30 min, most of the processes that presented changes in phosphorylation levels are recurrent in healthy neuronal cultures, such as processes of growth development, extension of the axon, organization of the cytoskeleton and recognition of neurons. Furthermore, cortical neurons were cultured for 7 days before the assay [29]. However, the substantial growth phase of this type of culture occurs between the 14th and 21st day in vitro [86]. This indicates that cytotoxic effects are in the initial phase and/or cells still act in order to adapt to the altered environment. The enriched biological processes are more related to the excitotoxic effect after 60 min of glutamate stimulus, according to Table 2.
Processes enriched in neurons submitted to overstimulation by glutamate for 60 min and pathways related to and affected by excitotoxicity
| Enriched process (glutamate overstimulation for 60 min) | Molecular mechanisms by affected by excitotoxicity |
|---|---|
| Protein localization at the membrane | Alterations in glutamate receptors (particularly NMDAR) and glutamate transporter functioning [9] |
| Organelle transportation along microtubule and regulation of cellular component movement | Changes in Ca2+ buffering and transposition of mitochondria [87] |
| Negative regulation of cellular amine metabolic | Over-recycling of glutamate in a dynamic pool, maintained through the glutamate-glutamine cycle by glutamine synthase in the cytoplasm of astrocytes and excitatory amino acid transporter dysfunction [88, 89] |
| Response processes to axon injury | Degeneration of axons and dendrites by a process associated with excessive increases in [Ca2+]i and mitochondrial dysfunction [90] |
Therefore, in view of the literature data analysis, scientists should review experimental conditions when designing their experiment. 1. Use a culture medium with low concentrations of glutamate or aspartate; 2. duration of exposure to excitotoxic stimuli would last at least 60 min; 3. stability of the agonist; 4. presence of glycine; 5. concentration-response curves using accurate methods for the quantification of the effects of excitotoxicity signaling.
Excitotoxicity triggered by the over activation of NMDA receptors always induces abnormal [Ca2+]i levels. Nowadays, there are varieties of methods to detect it. Here, some methods based on indirect measurements of free [Ca2+]i levels are described.
An accessible strategy to quantify [Ca2+]i concentrations within the cytosol is to use some [Ca2+]i indicators that upon binding to the ion can emit fluorescence or shift their emission spectrum wavelength. In this case, the sensors are based on the Ca2+ chelating properties of ethylene glycol bis (2-aminoethyl) tetraacetic acid (EGTA) [91] and detection using a charge coupled device (CCD) camera. Some sensors can be expressed by the cells using genetic engineering, such as GCaMP or loaded into the cells through membrane permeabilization like Fura-2 and Fluo-4 acetoxymethyl (AM), they vary in their affinity to Ca2+, fluorescence emission intensity and stability. Russell in 2011 provided a seminal overview about these techniques [91].
Fluorescent probes, which change their excitation or emission spectra upon Ca2+ binding, allow ratiometric measurements. Consequently, measurements are not affected by differences in dye loading between cells. The calculation of the dynamic range is the ratio of the fluorescence intensities recorded in Ca2+ saturated and depleted conditions. Although the most frequently used estimation is the signal-to-baseline ratio (SBR), which is the ratio of the change in peak-rest fluorescence (F-F0) over the baseline fluorescence. In order to define the absolute [Ca2+]i concentrations, calibrated values of Ca2+ standard levels are plotted against fluorescence intensities and ratios. As mentioned in the previous topics, it is important to use culture medium that does not have high concentrations of glutamate, and in the case of NMDA studies, it is important to supplement with glycine. We recommend recording Ca2+ for at least 10 min, because in conditions of excitotoxicity, [Ca2+]i levels do not return to the basal state.
Further to increase experimental data quality, an optional experimental control is the addition of divalent cation ionophores. Ca2+ ionophores are lipophilic molecules capable of permeabilizing Ca2+ through the plasma membrane. The most common options are ionomycin and Br-A23187 [92]. Both ionophore molecules have distinct kinetic properties that are linked to their affinity in different medium pH [92]. The use of Ca2+ ionophores allows the estimation of maximal [Ca2+]i within the cell, and consequently work as an experimental positive control for both fluorescence and luminescence assay, as well as mimicking the overt inflow of Ca2+ through NMDAR during excitotoxicity [93].
Besides Ca2+ imaging, researchers can use patch-clamping electrophysiology to evaluate excitotoxicity. More specifically, the whole-cell patch-clamp technique can estimate the activity of glutamate-gated ion channels and changes in the cell membrane potential. The whole-cell mode allows the evaluation, in excitable cells, of transmembrane voltage and current dynamics. In voltage-clamp mode, cells are “clamped” at a specific voltage, allowing the recording of currents that flow through ion channels. In current-clamp mode, cells have spontaneous or depolarizing current-induced action potentials recorded [94].
In excitotoxicity studies, the most common parameter analyzed with whole-cell patch clamp is the function of NMDA receptors. Through this assay, studies can explore what the effect of receptors, gene expression, neurotrophic factors modulation has on NMDA receptors [66]. In this assay researchers use for stimulation NMDA combined with glycine, which plays a co-agonist role [95]. Upon agonist binding to the NMDAR, an inward current of Na+ and Ca2+ flux occurs. For the specific evaluation of NMDAR currents, glycine, GABA subtype A (GABAA), AMPA, and kainate receptors plus voltage-gated Na+ channels are blocked through the application of specific antagonists. Glycine itself is an inhibitory neurotransmitter, acting via specific glycine receptors, and strychnine is applied as an antagonist to block this effect. Bicuculline is used to inhibit GABAA receptors, 2,3-dihydroxy-6-nitro-7-sulphamoyl-benzo(F)quinoxaline (NBQX) for AMPA and kainate receptors, and tetrodotoxin (TTX) for voltage-gated Na+ channels. Although NMDA receptors are physiologically activated by glutamate, they are also coupled to a voltage-dependent condition, carried out by Mg2+ blockage of current flow [96]. For this reason, whole-cell patch-clamping experiments aiming to evaluate NMDA currents must have a Mg2+ free bath solution.
When studying excitotoxicity, it is also possible to analyze miniature excitatory postsynaptic currents (mEPSC). The mEPSC assay evaluates spontaneous currents, rather than inducing the activity of specific receptors as mentioned before. In this whole-cell patch clamp analysis, it is possible to observe the spontaneous release of glutamate and the excitatory response on the neuron recorded. TTX and bicuculline are necessary to block voltage-dependent excitatory currents and GABAA receptor-mediated inhibitory currents [97].
One of the major hallmarks of excitotoxicity is mitochondrial dysfunction. Mitochondrial membrane depolarization, ATP quantification and mitochondrial membrane permeability or rupture are the most of them observed parameters when studying excitotoxicity [98]. As mentioned before, the substantial increase of intracellular Ca2+ is the key factor for cell death induced by excitotoxicity. Among several pathways that are activated in this process, many are involved with mitochondrial bioenergetics. Besides being responsible for cell respiration and ATP production, mitochondria are considered a major hub for Ca2+ homeostasis [99]. Whenever [Ca2+]i rises above the set-point (50–100 nmol/L in neurons), mitochondria promote Ca2+ buffering through its uptake, conducted by a specific MCU [100]. Later, concentrations are re-equilibrated between mitochondria and cytosol, through a slow Ca2+-Na+ exchange [101]. During excitotoxicity conditions, this Ca2+ overload induces depolarization of the organelle, triggering cell death mechanisms. Neuronal cells exposed to glutamate that show an ability to overcome this depolarization, recovering mitochondrial potential and energy balance, tend to survive early necrosis stages [102]. The partial inhibition of MCU is considered a possible neuroprotective treatment, but only when mitochondrial Ca2+ uptake reaches concentrations between 100 nmol/L and 5 μmol/L [61]. Indeed, the partial knockdown of MCU expression in hippocampal neurons impaired NMDA-induced Ca2+ overload in the mitochondria, leading to a neuroprotective effect [103]. On the other hand, a full inhibition of MCU would result in a large impairment of mitochondrial activity, which can lead to a bioenergetic collapse, causing the opposite effect and enhancing excitotoxicity [104]. Thus, mitochondrial depolarization is one of the major indicators of excitotoxicity by the continuous entry of Ca2+ in the cell.
Measurement of mitochondrial membrane potential in intact neurons is challenging. Most methodologies rely on fluorescent membrane-permeant cations, although this methodology can have an interference of plasma membrane potential [105]. The most frequently used indicators tetramethylrhodamine methyl ester (TMRM) [64, 106, 107] and rhodamine 123 [60, 61, 63, 64, 66] are accepted as the most reliable fluorescent indicators, with low interference rates in mitochondrial functions [105]. The fluorescent probe JC-1, also used to measure mitochondrial membrane potential in excitotoxicity studies [40, 43, 48], is the least indicated dye to evaluate these dynamics. The JC-1 dye emits fluorescence in the green light spectrum, and when aggregated within the mitochondrial matrix, shifts to a red fluorescence. First, it is inaccurate to calculate a ratio of red aggregated JC-1 and green JC-1 monomers. In addition, when mitochondrial membrane potential decreases, JC-1 aggregates may not dissipate completely back to the green monomer. This combined with a slow permeation compared to hydrophilic dyes, can give incorrect results [105]. There are also commercial kits available that use tetramethylrhodamine ethyl ester (TMRE), an ethyl ester closely related to TMRM [66], or the range of MitotrackerTM dyes [48, 58]. All these methods do not specifically determine the mitochondrial membrane potential. Yet they provide a semiquantitative estimative when comparing basal vs. treated values. For further reading, Nicholls [105] provided a seminal overview of mitochondrial membrane potential fluorescence methods in neuronal cell culture.
Upon depolarization, the loss of mitochondrial membrane potential can be dangerous for the cell metabolism, mainly due to ATP synthesis impairment. The evaluation of mitochondrial parameters is important to estimate damage extent, as it may lead to necrosis or apoptosis. Cell necrosis occurs when there is a sharp ATP synthesis decrease, resulting in a bioenergetic collapse. On the other hand, apoptosis is facilitated due to membrane rupture by mitochondrial swelling or by outer membrane permeabilization, causing the release of cytochrome c to the cytosol and activating the apoptotic caspase cascade pathway [108]. Mitochondrial activity in intact cells can be assessed by measuring oxygen consumption through high resolution respirometry [40, 61]. The use of specific inhibitors allows us to collect important information about mitochondrial function during these assays. Initially, basal respiration values are detected. After addition of the ATP synthase inhibitor oligomycin, the integrity of mitochondrial inner membrane can be observed, once respiration in this step occurs only due to proton leakage. Next, trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP) or carbonyl cyanide m-chlorophenyl hydrazone (CCCP) addition uncouples the electron transport chain to ATP synthase, in which we see maximal respiration capacity and can observe the function of enzymatic complexes I to IV. And finally, by the inhibition of complexes I (rotenone) and III (antimycin A), oxygen consumption reflects mitochondria-independent oxidative processes [61].
While high resolution respirometry is crucial for analyzing mitochondria activity, it is important to combine it with the measurement of intracellular ATP concentration to provide reliable results. Luciferase-dependent assays are the most common for ATP quantification [40, 48, 51]. It is noteworthy to mention that prolonged mitochondrial depolarization can reverse ATP synthase activity, which then starts hydrolyzing ATP, contributing to bioenergetic collapse. In addition to high resolution respirometry and ATP quantification, excitotoxicity-induced cell death can be detected by mitochondrial cytochrome c, which is released through the permeabilization or rupture of mitochondrial outer membrane. Cytochrome c can be detected by commercial ELISA kits, normally followed by the detection of caspase-3 cleavage to confirm if this release was sufficient to activate caspase cascades [109]. Lastly, although mitochondrial membrane potential, oxygen consumption and ATP quantification are the most used methods to measure mitochondrial impairment in excitotoxicity, there are other important mitochondria-related pathways to consider, such as the activation of nitric oxide synthase and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [98].
ROS participate in diverse biological events, including excitotoxicity. PSD-95 is a scaffold protein that interacts with c-terminal NR2A or NR2B subunits of the NMDAR and to the residues 108–111 of nNOS. Activation of NMDAR triggers nNOS to produce nitric oxide. During excitotoxicity, over-activation of nNOS promotes production of nitric oxide, ROS and peroxynitrites (ONOO–). ROS triggers apoptosis by facilitating cytochrome c release through expression and activity upregulation of pore-stabilizing proteins (Bcl-2 and Bcl-xL). ROS detection can be challenging, since ROS concentrations are often in nanomolar range [110].
The commonly used method is the application of fluorescent dyes like fluorescein and rhodamine that were previously modified to “dihydro” colorless, nonfluorescent leuco dyes. Upon oxidation by ROS they turn back to a fluorescent state, serving as an indicator. 2’,7’-dichlorodihydrofluorescein diacetate (H2DCFDA) is the most common one, and similarly to calcium sensitive dyes they are trapped in the intracellular space after lipophilic blocking groups removal by esterases [111]. Carboxy-H2DCFDA shows improved cellular retention while the fluorinated molecule (H2DFFDA) is more photostable, producing less artifacts. Although this is a simple and cheap technique, it is noteworthy to remember that any oxidative factor can increase the signal [112]. Another way to overcome this limitation, is the use of MitoSOX™ Red, which detects superoxides in mitochondria, emitting red light upon oxidation.
Additionally, some genetically encoded probes are available. These can detect ROS changes in real time, like redox sensitive green fluorescent protein (roGFP) [113]. The roGFP is a redox sensor due to the two cysteines added to the original GFP sequence. The oxidized state is a disulfide while dithiol is the reduced. The redox state determines the fluorescence spectrum of the sensor, 488 nm wavelength excites the oxidized form and 405 nm for the reduced form [113]. A good experimental condition for comparison of a positive signal is the addition of H2O2 to the sample.
NMDA-induced excitotoxicity triggers the apoptosis pathway [114]. This process, known as “programmed cell death”, is crucial for organism development and homeostasis [115, 116]. Many circumstances and signals regulate apoptosis through some canonical pathways, such as uncontrolled cell proliferation, DNA damage, cell stress and alteration of metabolic pathways [117]. Upon activation, apoptosis pathway leads to DNA fragmentation, chromatin condensation, cytoplasm compaction with plasma membrane bubbles, cellular swelling and apoptotic body formation. Later, immune cells phagocyte these bodies through recognition of the cell surface markers [118].
Furthermore, the three main apoptosis pathways are: (a) the intrinsic pathway begins with intracellular stimuli, such as viral infection and toxic agents, causing permeabilization of the mitochondrial intermembrane space. This event is followed by the release of pro-apoptotic elements to the cytosol, where cytochrome c can form a complex with Apaf-1 and caspase-9, known as apoptosome, that activates a cascade of caspases, resulting in cell death [119]; (b) another intrinsic pathway starts in the ER, where the accumulation of unfolded or misfolded proteins activates ER-proteins sensors, as protein kinase R-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1a) and activating transcription factor 6 (ATF6) that lead to transcription attenuation, decreased protein synthesis and proteolytic degradation [117, 120], causing cell death; and (c) the extrinsic pathway consists of the interaction between death ligands in specific immune cells and death receptors in the cell destined to die. This interaction leads to a recruitment of adaptor proteins, producing a protein structure called death inducing signaling complex (DISC) that mobilizes and activates procaspases, promoting apoptosis [121]. The published studies in the field of excitotoxicity usually evaluate the outcome, which is cell death. However, the field still lacks a standard method that reliably assesses cell death induced through NMDA excitotoxicity. Among the various methods, we highlight the most frequent ones, such as TUNEL staining, LDH, MTT, and WB for Bax and Bcl. Although all of them are suitable as a cell death detection assay, most powerful methods are here suggested to reliably identify and quantify excitotoxicity-induced apoptotic cell death, as well as the disadvantages of each technique.
Even though cell death can be qualitatively obvious by simple morphological microscopic observation, such as shrinkages, nuclear blebbing and apoptotic bodies [122, 123], this is not enough to measure the extension of the damage in the cell population. There are many quantitative methods to detect apoptosis and they are based on different characteristics or stages of the process. Dye exclusion assays are often used for apoptosis study due to their simplicity and inexpensiveness, and are based on membrane integrity. The most common dye is trypan blue, which enters and stains dead cells, but not the viable ones [124]. However, plasma membrane disruption is a late event of apoptosis; therefore, dye exclusion assays are restricted to later time points [125]. The scientist should account for some technical flaws that occur when cells absorb imperceptible amounts of dye, or cell loss during the process [124].
Since apoptosis frequently involves DNA fragmentation, a common strategy is to examine the different sizes of DNA fragments by agarose gel electrophoresis. In this method, the total genomic DNA samples containing the fragmented DNA are exposed to electric tension and separated by their sizes in agarose gel. The result is a DNA ladder pattern through the gel [126]. Nevertheless, this method is not very accurate, once the DNA fragmentation occurs later in the apoptotic process. In this case, DNA fragmentation should be evaluated at different time points to avoid false negative results [127].
Another technique to determine DNA fragments is the TUNEL. Basically, the method quantifies DNA strand breaks within a cell by the detection of the in situ insertion at free 3’-OH ends with fluorescent labeled nucleotides, such as BrdU and biotinylated deoxyuridine triphosphate (dUTP), mediated by terminal deoxynucleotidyl transferase enzyme activity [128]. Besides the modernity of the method, the TUNEL assay is costly and results in significant number of false positive results [129]. These unreliable outcomes emanate from the fact that the TUNEL assay is not specific for apoptosis, but for DNA damage in general, which can occur in necrosis cell death, highly proliferative cells and cells in specific processes, such as DNA repair and active gene transcription [130, 131].
During healthy and non-stressful conditions, phosphatidylserine (PS) location is within the cytoplasmic cell membrane leaflet surface. Upon apoptosis, two main events are responsible for PS translocation to the outer plasma membrane leaflet: active PS externalization and inhibition of PS internalization. Scramblases translocate PS to the external membrane leaflet surface in order to be engulfed by macrophages [132, 133], followed by inhibition of flippase enzymes responsible for homeostatic maintenance of PS in inner plasma membrane leaflet. A common approach to quantify apoptosis signal is the detection of fluorescent labeled annexin V [134]. Annexin V is a Ca2+-dependent phospholipid-binding protein present in humans that shows high affinity to PS. Incubation of fluorescence- or biotin-labeled annexin V with the intact membrane cells, followed by its detection through flow cytometer allows the quantification of the percentage of apoptotic cells [135]. As for the TUNEL assay, the cost of this technique is above the average. In addition, to ensure accuracy, some comparative controls that access basal PS externalization should be conducted in parallel.
The caspase activity assay for cell death analysis focuses on detection of executioner caspase activity, such as caspase-3 and caspase-7. The strategy is to use caspase substrates linked to fluorochromes. Upon cleavage of these substrate, the fluorescence emission rates are recorded and caspase activity is measured by flow cytometry. This technique is very accurate but more expensive than the simpler ones [136, 137]. The immune detection of cleaved caspases through WB is also possible, as well as the expression of other important proteins involved in the apoptosis process, such as Bcl-2, Apaf-1 and cytochrome c [138, 139]. Normally, this apoptosis WB assay is performed simultaneously with the application of a cocktail of apoptosis induction proteins such as p53, cytochrome c, Bcl-2, Apaf-1, PARP 1, CD95, CD261, CD262, CD120b, BAX, BAD, receptor-interacting protein kinase 3 (RIPK3), RIPK1, caspase-3, 7 and 9 and with DAPI staining to identify apoptotic nuclei.
Apart from this, another widely used method to measure neuronal cell death in excitotoxicity experiments is the quantification of cellular metabolic activity. This high throughput assay uses cellular metabolic activity as a proxy for cellular viability, proliferation and cell death. It relies on tetrazolium salt reduction to formazan crystals through mitochondrial dehydrogenases. Compounds for high throughput assays are available as MTT or other reduction salts as shown in Table 1 [140]. Despite their wide application throughout the literature, the use of oxidation-reduction (redox) reagents should be carefully evaluated, because MTT reduction is cytotoxic and its assay alone does not distinguish between proliferation/cell death and metabolic changes. Cells undergoing differentiation programs or under drug treatment may trigger “metabolic” shifts. In view of that, additional assays for verification of cell death induction are needed, like the ones previously mentioned here [141, 142]. Moreover, some drug interventions could potentially introduce experimental bias in the MTT assay, as demonstrated for ionizing radiation, which evoked mitochondrial proliferation and consequently increased MTT reduction, while it also increased the cellular death observed in other methods [142]. Similar effects were observed following drug administration with cholesterol, polyphenols and some protein inhibitors [142–145].
In the excitotoxicity field, the leakage detection of LDH is consistently used as an approach to detect cell death [146]. LDH is a soluble intracellular enzyme released by cells into the extracellular medium when the plasma membrane is disrupted. As in the MTT assay, the LDH method involves redox reactions. However, in this case, the reaction of converting tetrazolium salt into the colored formazan form is provided by the nicotinamide adenine dinucleotide (NADH) that is produced when LDH catalyzes the oxidation of lactate to pyruvate [147]. The extent of formazan formation MTT is colorimetrically measured. The essential point of this assay is detection of necrosis, once this depends on acute membrane permeability, but does not discriminate between primary and secondary necrosis. The difference between primary and secondary necrosis is that the last one occurs as a consequence of the apoptosis process. Considering all this, the LDH assay is capable of detecting apoptosis only in late stages. As for the MTT assay, reagents interfering with the redox reactions or with colorimetric patterns should be averted, such as compounds include oxidizing and reducing agents and even products that can modify the color of the cell culture medium. Common reagents used in cell culture, such as serum and medium, may contain factors, interfering with the MTT assay. Another limitation is that quantities of LDH formation differ between cell types. Therefore, cell death comparison between lineages based on LDH formation is a convoluted concern.
Taken together, the here discussed methods explore distinct mechanisms for assessing viability and quality of living or quantifying dead cells. Every protocol has considerable advantages and disadvantages that should be taken into account for each experiment. For this reason, combining methods based on different detection mechanisms may result in improved results.
In conclusions, our critical investigation of the state of art of excitotoxicity provided surprisingly interesting information: (a) Besides being widely studied, the excitotoxicity field lacks standardization regarding models, stimuli features and quantitative measurements; (b) the most described models in literature are primary cultures of neurons and stem cell-derived neurons; (c) phosphoproteomic data from mouse primary cultures of cortical neurons showed altered intracellular pathways are related to adaptation to the in vitro environment after 15 and 30 min of glutamate (100 μmol/L) stimuli, indicating that the culture time was insufficient for the cells to reach a state resembling excitotoxicity. Excitotoxicity-related pathways are activated just after 60 min of glutamate receptor overstimulation, as a minimum period for establishing a significant relationship of treatment with effects of excitotoxicity; (d) we suggested the best methods in our opinion for each feature of excitotoxicity (Figure 4) and we propose the employment of comparative controls and experimental design conditions for quantification of excitotoxicity.
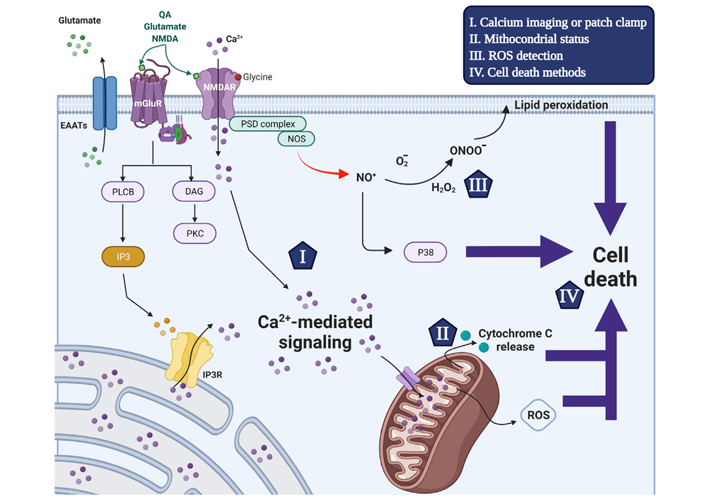
Schematic representation of the excitotoxicity mechanism that triggers neuronal cell death and used detection methods. Glutamate receptor agonists trigger intracellular [Ca2+]i increase through ionotropic and metabotropic glutamate receptor activation. The activation of NMDAR by NMDA, glutamate or QA and the co-activator glycine increases the plasma membrane permeability to Ca2+ ions. Alternatively, glutamate also induces transient [Ca2+]i increases through activation of G protein-coupled metabotropic receptors activating phospholipase C-β to cleave PIP2 into IP3 and DAG. In turn, IP3 triggers the release of ER-mobilizated intracellular Ca2+ ions through the IP3 receptor. The Ca2+ signaling is an important second messenger and disruption of its homeostasis is related to the excitotoxicity process in excitatory neurons. Following [Ca2+]i increase, MCU transports Ca2+ ions to the mitochondrial matrix (Ca2+ buffering) in a gradient favorable manner, which in turn disrupts mitochondrial membrane potential interrupting ATP production and initiating cell death mechanism. Moreover, The NMDAR interacts with nNOS in a PSD-95-dependent manner, and upon overstimulation the formation of nitric oxide, ROS and peroxynitrites that also contribute to cell death induction. The current accurate methods to evaluate excitotoxicity process are highlighted by items I-IV that are reviewed within the manuscript. EAATs: excitatory amino acid transporters; PLCB: phospholipase β; PKC: protein kinase
Together, these practice recommendations will ease the understanding of mechanisms of excitotoxicity, both under physiological and altered conditions, and facilitate the comparison between findings of various scientific groups. Improved methods for excitotoxicity detection will also help to standardize neuroprotection screening methods for obtaining lead compounds, which can be later on tested in animal models.
Aβ: amyloid-β
AD: Alzheimer’s disease
AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
BAX: B-cell lymphoma-2-associated X
Bcl-1: B-cell lymphoma 1
[Ca2+]i: intracellular Ca2+ concentration
DAPI: 4’,6-diamidino-2-phenylindole
DMEM: Dulbecco’s Modified Eagle’s medium
ER: endoplasmic reticulum
GABAA: γ-aminobutyric acid subtype A
HD: Huntington’s disease
HTT: huntingtin
IP3: inositol 1,4,5-trisphosphate
LDH: lactate dehydrogenase
MCU: mitochondrial Ca2+ uniporter
MEM: minimal essential medium
mGluRs: metabotropic glutamate receptors
mHTT: mutant huntingtin
MTT: methyl thiazolyl tetrazolium
NMDA: N-methyl-D-aspartate
NMDAR: N-methyl-D-aspartate receptor
nNOS: neuronal nitric oxide synthetase
NSC: neural stem cells
PC12: pheochromacytoma 12
PD: Parkinson’s disease
PI: propidium iodide
PS: phosphatidylserine
PSD-95: postsynaptic density protein-95
QA: quinolinic acid
ROS: reactive oxygen species
TUNEL: transferase deoxyuridine triphosphate nick end labelling
WB: western blotting
TG conceived the main hypothesis of the review. TG, JBS, GAJ, PNM, NT, LLP, LBM, DER, QY contributed to the manuscript writing and figures’ design. GAJ did the computational analysis. TG and JBS contributed to the formatting. TG, YT and HU reviewed the paper.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
HU acknowledges grant support from the São Paulo Research Foundation (FAPESP, Project Nos. 2018/07366-4 and 2018/08426-0) and the National Council for Scientific and Technological Development for fellowship support (CNPq, References Project No. 306392/2017-8). DER and TG are grateful for postdoctoral fellowships granted by FAPESP (Project Nos. 2018/17504-5 and 2015/13345-1-PD, respectively). LLP is grateful for a CNPq DTI-A fellowship (CNPq 380405/2020-2). The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

