 Review
Review

Affiliation:
1CiberNed, Network Center for Neurodegenerative diseases, National Spanish Health Institute Carlos III, 28029 Madrid, Spain
2Molecular Neurobiology Laboratory, Department of Biochemistry and Molecular Biomedicine, Faculty of Biology, Universitat de Barcelona, 08028 Barcelona, Spain
3School of Chemistry, Universitat de Barcelona, 08028 Barcelona, Spain
Email: rfranco@ub.edu
ORCID: https://orcid.org/0000-0003-2549-4919
Affiliation:
2Molecular Neurobiology Laboratory, Department of Biochemistry and Molecular Biomedicine, Faculty of Biology, Universitat de Barcelona, 08028 Barcelona, Spain
ORCID: https://orcid.org/0000-0001-9316-0721
Explor Neuroprot Ther. 2023;3:8–23 DOI: https://doi.org/10.37349/ent.2023.00035
Received: November 18, 2022 Accepted: January 31, 2023 Published: February 27, 2023
Academic Editor: Thomas Müller, St. Joseph Hospital, Germany
The article belongs to the special issue The Urgent Need for New Hypotheses to Develop Effective Therapeutic Tools Against Alzheimer's Disease
There is no approved drug capable of halting the progression of the most prevalent neurodegenerative disorders, namely Alzheimer’s disease (AD) and Parkinson’s disease (PD). Current therapeutic strategies focus mainly on the inhibition of the formation of protein aggregates and their deposition in the central nervous system. However, after almost a hundred years, proper management of the disease is still lacking. The fact of not finding effective management tools in the various clinical trials already carried out suggests that new hypotheses and strategies should be explored. Although vast resources have been allocated to the investigation of protein aggregates and the pathophysiology is now better understood, clues to the actual etiology are lacking. It is well known that brain homeostasis is of paramount importance for the survival of neurons. Drugs that target the periphery are often not subject to evaluation for their potential effect on the central nervous system. While acute treatments may be irrelevant, pills used for chronic conditions can be detrimental to neurons, especially in terms of progressive damage leading to a long-term decline in neuronal survival. Due to the lack of advances in the search for a curative treatment for neurodegenerative diseases, and the lack of new hypotheses about their etiology, a novel hypothesis is here proposed. It consists of assuming that the effects of the drugs most commonly used by the elderly, such as antihypertensive, hypoglycemic, and hypocholesterolemic, could have a negative impact on neuronal survival.
In the last two centuries, human life expectancy has increased remarkably. Natural selection has not had time in just 200 years to eradicate some diseases in older individuals. However, humans have managed to extend life in the most technically and scientifically developed countries, but with the risk of suffering serious age-related diseases. One of them, Alzheimer’s disease (AD), whose global burden on Earth is extreme, is among the most serious diseases of the elderly and scientists have found no cures or effective treatments [1, 2].
AD is included among the so-called “dementias”. The Alzheimer’s Association claims: “Dementia is an overall term for diseases and conditions characterized by a decline in memory, language, problem-solving, and other thinking skills.” [2–4].
Despite the fact that AD research is receiving a large percentage of the research budget in all Western countries [3], there is still no curative or palliative treatment. Unlike cancer research, in which several therapeutic interventions have been developed in the last decade, neurodegenerative diseases remain on the fringes of therapeutic advances. One of the main differences between research related to oncology and neurodegeneration is that, in the former, several hypotheses had been addressed, and some of them have been successful. In contrast, research on neurodegenerative diseases has focused primarily on one hypothesis, namely neuronal damage resulting from protein aggregation. Protein aggregations in AD lead to what is known as (extracellular) beta-amyloid plaques and (intracellular) neurofibrillary phospho-tau-protein-containing tangles. Taking into account the lack of advances when considering the hypotheses related to protein aggregates, it is necessary to look for novel hypotheses to get out of explored and failed paths.
The prevailing hypothesis is that patients accumulate proteins outside and inside brain neurons that, over time, kill cells necessary for cognition and memory retrieval. Cell death is progressive and sufferers deteriorate as neither cure nor delaying interventions are available [4].
Protein accumulation and aggregation in the extracellular medium is known as “plaque” and mainly contains “amyloid”, a degradation product of the amyloid precursor protein (APP). After investing extremely high resources to obtain a large amount of data on AD, it is quite puzzling why we still do not understand the function of APP [5, 6]. This has not deterred attention from the “amyloid” hypothesis for years and years, yet it is paradoxical that the question Drachman [7] posed in the title of his 1986 article: “Plaques and tangles in Alzheimer’s disease: cause, consequence or epiphenomenon?”, has not found an answer to this day.
From the start, the amyloid hypothesis was dismissed by some scientists, who have been silenced and not allowed to adequately test alternative hypotheses (see Conclusions). These scientists thought that the protein buildups were a consequence and not a cause.
At the moment, several data suggest that the negative effects caused by amyloid plaques include the so-called “neuroinflammation” which is supposed to impair the survival of neurons. Proinflammatory events in the central nervous system (CNS) may be caused by protein aggregations or may be due to the yet undiscovered pathophysiological mechanisms. The improvement in cognition observed in animal models of AD when plaques are diminished by the use of antibodies is not inconsistent with an amyloid-independent cause of disease onset [8]. The gold standard in the management of a disease is to address the cause(s). Therefore, if amyloid plaques are a consequence of a disease that aggravates symptoms, a decrease in amyloid load may be beneficial, but not because it addresses the cause(s). This notion is especially relevant if, as coined by Cacabelos et al. [9], AD is a “spectrum of syndromes”.
Since dogma often prevails and dissident visions are silenced by the system at large [10], everything possible was done to combat the “cause” of AD, namely dissolving amyloid plaques. Many of the most powerful research laboratories and many of the most powerful companies developing drugs to cure diseases began a race to dissolve amyloid plaques that ended in nothing. Extraordinary resources were wasted and thousands of patients were recruited for interventions that did not provide any encouraging results in terms of effective management of AD.
It is worth reading the article “On the origin of Alzheimer’s disease. Trials and tribulations of the amyloid hypothesis” [8]. Actually, the dogma has been translated into another neurodegenerative disease, Parkinson’s disease (PD), characterized by tremors and walking difficulties. PD and AD share chronicity, progression, and the occurrence of protein deposits. α-synuclein is the protein that accumulates in PD. In fact, many resources related to PD research are devoted to approaches to dissolving α-synuclein aggregates that have been used to dissolve amyloid [11]. The results are expected to be little or nothing. Fortunately, PD patients have a range of efficacious interventions helping them in addressing symptoms. It should be noted that in extreme cases, surgery allows electrodes to be inserted into the brain to stimulate brain areas that are involved in motor control. Therefore, PD patients have better prospects/hopes than AD patients.
A basic flaw underlying both diagnosis and research is the lack of biomarkers for neurological diseases. Clinicians rely on indirect means to make AD diagnoses. This is a major drawback that has not been resolved despite the resources allocated. Again, the search for amyloid-related biomarkers had a negative impact on the search for truly useful biomarkers. The old controversy about the role of amyloid is now translated into discovering whether increases in amyloid-derived products correlate positively or negatively with symptoms or whether they predict rapid or slow disease progression. Based on research developed in our laboratory [12–14] and data from the literature [15–17], we believe that adenosine 3’,5’-cyclic monophosphate (cAMP) and/or guanosine 3’,5’-cyclic monophosphate (cGMP) in cerebrospinal fluid may be better biomarkers for assessing AD status than the determination of the amount and ratio of amyloid species.
Diabetes has a key biomarker, glycemia, and other secondary biomarkers (e.g., insulin and glycosylated hemoglobin blood levels) that help direct clinical interventions. Professionals have good references and follow equivalent protocols to treat diabetes. There are good biomarkers for hypercholesterolemia, while high blood pressure can be directly addressed using a simple device that the patient can use at home. The lack of suitable biomarkers for almost any trouble affecting the brain has consequences. Probably the two most relevant are:
(1) The presence of different clinical symptoms is diagnosed under a single condition, for example, AD.
(2) Physicians do not have a reference to act consistently, that is, to prescribe the same therapy/intervention for a given clinical scenario.
Any intervention aimed to alleviate the symptoms of a disease first requires testing in animal models. In agreement with the amyloid dogma, one rodent model consists of overexpressing a mutant form of human APP. Over time the mouse develops memory impairment that is measured with a method consisting of a water maze also known as the “Morris water maze”. What is striking and correlates with the fact that the amyloid hypothesis is dubious is that neuronal death does not occur. Similar to the human condition, the “symptoms” in animals appear late in life. Unlike in humans, it is not due to any significant neuronal death. Surprisingly, these animal models improve cognitive decline when very different drugs are administered. The image in Figure 1, taken from an article entitled: “Successful therapies for Alzheimer’s disease: why so many in animal models and none in humans?” [18], reflects the fact that scientists are producing animals having “human diseases” in which a myriad of different molecules are tested for efficacy. The “Alzheimer’s disease” in rodents is relieved by hundreds of different molecules [18]. Various types of drugs have been shown to be effective in reversing cognitive decline in animal models of AD. These include ligands for G protein-coupled receptor (adrenoceptors, histamine receptors, etc.), cAMP/cGMP phosphodiesterase inhibitors, and molecules targeting enzymes involved in the epigenetic mechanisms [13, 19–22]. Among the more than 65,000 articles in PubMed (as of January 2023) on “therapy” and “Alzheimer’s”, there is even room for the “demonstration” that moderate consumption of Cabernet Sauvignon wine reduces amyloid pathology in the model of AD from Tg2576 transgenic mouse [23]. Unfortunately, drugs effective in animals become ineffective by the time they reach clinical trials in AD patients.

Cartoon depicting the many reported possibilities for “curing” animal models of AD [18]. Copyright retained by Nuria Franco (franconuria@gmail.com)
Neurons in AD die and the causes cannot be very convoluted. Either there is a lack of activity, a lack of nutrients, a lack of oxygen, or all together. The triggers may be different, but the steps towards cell death may have common denominators.
A possible culprit in AD, as in almost any other disease, is a viral infection. It has been suggested that a virus that cohabits with humans is to blame for AD. Herpes simplex virus types 1 and 2 [24] infects neurons when immune cell function is undermined due to disease, improper nutritional status, etc. Active infections are painful and obvious. Despite the impact of active infection on neurons located outside the brain, a similar infection in the brain would cause significant symptoms, which have not been reported as a step prior to the diagnosis of AD. Is the virus latent in the brain thus contributing to the neurodegeneration caused by the key factors that cause AD? Could some of the patients have had concurrent active infections that led to neuronal cell death? These are reasonable possibilities, but the illness cannot be attributed to a virus. As in cancer, AD is not primarily due to viruses, but exceptions can occur and viral infections can trigger neuronal death and/or accelerate AD progression.
Although the main risk factor for AD is age, all robust epidemiological studies agree that other relevant risk factors are diabetes, hypertension, and/or hypercholesterolemia, that is, people who have one or more of these conditions are more likely to develop AD-type dementia [1, 2, 25]. The hypothesis put forward in this article is that it is not the disease, but the chronic medication against diabetes, hypertension, and/or hypercholesterolemia that is the cause of the increased risk of AD. Could it be that the sooner these drugs are taken, the greater the risk of AD? There is evidence suggesting that communities using herbal-based remedies or reluctant to take pills have a lower risk of AD (see Distribution of patients with AD in different populations).
The nuns are religious women who live together, which means they have similar eating and social behavior. Why do only some of them get the disease? A report known as the “Nun Study” [26, 27] supports the idea that lifelong knowledge acquisition and mental activities help prevent AD.
An important piece of information that is often missing, but that is important is the profession of the AD patient. One wonders, for example, how many piano virtuosos suffer from the disease. Pianists must use both hands in two separate patterns, one for each hand. Music shapes the brain in a way that it may be refractory to neurodegeneration. It is also intriguing why we think the size and weight of their brains are quite similar. One may be surprised by the results of both meson-based telemetry and direct inspection at forensic offices. The weight of the human brain within a given population can differ very significantly. Apart from the extremes reported for renowned writers, 2.021 kg for Ivan Turgenev and 1.017 kg for Anatole France [28], the average brain weight within a Nepali homogeneous population aged 21 to 30 is 1.372 kg, with a range of > 400 g (1.150–1.588 kg) [29]. In a specific region of northwestern India, the brain weight of people aged 21–30 is reported to vary between 1.040 kg and 1.620 kg (average of 1.340 kg) [30]. This high variability is surprising and suggests that neural reserve and neural connectivity have not been adequately addressed. Cognitive reserve can be considered as an indicator of the number of connections between neurons. Although the number of connections cannot yet be measured, it has been shown that there are marked differences in the brains of musicians and non-musicians [31, 32]. A piano virtuoso is likely to develop many more neuron-to-neuron connections than the average individual. As a result, compromised neurons in a pianist’s brain will take longer to die than equivalent neurons in many other brains that have fewer connections. There is evidence that playing musical instruments that require the synchronization of both hands could protect against neurodegenerative diseases [33, 34]. An interesting study on twins (musician and non-musician) provides encouraging data suggesting a protective effect of pursuing a professional career in music. A meta-analysis by Walsh et al. [35] suggests that there is a trend, but more research should be done to confirm a relationship between being a professional musician and a lower dementia risk.
Taking into account all the possible scenarios, the still missing data, and the misleading dogmas, it is necessary to further explore why neurons can remain active and survive for years or, alternatively, decay and die.
A naive look is convenient to avoid falling into any dogma. For instance, in our opinion, data on caffeine is relevant. Taking caffeine prevents both AD and PD [36–48]. All civilizations on Earth know that keeping a person awake and alert can be achieved through the action of drinks containing caffeine and related molecules (methylxanthines). Although not directly related to neurons being more alert, physical activity is also considered good for preventing AD. But there is something in common when you consider playing a musical instrument, caffeine consumption, and physical activity. In all cases, the neurons in the brain get what they need to stay active and alive. In reality, this is a hormetic mechanism, a type of mechanism that is often overlooked in assessing human health and disease [8, 49–51].
In short, the blood supply and two main components in blood, oxygen, and glucose, are key to keeping neurons alive. These well-known facts support that:
(1) Physical exercise (including walking) prepares the cardiovascular system to react when the brain requires more blood to perform an activity (physical or mental).
(2) Playing musical instruments that require both mental activity and motor coordination prepares the cardiovascular system to allow adequate blood supply to the brain.
(3) Taking caffeine quickly produces more blood supply to the brain: if an individual is more alert, brain regions are better interconnected.
The different phases (I, II, III, and IV) of the clinical trials necessary to decide on the approval of a drug for human use are intended to confirm the efficacy and avoid side effects. However, such adverse events are usually addressed in short periods of time. There is very little information on the long-term effects of chronic treatments. It is fair to assume that the benefits outweigh the risk of any long-term side effects [52]. However, the increase in life expectancy and the overwhelming use of pills by the elderly make it necessary to review the benefit/risk balance. Except in the least westernized countries, older people take pill-based medicines. At least one pill in 60% of people older than 65 years and at least 2 or more pills in 85% of people older than 70 years [53]. Surely it is difficult to design a trial to assess the long-term side effects of drugs, but in our opinion, it is necessary to assess whether chronic therapies are influencing the appearance or worsening of other diseases, especially neurodegenerative diseases whose main risk factor is age.
The brain is the most sensitive organ in terms of oxygen and nutrient supply, so any drug that regulates vascularization or in situ nutrient uptake/synthesis is expected to affect brain homeostasis. Any drug that reaches the brain, even in a small concentration, will have an impact on the activity of the brain. Any peripherally acting drug that affects cerebral blood flow or the concentration of nutrients entering the brain can also affect brain performance and neuronal survival [54]. In this article we will focus on three diseases that chronically affect the elderly:
(1) Diabetes: characterized by increased glucose concentration in blood plasma due to problems getting glucose inside the cells. Medication aims at lowering the levels of glucose in the blood.
(2) Hypertension: characterized by increased pressure in blood vessels that may lead to breakage, blood leakage, and life-threatening situations. Medication aims at lowering the blood pressure.
(3) Hypercholesterolemia: characterized by increased levels in the blood of cholesterol-containing lipoproteins that may lead to blood vessel obturation and failure to deliver oxygen to the tissues. Medication aims at lowering the levels of lipoproteins in the blood, especially the level of low-density lipoproteins (LDL).
Now, the question is whether there are risks of taking medications for life. To answer it, one should be aware of two facts:
(1) Cognitive reserve is essential for neuronal survival when nutrient supply and physical activity decline with age.
(2) Levels of both glucose and cholesterol in healthy populations increase with age.
Consequently, the hypothesis to be validated would be: does aging combined with pills to reduce blood glucose, blood cholesterol, and hypertension facilitate neuronal death due to the lack of supply of essential molecules?
Let’s take a closer look at this possibility based on data/evidence.
The neuron-to-neuron connection is a dynamic process by which the contacts between neurons are subject to continuous plastic changes. Aging from a hectic life to a boring retirement is accompanied by a loss of connections, that is, of cognitive reserve. Reduced perfusion with a shortage of oxygen and glucose impacts neuronal fate. Glucose is essential for neuronal activity and survival. Can reduced activity plus the reduced supply of essential components lead to extreme cellular suffering and ultimately neuronal death?
When clinical chemistry was developed in the middle of the 20th century, the main textbook showed that older people have more cholesterol and more glucose than young people [55]. Linking high cholesterol to heart problems led to the development of serum cholesterol-lowering pills. The question is where to put the threshold, first set at 220 mg/100 mL and now at 200 mg/100 mL.
Blood cholesterol-lowering pills can lead to a shortage of cholesterol in neurons and glial cells (Figures 2 and 3). Neurons need cholesterol and it is synthesized in situ, that is, “peripheral” cholesterol does not reach the brain [56]. Therefore, drugs that lower blood cholesterol can become harmful by crossing the blood-brain barrier. Two of the most common cholesterol-lowering drugs, simvastatin and atorvastatin are able to cross the blood-brain barrier [57, 58]. Mechanisms have been proposed to explain the cognitive impairment induced by statins [59] or how alterations in cholesterol metabolism lead to an increased risk of AD [60]. Furthermore, temporary cognitive impairment due to statin treatment has been reported in 2 women [61].
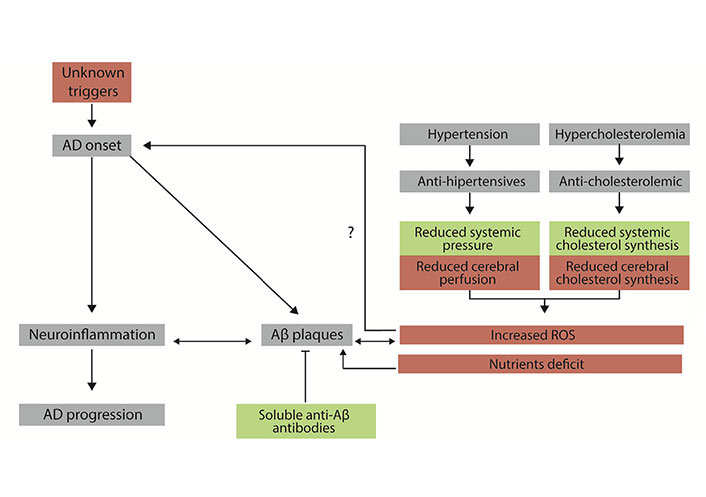
Diagram showing the basis of the drug-focused neurodegeneration hypothesis. Events are useful for patients with high blood cholesterol and/or high blood pressure are in green. Events that may be harmful to patients’ CNS neurons are in red. Mechanisms associated with the initiation and progression of AD are in grey. Aβ: β-amyloid; ROS: reactive oxygen species; →: implication; ?: hypothesized implication
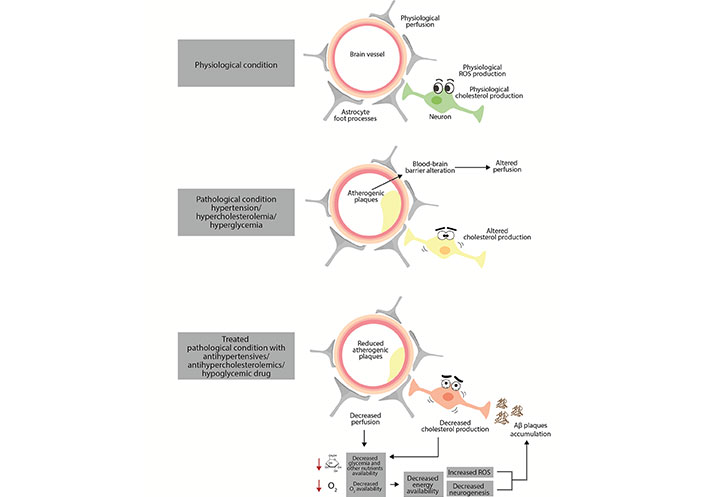
Mechanisms potentially involved in drug-related alteration of neuronal function in the central nervous system. Antihypertensives in combination with reduced glucose availability and antihypercholesterolemic drugs can compromise metabolism and key cellular functions, thereby compromising neuronal survival. →: implication; →: decrease
The 90+ Study led by Kawas and Corrada [62] is continuously providing relevant information on a variety of factors and molecules related to AD pathophysiology. One of the most recent reports consists of a case of cognitive resilience despite the detection of three dementia-related neuropathologies at autopsy [63, 64]. Regarding apolipoprotein E (ApoE), whose allele composition is a risk AD factor [65] and that is involved in lipoprotein transport and cholesterol homeostasis [66], one of the major conclusions of the 90+ Study is that “people aged 90 and older with an ApoE2 gene are less likely to have clinical Alzheimer’s dementia, but are much more likely to have Alzheimer’s neuropathology in their brains.” [62, 64]. This means that the finding of “AD neuropathology” does not necessarily imply AD-like dementia and that cholesterol in the brain is a factor that deserves close scrutiny to advance the fight against the disease.
Plasma glucose-lowering pills lead to a shortage of glucose availability for neurons. Neurons depend to a large extent on glucose to survive and perform their function correctly. Unlike many other cells, neurons cannot use fatty acids for energy production. Diabetes can lead to serious, life-threatening conditions. Lowering blood glucose levels in diabetes is necessary, but without reducing glucose supply to the brain. Blood glucose levels must be higher in older populations and diabetes pills should not compromise the glucose supply to the brain. In this sense, the existence of medication that inhibits the synthesis of glucose from proteins, but that does not modify the entry of glucose into cells, is puzzling. Lowering plasma glucose is good, but only if glucose uptake is sufficient, especially for the most important cells (neurons, heart cells, etc.). In short, diabetes medication often leads to a shortage of fuel for cells, and the cells most affected are the ones most dependent on glucose: neurons.
The glucose used by the neurons is completely oxidized and this requires oxygen and the integrity of the so-called cellular respiration, which occurs in the mitochondria. If the reduced perfusion leads to a shortage in the oxygen supply to the brain, the mitochondria in neurons suffer. Sufficient evidence already exists in various neurodegenerative diseases pointing to alterations in mitochondrial function [67–74]. In short, would it make sense that a shortage of blood would affect the mitochondria and ultimately lead to neuronal death?
The hydrodynamic pressure combined with the so-called gravity and the position of the head in the human body does not help cerebral perfusion. It is puzzling, then, why blood flow is reduced in hypertensive people. Actually, the result may be better understood if the decrease in blood flow to the brain result from the drug-induced reduction in blood pressure [75]. In fact, the experiments were conducted on people taking antihypertensive drugs [75]. According to Ockham’s razor rule, a plausible interpretation is that hypotension produced by antihypertensive medication is to blame (Figures 2 and 3) [76]. This rule, also known as the law of parsimony, is defined in Britannica Encyclopedia as “of two competing theories, the simpler explanation of an entity is to be preferred.” [77].
The control of blood pressure by the elderly is progressively lost and it is common to find a tendency to arterial hypertension. Blood pressure pills are essential because high blood pressure can lead to life-threatening conditions. However, clinicians need to understand that chronic hypotension is not good for older people. It also happens that hypertension can occur only during working hours and disappear on weekends. Then the dose and frequency of medication must be chosen very carefully. Antihypertensive medication may lead to hypotension, and if hypotension is sustained and/or leads to frequent and/or prolonged unconsciousness/hypoxia, neurons suffer and have no choice but to adapt to the new condition by spending less glucose/oxygen, something that should affect neuronal performance/resilience. Can antihypertensives lead to an “essential hypotension-like” state in some people? In 1925, an editorial in the Journal of the American Medical Association stated that essential hypotension “has heretofore received little consideration from writers on clinical medicine” [78]. In our opinion, the more or less prolonged periods of hypotension that can occur in some individuals taking antihypertensives have not attracted the attention they deserve.
A non-negligible percentage of antihypertensive drugs have effects on CNS, these include hallucinations and depression [79–82], and those findings suggest that these types of drugs may have long-term effects on the CNS by mechanisms that have not yet been deciphered.
Unlike hypocholesterolemic drugs, which are basic inhibitors of the enzyme that catalyzes the rate-limiting step in cholesterol biosynthesis, there are different classes of drugs that can lower blood pressure and are approved for human use. Some classes of antihypertensives can enter the brain, likely affecting the regulation of neurotransmission in several ways. Candesartan is a prototype of the “sartans” type of hypotensive drug. They act by antagonizing the angiotensin II (AT1) receptor. It happens that this receptor, expressed in neurons, is a potential target of neurodegenerative diseases [83–92]. Animal models of neurodegenerative diseases have been instrumental in demonstrating that candesartan can cross the blood-brain barrier and act on receptors expressed in neural cells. As with cholesterol-lowering drugs, the possibility of hypotensive drugs entering the brain has not been considered in clinical practice. Unlike the likely undesirable effect of cholesterol-lowering drugs, there is evidence in both animal models of AD and in AD patients that candesartan provides pleiotropic benefits by acting in cells in the central nervous system [93–98]. In summary, the effect of antihypertensive medication on AD risk may be different depending on the drug’s ability to cross the blood-brain barrier and on the target of the drug. Longitudinal studies evaluating cognition and other neurological parameters depending on the type of drug used to lower blood pressure could be very informative.
Experience shows that dogma has not been useful in helping AD patients. A new dogma in the field may come from a “systematic analysis” that estimates that 22.3% of dementias are attributable to these “modifiable” risk factors: “high body-mass index, high fasting plasma glucose, a diet high in sugar-sweetened beverages, and smoking” [1]. Our hypothesis is that risk factors are being misinterpreted, and if this is correct, this new dogma will be just as ineffective as the one that focused on the amyloid.
There is no doubt that age is the main risk factor for AD and that AD is prevalent in elderly populations who chronically take antidiabetic, antihypertensive and/or hypocholesterolemic drugs. Despite i) the impossibility of reliably comparing the parameters related to patients with AD in different populations and ii) the bias due to genetic factors, it is relevant to find that individuals separated from one society and located in another rapidly acquire the tendency to the disease of the new environment.
There is an isolated population among one of the greatest civilizations on Earth that live without access to modern technology and are very reluctant to take pills. Known as Amish, their lower incidence of AD may be due to genetic factors (they inbreed), reduced prevalence of some diseases and/or lack of medication. This idea was masterfully conveyed in 1997: “The lower level of cognitive impairment among the Amish could reflect a lack of inherited susceptibility to dementing diseases, or environmental factors characteristic of their traditional lifestyle.” [99]. A naive view of this data is that the decreased risk of AD is due to the reported low prevalence of diabetes, hypertension, and hypercholesterolemia in Amish communities [100]. However, a rigorous view would also consider that the risk factors for AD may not be the diseases, but the pills currently used to control diabetes, hypertension, and/or hypercholesterolemia. It is intriguing that Amish aware of having the disease (diabetes, hypertension, and/or hypercholesterolemia) are “less likely to report that they were currently taking medications to treat their disease compared with European Caucasian subsample of the 2013–2014 US National Health and Nutrition Examination Survey (NHANES) patients” [100]. Why would the Amish want to hide that they take medication? Is it because they actually take less medication than non-Amish communities?
Yet another different type of hint comes from a populated area of the planet, Asia, where pill-taking was not prevalent until recently. Despite the lack of definitive studies, the key is that alternative curative interventions are correlated with a lower incidence of AD. It could be hypothesized that the lack of pill-taking is, at least in part, the cause of the lower risk. For example, in the so-called traditional Chinese medicine, there are-pill independent approaches to hypertension, including acupuncture, moxibustion, Qigong, or Tai Chi [101]. A cohort study showed a decreased risk of dementia in migraine patients using traditional Chinese medicine [102].
In addition, the traditional societies that have incorporated the so-called western lifestyle (which includes taking pills) are the ones that have increased the incidence of AD the fastest [103, 104].
In PD, some antihypertensive drugs capable of crossing the blood-brain barrier are protective [105] raising the suspicion that some drugs may be beneficial by entering the brain. They would regulate the neuronal activity, that is, they would not act by reducing blood pressure, but by favoring neuronal survival. In fact, among the approved antihypertensive drugs, some can enter the brain while others do not cross the blood-brain barrier.
Antihypertensives capable of penetrating the brain to regulate neurotransmission and neuronal plasticity may play a beneficial role in preventing and/or delaying the progression of AD. The suggestion that potential benefits are due to actions on the hydrostatic pressure of blood vessels in the brain makes little sense. The benefits, if any, probably come from the well-proven fact that there are receptors, e.g., angiotensin receptors, which are targeted by antihypertensives and regulate neural cell fate and higher functions in various areas of the brain [83, 84, 106–108].
So far much is known about the functioning of the brain, but it must be recognized that we are lost in the fight against alterations in brain function (not only Alzheimer’s, but others—“depression”, “autism”, etc.). Even without knowing all the clues, it is tempting to speculate that the brain would benefit from i) sufficient connections, the higher the better; ii) adequate blood perfusion; iii) adequate glucose supply; iv) good ability to synthesize cholesterol, a molecule that, in order to be used by neuronal cells, must be generated in the brain cells themselves. We believe it is convenient to design longitudinal studies (measurement of cerebral perfusion, glucose consumption, etc.) to:
(1) Correlate pill use and the worldwide regional incidence of AD in elderly populations (age-adjusted).
(2) Assess whether older individuals who take pills are at a higher risk of developing AD, taking into account both the target of the drug(s) and whether or not the medication crosses the blood-brain barrier.
(3) Measure cerebral perfusion and glucose consumption both before and during the performance of intellectual tasks (ideally comparing data in young and old people).
(4) Measure cerebral perfusion and glucose consumption both before and during the performance of intellectual tasks (ideally comparing data in young and old people).
(5) Follow parameters over time, before and after taking any pill and before and after the appearance of the neurodegenerative condition.
Not all drugs are created equal. To control peripheral parameters, drugs must not reach the brain. However, drugs that reach the brain can be tested for their usefulness in preventing and ultimately fighting diseases that affect the brain.
At last, an important observation: the parameters and protocols to assess neuroprotection, are not yet available. It is urgent to discover neuroprotection biomarkers and/or protocols to detect neuroprotection in humans.
AD: Alzheimer’s disease
APP: amyloid precursor protein
CNS: central nervous system
PD: Parkinson’s disease
RF, JSM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Supervision, Validation, Visualization, Writing—original draft, Writing—review & editing. RF: Project administration, Resources.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Karolina Armonaite ... Luigi Laura
Jesús Avila ... Félix Hernández
Glòria Salort ... Jesús A. García-Sevilla
Milind Watve, Ashwini Keskar Sardeshmukh
Iyawnna Hazzard ... Forshing Lui
