 Review
Review

Affiliation:
Centre of Excellence in Healthcare Technologies and Informatics (CEHTI), Department of Biotechnology and Bioinformatics, Jaypee University of Information Technology (JUIT), Solan 173234, Himachal Pradesh, India
Affiliation:
Centre of Excellence in Healthcare Technologies and Informatics (CEHTI), Department of Biotechnology and Bioinformatics, Jaypee University of Information Technology (JUIT), Solan 173234, Himachal Pradesh, India
Email: tiratharaj.singh@juitsolan.in
ORCID: https://orcid.org/0000-0003-1109-5626
Explor Neuroprot Ther. 2023;3:90–104 DOI: https://doi.org/10.37349/ent.2023.00039
Received: July 05, 2022 Accepted: January 20, 2023 Published: April 03, 2023
Academic Editor: Jose Miguel Soria, CEU Cardenal Herrera University, Spain
Alzheimer’s disease (AD), which affects around twenty-seven million people globally, is an aging-related neurodegenerative condition characterized by the extracellular deposition of misfolded amyloid-β (Aβ) peptides and the intracellular production of neurofibrillary tangles (NFTs) AD results from the death of certain groups of neurons in the brain while appearing to have no impact on neighboring neurons. It is progressive and incurable. Therefore, the pathophysiology of afflicted populations and the development of intervention measures to stop neuronal cell death have been the main areas of attention for delineating therapeutic options. Proinflammatory cytokines are responsible for the stimulation of inflammatory responses and are mostly generated by activated macrophages in the brain. This review discusses how glial cells and innate and adaptive immune responses have a critical role in AD. It also provides information about microglial activation through the cluster of differentiation 40 (CD40) ligation and CD40L. CD40L ligation of microglial CD40 results in a large increase in tumor necrosis factor-α (TNF-α) production. Cultured cortical neuronal injury is caused when microglia are activated by CD40 ligation in the presence of interferon-γ (IFN-γ). This injury is significantly reduced by blocking the CD40 pathway or neutralising TNF-α. The management of AD would require integrating all available information about the innate and adaptive immune response affecting AD-related neuronal death.
Alzheimer’s disease (AD) is a neurodegenerative ailment that is irreversible. It is characterized by the progressive degradation of brain areas necessary for memory and learning. The condition gradually worsens over months or years, affecting one’s memory, reasoning, judgment, communication ability, and even the ability to perform simple chores [1]. AD is thought to have been discovered for the first time in 1906, when a German psychiatrist, Alois Alzheimer, performed an autopsy on a woman with language and memory difficulties [2]. Abnormal structures of senile plaques and neurofibrillary tangles (NFTs) across the cerebral cortex of the patient were the first signs of AD [2]. As people grow older, their cognitive abilities change more frequently, and being older is the leading cause of cognitive decline, mild cognitive impairment, and age-related dementias like AD. It is believed that specific AD pathologies, such as the development of intracellular NFTs made up of tau and naturally occurring proteins that clump abnormally to form plaques that accumulate between neurons and impair cell function, start to form decades before symptoms appear [3], and lead to cortical atrophy and ventricular enlargement, which results in around a 35% reduction in the total mass of the brain. Early in the disease, the para-hippocampal areas are essential for the formation of new memories, but as it worsens, patients suffer from neuronal and synaptic loss [4]. AD is the most common cause of dementia in the world, it follows by frontotemporal lobar degeneration (FTLD), and cerebrovascular illness. Lewy bodies are protein accumulations that form in nerve cells in the parts of the brain responsible for thought, movement, and memory [5]. At least 60–70% of cases of dementia are caused by AD. Research suggests that AD appears to affect women more frequently than men [6, 7]. However, the reason for this fact is not known. AD is the sixth-largest cause of mortality in the USA and one of the most prevalent neurological conditions worldwide [8, 9].
One of the most complex organs in the body is the brain. Aging is a significant cause of neurological conditions like AD [10, 11]. Most research into diseases caused by protein misfolding has been concentrated on the nervous system for many years. Glial cells are increasingly recognized as significant participants in neurodegeneration. Several diseases caused by protein misfolding [including amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), and AD] show glial cell activation along with the deposition of misfolded proteins, yet their exact function in the neurological disease phase is unidentified. A large body of evidence indicates that misfolded proteins alone will not cause neuronal damage [12]. Glial cells were discovered to play a role in neurodegenerative diseases as early as 1910 [13]. As a result, it’s only recently been found that a significant study focus has switched back to improving the understanding of glial cells. Recent developments in identifying the stages involved in dementia have revealed that glial cells play a significant part in the process, even though much of the early research concentrated on the clinical stages of the condition [11].
In the central nervous system (CNS), the innate immune system comprises microglia and astrocytes. Microglia act as scouts for potentially hazardous substances or events, whereas astrocytes are primarily responsible for neurotrophic support and tissue homeostasis [14, 15]. Microglia become activated when immune-suppressive signals are blocked and pathogen-associated molecular patterns or endogenous damage-associated molecular patterns (DAMPs) are present. Activated microglia can develop various phenotypes based on environmental inputs [15]. According to preclinical studies in animal models, microglia are linked to the progression of neurodegenerative disorders such as PD and AD [16]. Based on environmental factors, microglia can become traditionally activated, causing a higher or smaller inflammatory process [17]. After activation, microglia can help remove the primary cause of inflammation by controlling the inflammatory response and promoting healing by secreting neurotrophins. Astrocytes, on the other hand, might increase inflammation, causing neurotoxicity [18–20]. In neurological illnesses, microglial heterogeneity appears, and it may affect disease activities [21, 22]. Activating toll-like receptors (TLRs) results in phagocytosis, cytokine synthesis, and cell activation. These receptors are hypothesized to contribute to innate immunity and increase activity within the nervous system [23]. TLRs-2, 4, and 9, have been linked to the approval of abnormal protein collection and may play defensive microglia roles in the early phases of AD. Also, the form of inflammatory response that such receptors trigger depends on the stage of the illness, which is frequently benign in the initial phases but harmful later on [24, 25]. In AD, the activation of three complement pathways produces complement 5 (C5) convertase, which cleaves C5 into C5a and C5b. While C5a acts as an anaphylatoxin, C5b binds to C6, C7, C8, and multiple copies of C9 to form C5b-9, also known as the membrane attack complex [26], which co-localizes with amyloid plaques and tangles [27]. Microglial activation is also aided by neurons. The neuron-microglia connection includes the fractalkine [CX3C chemokine ligand 1 (CX3CL1)]-fractalkine receptor (CX3C chemokine receptor 1 (CX3CR1)] route, which restricts microglial activation. The microglia become active and hyperresponsive after the neuron is injured [28]. Microglial and astrocyte activation orchestration of non-cell-autonomous death is a common pathogenic feature of neurodegenerative diseases [29]. Hence, it is evident that microglia and astrocyte cells maintain the innate immune system in the CNS. Microglia cells are activated by a number of molecules and may contribute to neurological disorders.
Cytokines released by the activated glia, such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), increase the endothelial production of adhesion molecules and enable lymphocyte infiltration in an inflammatory environment. When cells reach the injury site, they contribute to the inflammatory response and increase neuronal death [30]. Since B lymphocytes are found in the CNS, cells of patients with neurological disorders, and experimental animals with these ailments, it appears that humoral immunity plays a supportive role in neurodegeneration [31]. In a study, the humoral immune response in ALS patients was examined. Serum immunoglobulin G (IgG) concentrations increased significantly, while IgM levels remained below normal limits. According to the scientists, ALS serum antibody levels “show distinct immunological profiles associated with predominant humoral or cellular immune responses” and could indicate an autoimmune phase in disease development [32]. T regulatory cells (Tregs) are a group of T cluster of differentiation 4+ (CD4+) cells that help to moderate or reduce inflammation [33]. Tregs have the power to restrict T effector cell function and potentially start the apoptotic process. CD4+ CD25+ Tregs support the survival of neurons while suppressing microglial activation. This impact may be caused by blocking T helper type 17 (Th17) pathways. Upregulation of neurotrophic factors, including glial cell-derived neurotrophic factor (GDNF) and brain-derived neurotrophic factor (BDNF), is one of the other hallmarks of Tregs [34]. Reduced inflammation protects neurons but also makes it more difficult to remove debris, misfolded proteins, and viral reservoirs [35]. Activated glial cells release IL-1β and TNF-α which promote lymphocyte infiltration and neuronal death. In ALS, IgG levels rise while IgM levels remain stable, indicating an autoimmune phase. Tregs restrict the function of T effector cells and start apoptosis, which is regulated by cell-derived factors and BDNF.
AD patients’ blood and cerebrospinal fluid (CSF) contain antibodies that can be used to identify human senile plaques in the CNS. These plaques, also known as amyloid-β (Aβ) deposition and NFTs, cause an innate immune response that may be pathogenic, as opposed to the inflammatory response observed in AD [36, 37]. The innate immune system recognizes non-self-initiated ligands that express highly conserved pattern recognition receptors (PRRs). Complement receptors, TLRs, and scavenger receptors (SRs) are glial cells’ most frequently expressed molecules. To aid in the removal of debris, the glial cells of the immune system contain a large number of receptors, of which it is important to be aware [38–41]. The complement system partly mediates the local inflammatory response brought on by Aβ deposition and NFTs in AD. Astrocytes, microglia, and neurons produce complement system components in the CNS. The majority of amyloid deposits in AD patients’ brains improve with complement component 1q (C1q) [42–45]. The C1q gene has a distinct mechanism for attracting reactive glial cells by producing pro-inflammatory cytokines and oxidative byproducts like nitric oxide. It is found near the Aβ plaque, causing inflammatory processes, neuronal malfunction, and, eventually, degeneration. Reduced neuropathology is caused by the deletion of this gene in transgenic AD mice [46, 47]. The complement system protects against neurological disorders by removing aggregated and toxic proteins linked to them. As a result, increased phagocytosis of Aβ by microglial cells can be induced by activating the complement system [48]. For the efficient removal of Aβ without causing brain injury and, hence, for successful treatment outcomes, balanced complement activation is probably necessary [49].
Chemokines are essential for controlling the innate immune response in the CNS. Chemokines draw in immune cells from the blood or brain. Chemokines may be produced by glial cells, particularly microglia and astrocytes in the CNS [50]. In the CNS, glial cells, mainly microglia, and astrocytes, are thought to be a key source of chemokines. Some chemokine receptors have already been found to be up-regulated in the AD brain, and studies have connected their activity to the activation of glial cells [50]. Antibodies found in the blood and CSF of AD patients may trigger a pathogenic immune response similar to the inflammatory response seen in the disease. TLRs and SRs are found on glial cells and many others to aid in the removal of debris. The CNS complement system is in charge of AD C1q—pulling reactive glial cells found near Aβ plaques, which cause neuronal malfunction and degeneration. Chemokines are also important molecules produced by glial cells that are found to activate them.
When bacterial infections associated with molecular patterns activate innate immune sensors in microglia, this sets off a series of signaling pathways that culminate in a planned response to the pathogens discovered. Microglia might not be able to distinguish between oligomeric or fibrillar Aβ and an invading pathogen. Fibrillar and aggregated Aβ can activate TLR2, TLR4, and TLR6 as well as their co-receptors to support signaling through the innate immune system. These receptors are present on microglia and participate in the activation of cells by various Aβ-types. Additionally, the development of DAMPs as well as the activation of microglia by Aβ have been linked to the SR-A and the ion channel Ca2+-activated K 3.1 (KCa3.1) [51]. The host uses the majority of the signaling routes that are triggered by neurodegenerative stimulation to combat infections. The immune system reacts to microorganisms by starting a proteolytic cascade that controls the synthesis of cytokines from the highly pro-inflammatory IL-1β family. The inflammasome apoptosis speck-like protein (ASC) binds to sensor molecules from the pyrin and hematopoietic expression, interferon (IFN)-inducible nature, and nuclear localization domain-containing (PYHIN) or nod-like receptor (NLR) families, which subsequently multimerize and activate caspase-1 to produce the inflammasome [52, 53]. Amyloid precursor protein (APP)/presenilin 1 (PS1) mice lacking NLR family pyrin domain containing 3 (NLRP3), caspase-1, or ASC are protected from developing AD by a protective mechanism that involves the production of IL-1β in the brain triggered by Aβ and increased Aβ clearance by microglia. The reduction of synaptic plasticity is entirely prevented by Aβ, which can affect the development of dendritic spines regulated by neurotrophic factor and tropomyosin-related kinase B (TRKB) [54, 55]. The NLRP3 inflammasome is a key player in both brain inflammation and typical age-related systemic inflammatory responses [56]. The brain’s inflammatory reaction can create a deadly feed-forward cycle. The immune system’s activation in response to Aβ and proinflammatory cytokines has been shown to hinder the clearance of Aβ and neuronal debris [57]. Phenotypic alterations in microglia work together to cause cognitive impairment in patients. The finding that IL-1 receptor-associated kinase 4 (IRAK4), a crucial kinase downstream of TLRs, can be disrupted to improve Aβ clearance and change microglial cells’ inflammatory phenotype from pro- to anti-inflammatory, is in line with the idea that pro-inflammatory molecules like the IL-1β family of cytokines or substances that mimic TLRs can impair the release feature of microglia [58]. When microglia cells come into contact with bacterial molecular patterns, they activate a number of signaling pathways. Certain receptors are present on microglia cells for different Aβ-types. A decrease in synaptic plasticity may prevent the development of dendritic spines. IRAK4 can be disrupted to improve Aβ clearance and change the inflammatory phenotypes of microglial cells. A family of cytokines that mimic TLR can impair the release features of microglia.
The health and regulation of the brain depend heavily on glial cells. The blood-brain barrier (BBB) can become more permeable in response to stress, allowing T cells and other specialized immune cells to be drawn in from the periphery. The effectiveness of any possible immune intervention therapy for neurodegeneration depends on understanding the communication with glial and peripheral immune cells [36]. Both innate and adaptive immune responses linked to the damage and repair processes in AD are tabulated in Table 1 [59].
Innate and adaptive immune response in AD
| Disease | Innate immunity | Adaptive immunity |
|---|---|---|
| AD | Chemokines, cytokines, and complements are all expressed in amyloid plaques. In vitro, Aβ induces innate immune expression. Aβ clearance is aided by microglia | As a therapeutic approach, activation of the adaptive immune system against Aβ peptide will aid clearance |
BBB is supported and maintained through the connections with glial cells, astrocytes, and endothelial cells. For mammalian brain homeostasis, the BBB is essential. The low-density lipoprotein (LDL) receptor-related protein 1 (LRP-1) and receptor for advanced glycation endproducts (RAGE) are the two major receptors used by BBB to control the transit of Aβ to and from the brain. RAGE transmits Aβ into the brain, and LRP carries Aβ out of the brain [60–62]. The balance between the pathways for Aβ synthesis and clearance depends heavily on LRP. Reactive astrocytes and the endothelium of the capillaries in the brain express LRP; apolipoprotein E (ApoE)-containing lipoproteins are secreted by glial cells and play a crucial role in maintaining the CNS’s lipid homeostasis. The presence of ApoE3, which is a risk factor for AD, was found to be more protective against cognitive decline than Apo4, suggesting it may be responsible for LRP’s upregulation [63–67].
Abnormal angiogenesis, dysregulated LRP1/RAGE-mediated movement, and arterial dysfunction may lead to Aβ formation, neurovascular inflammation, brain hypoperfusion, and cerebrovascular regression [68]. After crossing the BBB, the circulating Aβ is quickly taken up by neurons, causing cellular stress. To suppress blood flow, endothelin-1, a potent vasoconstrictor, is expanded as a result of the RAGE-Aβ interaction [60]. In 80% of cases of AD, there is amyloid buildup around cerebral blood vessels, which can result in cerebral amyloid angiopathy (CAA). Aβ deposits in the cerebrovasculature are connected to changes in smooth muscle cells, endothelial cell thinning, and mitochondrial loss. Aβ stroke with hemosiderin deposits, numerous infarcts, and cognitive impairment are caused by the deposits, which also cause intraparenchymal and subarachnoid hemorrhage [69].
Middle-aged people with AD have a weaker T cell response to Aβ than healthy older people and patients with AD. It has become essential for AD immunotherapy to understand T cell responses and adaptive immunity, even though there is no detectable T cell reaction in patients with AD [70]. Glial cells are crucial in promoting the T cell response and controlling how the immune response develops. Contact between glial cells and T cells occurs through a variety of receptors, and the first type of signal regulates how T cells mature [71]. Recent studies have demonstrated that the interaction between the T cell receptor CD40L and CD40, which is expressed on the surface of microglia, enhances pro-inflammatory microglial cell activation in reaction to Aβ [72]. Decreased levels of Aβ and Aβ plaque deposition are seen in transgenic mice overproducing Aβ but lacking CD40L. This discovery confirms the CD40-CD40L relationship for future therapeutic aims in AD and points to several potential mechanisms behind the reduction of AD pathology in response to CD40L. Signaling pathways that are started by CD95L, CD95L [Fas ligand (FasL)]/CD95 (Fas), perforin, and the TNF receptor 1 pathway help prevent brain inflammation as part of the defense mechanism. Glial cells may promote the death of the infiltrating T cells [73, 74]. T cell responses could be advantageous by assisting in the removal of Aβ, most likely through activating microglial cells. This glial and T cell connection could point to a new target for reducing brain inflammation [36]. This association may aid in the production of innovative immunotherapeutic treatments for AD.
Non-steroidal anti-inflammatory drug (NSAID) use is also connected to a lower incidence of AD in humans, and administering NSAIDs to AD mice shows less amyloid plaque load and improved microglial activation. Because activated microglia secrete pro-inflammatory innate cytokines like TNF-α and IL-1β, which have been shown to increase the risk of neuronal damage, they may play an important role in the etiology of AD [75–78]. When microglia are challenged with tagged Aβ-peptides, phagocytosis is promoted but soluble or fibrillar Aβ-peptide breakdown via SRs is low. Maxfield’s laboratory discovered that the primary SRs (type I and II) are responsible for Aβ absorption by microglial cells [79–83].
The CD40-CD40L connection may contribute to AD pathogenesis by boosting brain inflammation after excessive amounts of astrocyte-derived CD40L and microglia-associated CD40 were recently discovered in and around Aβ plaques in the brains of AD patients. In animal models of AD, genetically eliminating CD40 or administering an antibody significantly lowers amyloid plaques [84–86]. The phagocytic response to Aβ alone was not associated with the production of the pro-inflammatory innate cytokines TNF-α, IL-6, or IL-1β, a finding that is consistent with when microglia are challenged with apoptotic cells and produce an anti-inflammatory, pro-phagocytic innate response [87, 88]. CD40L reduced the speed of microglia phagocytosis and chased them away from cells after an hour of culture in the absence or presence of CD40L stimulation, preventing them from breaking down Aβ peptides. This reaction was enhanced by pro-inflammatory cytokines known to enhance effector T cell function. IFN-γ and TNF-α both pro-inflammatory Th1 cytokines, inhibited Aβ phagocytosis, whereas pro-inflammatory IL-4 and IL-10 increased its clearance. By using this experimental method, it can be concluded that CD40L also slowed down the microglial clearance of the peptide [80, 82].
CD40 ligation raises increases Th1-promoting cytokines such as IL-6, TNF-α, IL-2, and IFN-γ in co-cultured cells while decreasing innate (phagocytic) activation of microglia stimulated with Aβ. An antagonistic CD40 antibody could be added to stop these effects, proving that the CD40-CD40L interaction itself is what causes these effects. The activation profile of microglia is biased away from innate, phagocytic activation and towards adaptive, APC activation when they come into contact with Aβ in the setting of co-simulation (e.g., CD40L) [80, 89].
To better understand the complicated glial responses in disease, animal models of developing neurodegeneration will be used to study the relationships between numerous brain area alterations. Glial cell interactions in the context of neurodegeneration have been extremely useful in learning prion disorders. Prions cause a predictable, advancing neurological disease in mice, with symptoms similar to those seen in human prion diseases [11]. One critical study used laser microdissection to separate astrocytes from distinct areas of the brain at various phases of AD, as characterised by the Braak neurofibrillary stages [90]. Many changes in gene expression were associated with astrocyte cytoskeletal proteins, which are compatible with the progressive reactive astrogliosis seen in AD. Apoptosis and ubiquitin-mediated proteolysis-related genes were revealed to be differently activated earlier in AD [91–93]. Glial cell activity changes as the disease progress in brain areas susceptible or resistant to neurodegeneration. Understanding the glia’s therapeutic potential should improve on gaining a better understanding of the glial response in various brain areas as the illness progresses [12]. The microglia in areas of the brain where the activity of a range of microglial markers is affected by neurological diseases such as AD and prion diseases differ from those in regions with no signs of dementia [94–96]. The stages of glial activation may influence the initiation of neurodegenerative diseases as well as the severity of dementia. An IFN-related phenotype of microglia was found in the afflicted brain regions of a recent meta-analysis of AD [97].
In the developmental stage of illness, microglia express a variety of innate immune activation genes. The impact of different microglial activation states on the neurodegenerative process has been discussed. Even though astrocytes exhibit a wide range of functional abnormalities as the disease progresses, the impact of astrogliosis and astrocytic dysfunction on neuronal health has been restricted for many years. It is well recognized that astrocytes express a variety of immune-related genes, suggesting that they may be a part of an organized immune-activated response to a neurodegenerative ailment. The process through which this glial communication occurs has been discovered in prion diseases. Several cytokines lead to astrogliosis, secreted by activated microglia in the early stages of illness.
Consequently, chemokine expression is enhanced by astrocytes, and this, along with local microglial proliferation, causes microglia to multiply and become more active. Progressive astrogliosis results from reactive microglia secreting more cytokines into the surrounding environment. Glial activation doesn’t seem to decrease over time during disease; rather, it seems to gradually increase with time, showing that this feedback loop is a glial-specific perpetual cycle. According to the latest research, AD neurodegeneration is caused by the astrogliosis condition induced by cytokines like IL-1β. The therapeutic potential of focusing on the connection between glial cells in AD is highlighted by investigations that have found proof of microglial-mediated astrogliosis resolution (Figure 1) [11, 95]. A previous study found that when injected into mice, a prion-infected neuron or astrocyte culture had chemotactic traits that weren’t seen in control cultures [98]. This finding suggests that neurons, astrocytes, or both mediated microglial recruitment to the injection site. One other study found that the proinflammatory cytokine IL-1β is crucial for activating astrogliosis and that, during illness, IL-1β production is powerful in microglia populations (including NLRP3 inflammasome pathway activation) [99, 100]. Microglia express various genes for the cultivation of the innate immune system. Astrocytes express a variety of immune-related genes, so they are part of the immune-activated response to a neurodegenerative ailment. The astrogliosis condition induced by IL-1β causes AD neurodegeneration. Either neurons, astrocytes, or both mediate microglial cell recruitment to the infection site. IL-1β is essential for astrogliosis activation, and its production greatly increases during illness.
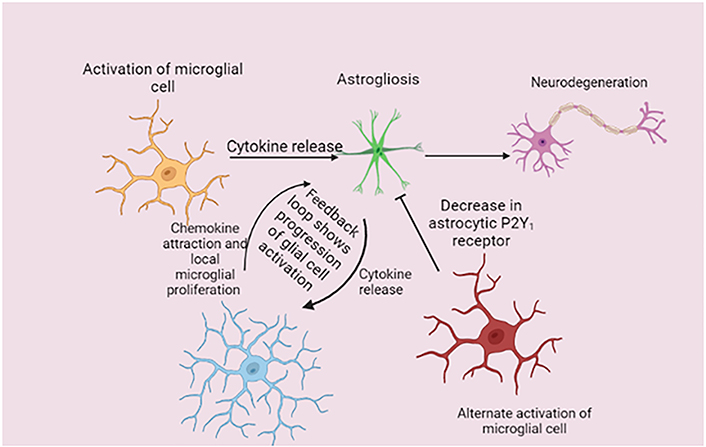
Mechanism of disease-related glial cells. Microglial activation causes the release of several cytokines that cause astrogliosis in the early stages of the disease. In response, astrocytes upregulate chemokine expression, which, together with local microglial growth, increases microglia numbers and reactive activity. This feedback loop is a glial-specific perpetual cycle because activation states of the glia do not appear to resolve over time. P2Y1: purinergic receptor
Note. Adapted from “Unravelling the glial response in the pathogenesis of Alzheimer’s disease,” by Alibhai JD, Diack AB, Manson JC. FASEB J. 2018;32:5766–77 (https://faseb.onlinelibrary.wiley.com/doi/full/10.1096/fj.201801360R). CC BY.
Microglial activation is important for astrogliosis because it triggers the cytokine that releases IL-1β, which in turn triggers a chemokine response that attracts more microglia, during illness, working as a feedback connection loop with glial cells. TNF-α, C1q, and IL-1β can be secreted by activated microglia and cause astrogliosis implying that the glial link processes underpin AD and other neurological disorders [101, 102]. Additionally, functional impairment in activated astrocytes can result in neuronal cell death. Such a type of astrogliosis is known as annexin 1 (A1) because the activation is functionally similar to a “classic” microglial activation state. A1 astrocytes have been found in a variety of neurodegenerative diseases including AD. This research shows that resolving the activation of glial cells or disrupting glial cell contact during illness could be a valuable treatment strategy. Recent research suggests that microglia can create neuroprotection in the astrocytic phenotype by down-regulating the P2Y1 receptor, opening the possibility of glial-targeted therapeutic intervention [103].
Heavy metal intake alters astroglial homeostasis and neuroprotective cascades such as the glutamate/gamma-aminobutyric acid (GABA)-glutamine shuttle, antioxidative machinery, and energy metabolism. Astrocytes are the main homeostatic cells in the CNS that defend neurons from all kinds of insults, particularly heavy metal accumulation. This makes them the primary target of heavy metal neurotoxicity [104]. There is no significant difference in cuprum (Cu), iron (Fe), or zinc (Zn) concentrations in large structures such as the corpus callosum, cortex, and striatum in Mouse Toronto KnockOut mice (MTKO) compared to age-matched controls [105]. MTKO mice retain approximately one-third as many Cu-rich aggregates, resulting in a decrease in periventricular Cu concentration. As a binding protein, metallothionein, a cysteine-rich protein important for metal homeostasis, was proposed [105]. Astrocytes have strong pro-inflammatory abilities. Astrocytes are thus emerging as key regulators of CNS inflammatory responses. Astrocytes play an important role in attracting and restricting CNS inflammation, which has significant implications for a variety of CNS disorders [106]. Astrocytes are the most abundant cell type in the brain and contribute to brain homeostasis in a variety of ways, such as buffering extracellular K+ and controlling neurotransmitter release. Astrocytes can also create the BBB and regulate the brain’s immune response [107].
AD is a neurodegenerative disease caused by gene mutations that influence amyloid formation, such as PS1, PS2, and APP. A small proportion of AD is autosomal dominantly inherited within families and appears as early dementia onset, often between the ages of 30 and 60 [108]. Over the age of 30, AD risk loci have been identified thus far through a combination of candidate genes, genetic linkage, whole genome/exome sequencing studies, and genome-wide association studies. Over ninety-nine, percent of cases of AD occur in people over the age of 65 and are known as late-onset AD. Genetics and age are the two most significant risk factors [109]. Over half of the different genes proven by functional genomics are involved in innate immune cell function and microglial function, such as the top 2 risk genes, triggering receptor expressed on myeloid cells 2 (TREM2) and ApoE. The epigenomic analysis revealed that AD genome-wide association study loci are selectively enriched in promoter sequences associated with innate immune mechanisms [110].
Immunotherapy has attracted the attention of pharmaceutical companies, clinicians, and researchers seeking to develop an effective treatment for AD. Several immunotherapies have been developed and tested in clinical trials to combat the pathology of amyloid or tau [111].
The presence of extracellular senile plaques, which are made of Aβ peptide, and intracellular NFTs, which are made of tau protein, is a hallmark of AD. Targeting the aggregation process of Aβ and tau has been an active strategy for designing and developing therapeutic agents for pharmacological intervention in AD [112]. Researchers found that synaptic pathology and microgliosis may be the first signs of neuronal dysfunction caused by tauopathy in P301S mutant tau transgenic mice. The activation of glycogen synthase kinase-3β (GSK-3β) or cyclin-dependent kinase-5 (CDK-5) kinases resulted in the stimulation of microglial cells and the release of IL-1β [113]. The CNS is affected by various neurodegenerative and neuroinflammatory disorders that affect GSK-3β function [8].
Chronic inflammation, which is a hallmark of neurodegenerative disorders, is stimulated in both directions by innate and adaptive immunity. Inflammation always turns pathogenic and causes neuronal death, although it typically acts as a preventive strategy in the initial phases of the disease. Glial cells are the first line of defense against pathological anomalies that develop in neurodegenerative illnesses. Glial cells are crucial for keeping a healthy brain, additionally, they also safeguard and support the brain’s ability to recover from damage. Glial cell activation can be mediated and affected by a variety of signals and is one of the most important functions of the CNS. Glial cells’ aberrant behavior throughout aging may be caused by alterations in their proliferating process and may provide light on the development of fatal disorders like AD. Both microglia and astrocytes appear to provide a dual function in the disease process by repairing CNS damage. Knowledge of these long-lasting neurological illness mechanisms has advanced with the discovery of commonalities among the many protein-misfolding diseases. However, because each glial family exhibits such diversity, they continue to remain a riddle that holds the secret to their alteration. In summary, inflammation and its pathology could be targeted to manage neurological conditions, and more specifically, AD. The glial response could be a crucial factor in accomplishing this future goal.
AD: Alzheimer’s disease
ALS: amyotrophic lateral sclerosis
ApoE: apolipoprotein E
Aβ: amyloid-β
BBB: blood-brain barrier
C1q: component 1q
C5: complement 5
CD40: cluster of differentiation 40
CNS: central nervous system
Cu: cuprum
IFN: interferon
IL-1β: interleukin-1β
LRP-1: low-density lipoprotein receptor-related protein 1
NFTs: neurofibrillary tangles
NLRP3: nod-like receptor family pyrin domain containing 3
PS1: presenilin 1
RAGE: receptor for advanced glycation endproducts
SRs: scavenger receptors
Th17: T helper type 17
TLRs: toll-like receptors
TNF-α: tumor necrosis factor-α
Tregs: T regulatory cells
Thanks are due to CEHTI and the bioinformatics lab of the Department of Biotechnology and Bioinformatics, Jaypee University of Information technology, Solan, Himachal Pradesh, India for the technical help.
TRS: Conceptualization, Writing—review & editing. AS: Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

