 Review
Review

Affiliation:
1Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas–Universidad Autónoma de Madrid (CSIC-UAM), 28049 Madrid, Spain
2Networking Research Centre on Neurodegenerative Diseases, Centro de Investigación Biomédica en Red Enfermedades Neurodegenerativas (CIBERNED), 28031 Madrid, Spain
Email: javila@cbm.csic.es
ORCID: https://orcid.org/0000-0002-6288-0571
Affiliation:
3Departamento de Anatomía Histología y Neurociencia, Facultad de Medicina, Universidad Autónoma de Madrid (UAM), 28029 Madrid, Spain
ORCID: https://orcid.org/0000-0002-8477-9554
Affiliation:
4Departamento de Psicología Experimental, Procesos Cognitivos y Logopedia, Universidad Complutense de Madrid, 28040 Madrid, Spain
ORCID: https://orcid.org/0000-0001-8537-2046
Affiliation:
5Facultad de Ciencias Experimentales, Universidad Francisco de Vitoria, 28223 Madrid, Spain
ORCID: https://orcid.org/0000-0003-2801-5020
Affiliation:
1Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas–Universidad Autónoma de Madrid (CSIC-UAM), 28049 Madrid, Spain
ORCID: https://orcid.org/0000-0001-8753-8249
Explor Neuroprot Ther. 2023;3:151–163 DOI: https://doi.org/10.37349/ent.2023.00044
Received: December 02, 2022 Accepted: March 17, 2023 Published: June 30, 2023
Academic Editor: Rafael Franco, Universidad de Barcelona, Spain
The article belongs to the special issue The Urgent Need for New Hypotheses to Develop Effective Therapeutic Tools Against Alzheimer's Disease
Currently, the predominant targets for the treatment of Alzheimer’s disease (AD) are the main components of the two pathological structures: senile plaques (composed of amyloid beta peptide aggregates) or neurofibrillary tangles (constructed of tau protein polymers). However, the existence of adequate disease modifiers based on such targets is discussed. In this special issue, it has been suggested to search for new possible targets for AD therapy. This contribution tries to analyze non-neuronal tissues (periphery) to identify potential factors (target) involved in the development of AD.
A strategy to prevent a disease is to determine the primary cause of that disease. In the case of Alzheimer’s disease (AD), there are two main types: familial AD (FAD) and sporadic AD (SAD). It is mainly assumed that the primary cause for FAD is the presence of mutations in some genes like presenilin 1 (PSEN1), presenilin 2 (PSEN2) , precursor of beta amyloid peptide (APP) [1] and, probably, sortilin related receptor 1 (SORL1) [2, 3] that could favor the presence of beta amyloid (Aβ) peptide aggregates, being that presence the first step for AD development. In this way, FAD could be similar to other neurodegenerative disorders, like Huntington’s disease (HD), that results from a genetic mutation [4]. However, even assuming that it was the primary cause, there is not suitable therapies for FAD or HD.
For FAD, the great majority of the cases are the consequences of PSEN1 gene mutations and it has been described that those PSEN1 mutations may produce alterations appearing in the brain previously to Aβ over-production, in a mouse FAD model [5]. Thus, the presence of Aβ could not be the first step in FAD due to mutated PSEN1. Nevertheless, the prevalent view is that in FAD, the increase in Aβ is a very early factor in the development of the disease and that it could be, also, for SAD (amyloid hypothesis) [6].
However, in SAD, there are other additional hypotheses suggesting primary causes for the disease [7]. Examples of those hypotheses are: cholinergic [8], calcium [9], oxidative stress [10], neuron inflammation [11], microbial (viral) infections [12], glucose hypometabolism [13] or the presence of an excess of tau aggregates [14], among others.
On the other hand, the main risk factor for SAD is aging [15] and healthy longevity could prevent the appearance of SAD.
Aging can be observed at different levels: the whole organism, organs (or tissues), cellular, and molecular levels. At cellular level, after the cells become old could be an additional step, senescence, with the senescence-associated secretion phenotype (SASP) [16] in which senescent cells could secrete toxic substances to the surrounding cells, with or without the same origin. Thus, the presence of a tissue with senescent cells could facilitate the toxicity and aging of neighboring tissues. Recently, it has been described an example, showing that skin cell aging drives age related bone loss via secreted cystatin A [17], or that knee osteoarthritis (OA), in old people, may accelerate Aβ deposition and neurodegeneration in the central nervous system (CNS) [18], although the presence of the brain blood barrier (BBB) may difficult the crossing of secreted compounds from the periphery to CNS. In any way, it has been proposed that AD may be a multiorgan disorder [19].
In addition to being a neurodegenerative disorder, AD pathology may include immune system dysfunction [20], inflammation, dysbiosis, insulin resistance, heart problems, kidney, and hepatic dysfunction, among others [19].
However, the main pathology is related to CNS disorders and the disorders in other tissues and organs may be used, or not, as putative biomarkers of the disease, although further work should be done to know if those peripheral disorders are cause or consequence of the CNS.
The most well-known neuronal hallmarks in AD are senile plaques [21] and neurofibrillary tangles [22], coming from two proteins, APP and tau protein, mainly expressed in neurons [23–25]. In addition, glia cells, present in CNS, may play an important role in the development of AD. In this way, microglia can have harmful actions in AD [26]. Activated microglia or disease associated microglia (DAM) with the new nomenclature [27] may be one of the key factors involved in the neuroinflammation found in AD [28]. Also, the activation of microglia could be the consequence of a change in the cross-talking neuron-resting microglia [29] and could be the consequence of microglia change of status specific features found in AD mouse models. Parabiosis experiments in mouse models indicate that microglia account for the increased myeloid cell number observed in brains with plaque pathology [30]. Additionally, astrocytes may also play a role in neuroinflammation [31].
AD is a CNS disorder but it could correlate with some changes in the peripheral nervous system (PNS) that could be used as biomarkers of the disease. An example is the lower degree of pain observed in AD patients [32].
About peripheral biomarkers, in human fluids, in addition to looking at the levels of Aβ or tau protein in cerebral spinal fluid (CSF) [33, 34], other peripheral fluids, like plasma, saliva, or urine, have been used as possible markers of compounds, like proinflammatory cytokines, or others, that could be altered in AD patients [35]. The kinetics for the appearance of these markers could be important since earlier markers could facilitate the earlier prevention of the disease [35].
Briefly, it will be described some peripheral features that are present in AD patients or in models for the disease, although, probably, in many cases the features could be more the consequence than the cause, that facilitate the development of the disorders. On the other hand, the use of palliative pharmacological treatments, for the disease, could result in damage to organs like kidney or liver.
Here, it is commented on the possible relation of AD development with seven different organs: heart, blood, gut, muscle, bone, kidney, and liver (Figure 1). Although other organs like lungs have been suggested to get a link with dementia [36], little is known about molecular factors involved in that interaction and they are not mentioned here.
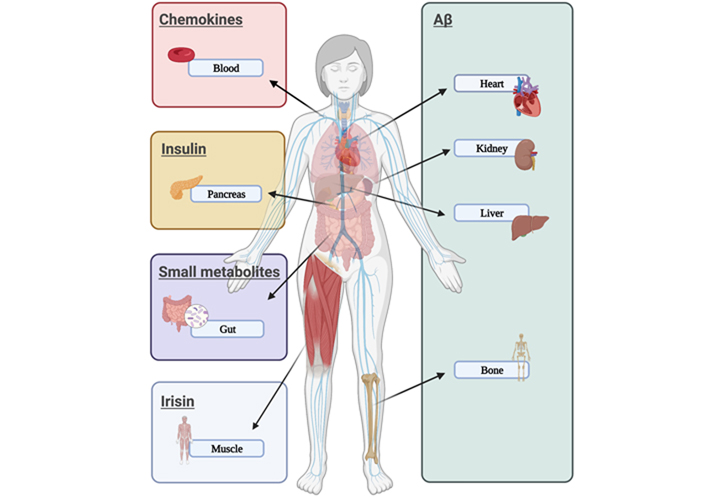
Peripheral disorders related to AD. The figure showed different organs that could express or secrete compounds associated with features found in AD (like Aβ aggregates, proinflammatory cytokines, insulin dysfunction, or small fat-derived metabolites) or compounds that could act as protective factors (like irisin)
Links between cardiovascular abnormalities and neurological disorders in AD patients have been reported [37] suggesting that “mind the heart in Alzheimer disease” [38, 39]. Also, a long time ago, the term “cardiogenic dementia” was indicated [40], being, actually, a possible therapeutical treatment, systolic blood pressure intervention trial memory and cognition in decreased hypertension (SPRINTMIND), based on the fact that a healthy brain starts with a healthy heart and that control of intensive blood pressure could reduce dementia [41].
Aging, but mainly a development of AD, results, for example, in a decline of adult hippocampal neurogenesis and cognitive function [42]. To avoid that aging cognitive decline, heterochronic parabiosis, mixing the blood of young and old mice, could help the recovery of cognitive function in old mice [43]. The levels of some compounds, like the chemokine eotaxin, are increased in old mice or aging humans, and those compounds could impair memory or cognitive functions [43]. Possible therapies based on blood exchange have been proposed as a possible treatment of neurodegenerative disorders [43]. Also, related to AD, it has been described that clearance of brain Aβ burden may take place by blood exchange, suggesting that defects in peripheral circulation may play a role in AD pathogenesis [44]. In this way, it has been indicated that blood-derived amyloid peptides could induce AD-like pathology [45] and behavioral deficits [46].
About the role of brain-gut microbiota axis dysregulation, many works have been published. Indeed, a whole long review article should deserve this subject, but, in this review, only a brief comment should be included to indicate the gut changes related to brain pathogenesis [47]. Changes in gut microbiota (dysbiosis) may induce increased permeability of the intestinal barrier leading to systemic inflammation. Curiously, Aβ has been described as an antimicrobial peptide [48] that could participate in that immune response [49]. It should be noted that intestinal inflammation triggers C/EBPβ/δ-secretase-dependent spread from gut to brain of Aβ in AD, suggesting that the gut is likely a critical source of Aβ in the brain [50]. In addition, there is a gut-derived amyloid that may contribute to Alzheimer’s pathogenesis [51]. Also, intra-gastrointestinal Aβ oligomers perturb enteric function and may facilitate the development of AD pathology found in another mouse model [52].
On the other hand, a high proportion of patients with inflammatory bowel disease show mental disorders, perhaps due to changes in brain choroid plexus modulated in response to intestinal inflammation [53]. Additionally, immunotherapy targeting tau can modulate gut microbiota in a mouse model for AD [54].
The presence of specific bacteria strains in gut microbiota could facilitate or decrease serotonin secretion and could promote neurotransmission dysfunction [55], which could result in depression, a risk factor for AD [56, 57].
The level of other metabolites like butyrate or propionic acid [58] should be also modulated by gut microbiota. By dietary source of tryptophan, some bacteria types could be recruited to the gut, increasing serotonin levels [59].
On the other hand, microbiota-dependent progression of neurodegeneration in a mouse model of tauopathies has been reported [60].
Finally, linking with muscle functions (see below), gut microbes shape athletic motivation [61], that could favor physical exercise, a protective factor for dementia.
As previously indicated, depression is a risk factor of dementia and decreased levels of brain-derived neurotrophic factor (BNDF) are related to depression, but there is a peptide, irisin, induced by physical (muscular) exercise that increases the level of BNDF and cognition in mouse models for AD. Recently, low levels of irisin have been found in CSF from old humans with depression [62].
Irisin is a cleaved peptide from transmembrane fibronectin type II domain-containing protein 5 (FNDC5) that has been linked to adult neurogenesis, being a possible mediator of muscle-brain crosstalk, that could explain the therapy based on physical exercise for patients with dementia [63]. In any case, irisin may be suggested as a possible biomarker for dementia [62].
It has been suggested that Aβ peptides produced in the liver can be involved in the formation of senile plaques [64]. Based on that, it has been proposed that peripheral (liver) Aβ clearance could be a good therapy for AD [65–67].
In addition, an association of altered liver enzymes with the diagnosis of AD has been proposed, implying changes in cognition, neuroimaging measures, or in the level of cerebrospinal fluid markers [68]. In addition, brain changes related to hepatic apolipoprotein E4 (ApoE4) expression have been reported in mice with humanized livers [69]. These changes are probably due to the role of peripheral ApoE4 in cognition impairment, by compromising cerebrovascular function [70]. Indeed, liver-restricted expression of human amyloid results in an AD-like neurodegenerative phenotype [71].
OA and AD are two main genetic diseases that could have common features like the presence of chronic inflammation [72]. Also, the prevalence of dementia in OA patients is higher than in non-OA subjects and treatment with etoricoxib and diclofenac may reduce the risk of dementia in patients with OA [73].
A possible link between OA and AD is that OA accelerates amyloid beta deposition and neurodegeneration, as it has been shown in a mouse model of AD [18].
Physiological clearance of the Aβ peptide by the kidney has been reported [74]. On the other hand, tau, the main component of neurofibrillary tangles in AD, is usually considered as a specific neuronal protein [12] but its possible presence in peripheral tissues was proposed a long time ago [75]. However, recent studies have indicated that tau could be present in kidney, playing a role in the morphology of podocyte architecture [76]. Also, in some tauopathies, like frontotemporal dementia, some patients report urinary incontinence [77] and, more recently, the association of impaired kidney functions with dementia and brain pathologies has been indicated [78, 79]. Curiously, AD patients with chronic kidney disease (CKD) treated with cholinesterase inhibitors showed a lower risk of CKD [80]. Due to this link between brain and kidney pathologies and formic acid, a toxic compound associated with several cognitive disorders, may be present in the urine of AD patients. It was found an increase in formic acid level measured in the urine of AD patients and it could be used as a biomarker for the dementia and used as a diagnostic tool in the future [81].
Insulin is a peptide hormone expressed in the beta cells present in pancreas. Insulin has a role in the regulation of the metabolism of carbohydrates, lipid, and proteins through the absorption of glucose from blood to some organs. The action of insulin is through its receptors present in different cell types [82].
Insulin receptors are expressed in cells from both PNS and CNS [83], playing some functions related, for example, to food intake or memory consolidation [84].
Defects in insulin function may result in disorders such as diabetes mellitus. There are two types of this disorder, type I and II. In type I, no insulin is made and in type II, the insulin pathway initiated by the binding of the hormone to its cellular receptors is not working and, as a consequence of that, the expressed insulin does not have the desired functional effect in the body.
Diabetes mellitus type II accounts for more than 90% of all diabetes mellitus cases and a correlation between type II diabetes and dementia has been reported, with diabetes being an important risk factor for the development of AD [84, 85]. Thus, some authors have described AD as diabetes type III [86].
Actually, the main targets for AD therapies are the components of senile plaques, Aβ peptide aggregates; or the component of neurofibrillary tangles, tau protein, since the amyloid [6] and tau protein [14] hypothesis are now the most popular hypothesis for AD development. Mainly, antibodies against Aβ aggregates have been tested, as a possible therapy for amyloid pathology. Although some of those antibodies like aducanumab or lecanemab have been raised some good expectations, no suitable results were obtained and, in addition, some side effects, like cerebral hemorrhages, were found in some treated patients [87]. More recently, it has been also indicated an accelerated brain volume loss is caused by these anti Aβ drugs [88]. Indeed, almost all of clinical trials for AD have failed to show positive results as indicated in the commentary “The long road to a cure for AD is paved with failures” recently published in Nature Medicine [89], discussing the last data in clinical trials for multiple targets like Aβ, tau, inflammation components, and other ones, see also [90]. Also, Aβ and tau have been mainly used as early biomarkers for AD, looking at fluids like CSF, or blood, see for example [91]. Also, from glia (microglia) cells, triggering receptor expressed on myeloid cells 2 (TREM2) level could be associated with the transition from mild cognitive impairment to dementia, during the development of the AD continuum process [92].
On the other hand, it has been discussed why successful results have been obtained in animal models but not in humans [93]. Also, some compounds that could result in a neurodegeneration or cognitive improvement have been proposed as possible palliative therapies [94].
The use of other compounds related to some features found in AD has been reported. For example, the small compound “simufilam”, a stabilizer of filamin A [95], has been proposed as a possible therapy for AD, although that use is under discussion [96].
Now, new targets or new early biomarkers for AD are being looked for. About new possible biomarkers for AD, from the periphery, some examples related to blood are included. A possible link between platelet biomarkers and cognitive functions has been proposed [97] and between plasma β2 microglobulin and N-methyl-D-aspartate (NMDA) receptors, since β2 microglobulin could act as an antagonist for NMDA receptors, resulting in cognitive impairment [98]. Also, related to blood, associations of plasma cluster of differentiation 22 (CD22) level with cognitive decline in AD have been reported [99].
Biomarkers in other fluids like saliva and urine, for AD diagnosis, have been also proposed. Since chronic stress is a risk factor for the disease, the level of cortisol (related to stress) could be used as a prodromic marker for the AD continuum process [100]. Additionally, as indicated in kidney section, the presence of formic acid in urine could be used as a possible marker for dementia [81].
This brief review is mainly focused on the contribution of peripheral tissues in AD development, although, in many cases, the observed peripheral pathology could be more the consequence than a possible cause for AD.
It is well-known that changes in life style, or improving the quality of nutrition, could result in a decrease in the incidence of AD development [101, 102]. Treatment of risk factors like hypertension or diabetes or playing physical exercise could retard the appearance of dementia, decreasing the fast progression of AD at world level [103]. Additionally, since aging is the main risk factor for neurodegenerative disorders, a possible therapy to reprogram (rejuvenate) old into young neurons has been proposed [104]. Also, the presence of senescent cells, like senescent astrocytes, with SASP, may contribute to an increase in Aβ aggregation or tau phosphorylation [105] and, in that way, the use of senolytic drugs has been proposed in clinical trials for AD [106]. Other possible therapeutical ways could be based on microbiota changes, since it has been suggested that the presence of some bacteria in the gut of aged people may contribute to AD progression [107, 108], affecting Aβ pathology. However, although Aβ accumulation is a factor for AD pathogenesis, in the absence of tau, its presence is insufficient for cognitive decline. Now, it has been described on microbiota-dependent progression of neurodegeneration in a mouse model of tauopathy, being that effect modulated by ApoE4 isoform [60]. In that way, reshaping or eliminating gut microbiota may decrease the progression of neurodegeneration.
About new biomarkers, from the periphery, testing the level of formic acid in patients’ urine [81] or measuring the lower degree of pain, related to PNS, observed in subjects with mild cognitive decline, previous to dementia onset, could be suggested [32].
Thus, to get a whole picture of the clinical features of a patient, at the early phases of AD continuum, the suggested Alzheimer’s precision medicine initiative could be a good approach to retard the progression of AD [109]. Nevertheless, more work should be done to look for modifiers of the disease that may prevent, decrease or stop the development of the disease.
AD: Alzheimer’s disease
ApoE4: apolipoprotein E4
Aβ: beta amyloid
CNS: cerebral nervous system
CSF: cerebral spinal fluid
FAD: familiar Alzheimer’s disease
OA: osteoarthritis
PNS: peripheral nervous system
PSEN 1: presenilin 1
SAD: sporadic Alzheimer’s disease
We thank Ms. Nuria de la Torre Alonso for technical and editorial assistance. Figure 1 was created with Biorender (BioRender.com).
JA: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. MP, MAV, ISM and FH: Conceptualization, Investigation. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work has been supported by grants from the
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Rafael Franco, Joan Serrano-Marín
Karolina Armonaite ... Luigi Laura
Glòria Salort ... Jesús A. García-Sevilla
Milind Watve, Ashwini Keskar Sardeshmukh
Iyawnna Hazzard ... Forshing Lui
