
Abstract
Aim:
The sequential phosphorylation of mitogen-activated protein (MAP) kinases MEK-ERK is the most relevant cellular signaling pathway. This study quantified the parallel in vivo regulation of brain phosphorylation-MEK1/2 (p-MEK1/2) to p-ERK1/2 by mechanistically different cannabinoid 2 (CB2) receptor ligands, i.e., direct (and endogenous) agonists and inverse agonists.
Methods:
Groups of Swiss albino CD1 IGS male adult mice were treated (i.p.) with the CB2 agonist JWH133 (1 mg/kg and 3 mg/kg, 1 h, n = 8) or the CB2 inverse agonist/antagonist AM630 (0.3 mg/kg and 1 mg/kg, 1.5 h, n = 8–9), and 0.9% NaCl (2 mL/kg, 1 h, n = 4–10) as vehicle control. Transgenic male mice overexpressing cortical CB2 receptors [messenger RNA (mRNA) and protein] on a Swiss ICR congenic background (CB2xP) and the corresponding littermates age-matched wild-type (WT) controls were used. Protein forms (total MEK and ERK p-kinases) were resolved by electrophoresis [sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) minigels] followed by immunoblotting standard procedures.
Results:
The selective CB2 agonist JWH133 (1 mg/kg and 3 mg/kg, i.p., 1 h) modestly decreased MEK (17%, n = 8) and upregulated ERK (25%, n = 8) activities. The endogenous CB2 agonists (acting on promoted overexpressed receptors) decreased MEK (44%, n = 9) and upregulated ERK (67%, n = 10) activities. The inverse agonist/antagonist AM630 (0.3 mg/kg and 1 mg/kg, i.p., 1.5 h) increases MEK activity (27%, n = 8) without significantly altering that of ERK (5%, n = 9).
Conclusions:
Acute treatments of mice with mechanistically different CB2 receptor ligands (i.e., direct agonists, endogenous agonists, and inverse agonists) resulted in disruption of MEK (p-MEK/total-MEK ratio) to ERK (p-ERK/total-ERK ratio) signals in the brain cortex. This striking disruption of MEK to ERK parallel regulation in the cannabinoid CB2 receptor system in the brain could be relevant to the postulated role of CB2 receptors in various central nervous system (CNS) diseases.
Keywords
Cannabinoid 2 receptor, JWH133, cannabinoid 2 receptor overexpression, constitutive cannabinoid 2 receptor activity, cannabinoid 2 inverse agonist/antagonist AM630, MEK-ERK phosphorylation/activation, mouse brain cortexIntroduction
Mitogen-activated protein (MAP) kinases are ubiquitous cell regulatory enzymes, which are grouped into three families: extracellular signal-regulated kinases 1/2 (ERK1/2), c-Jun NH2-terminal protein kinases 1/2 (JNK1/2) and p38 kinases [1]. In particular, ERK1/2 cascades [2–4] are initiated by the phosphorylation of their upstream kinases MAP kinases kinase/ERK1/2 [MEK1/2 (i.e., ERK kinases [3, 4])] leading to the sequential basal activation of phosphorylation-ERK1/2 (p-ERK1/2), the only known substrates of p-MEK1/2 enzymes [1, 3–5].
The endocannabinoid system consists of two major types of G-protein (Gi/o)-coupled receptors, the cannabinoid 2 (CB2) and CB1 receptors that are stimulated by the endogenous ligands anandamide and/or 2-arachidonoylglycerol [6]. In the context of MEK-ERK signaling, the CB1 receptor agonist WIN55212-2 was shown to induce, as expected, the parallel upregulation of p-MEK1/2 and p-ERK1/2 in rat and mouse cerebral cortex [7, 8]. Moreover, the non-selective CB1/2 receptor agonist CP55940 was also reported to stimulate p-ERK1/2 in transfected Chinese hamster ovary (CHO) cells expressing the human CB2 receptor (CHO-CB2), through pertussis toxin (Gi/o protein-dependent) mechanisms [9].
Notably, the CB2 receptor (whose expression was initially believed to be limited to peripheral immune cells with low levels in the brain, mainly in microglia) was also recently shown to display constitutive activity (i.e., tonic control or ligand-independent receptor activity [10]), both in CHO-CB2 membranes [11, 12] and in mouse brain tissue (glia and neurons under physiological conditions, see [13]) which allowed the assessment of various inverse agonists (AM630, JTE 907, and raloxifene) on the in vivo signaling of pro-apoptotic JNK1/2 MAP-kinases [13]. Thus, an inverse agonist at CB2 receptors is a ligand (e.g., AM630) that exerts the opposite pharmacological effects to a full receptor agonist (e.g., JWH133 [13]).
However, little is known about the sequential in vivo regulation of brain p-MEK1/2 to p-ERK1/2 by mechanistically different CB2 receptor ligands, i.e., direct agonists, inverse agonists, and endogenous agonists. Therefore, the aim of this study was to quantify the acute effects of CB2 receptor selective agonists (JWH133), the sustained effects of endogenous agonists (acting on overexpressed receptors) and the acute effects of inverse agonist/antagonist (AM630) on p-MEK to p-ERK signaling in mouse brain cortex.
Materials and methods
Experimental animals and ethical guidelines
Swiss albino CD1 IGS male adult mice (Charles River, 7–9 weeks old, 30–40 g) were used. All efforts were made to minimize the number of mice used and their suffering. The present groups of mice and cannabinoid drug treatments (see below) were initially reported in a previous study that dealt with various aspects of the in vivo regulation of CB2 receptor constitutive activity in the brain [13].
Treatments with cannabinoid CB2 receptor ligands
Groups of mice were treated (i.p.) with the CB2 agonist JWH133 (1 mg/kg and 3 mg/kg, 1 h, n = 8, 259869-55-1, Tocris Bioscience, UK) or the CB2 inverse agonist/antagonist AM630 (0.3 mg/kg and 1 mg/kg, 1.5 h, n = 8–9, 164178-33-0, Tocris Bioscience, UK), and 0.9% NaCl [saline 2 mL/kg, n = 4–10, 131659, PanReac AppliChem (ITW Reagents), Spain] was used as vehicle control [13]. In the present study, these two low doses of JWH133 (1 mg/kg and 3 mg/kg) and AM630 (0.3 mg/kg and 1 mg/kg) only induced small differences (between doses) in MEK1/2 and ERK1/2 contents and enzyme activities [i.e., p-kinase/total-kinase ratios], and therefore the final data were combined into one experimental group for each MAP kinase to gain visual clarity in drug effects and power in the statistical analyses. Other groups of transgenic male mice overexpressing cortical CB2 receptors [messenger RNA (mRNA) and protein] on a Swiss ICR congenic background (CB2xP, n = 9–10) and the corresponding littermates age-matched wild-type (WT, n = 10) controls [13] were also used to assess the basal regulation of MEK and ERK (WT vs. CB2xP) and for comparison with the effects of prototypes CB2 ligands (agonist JWH133 and inverse agonist/antagonist AM630) in normal CD1 mice. The CB2xP mouse is a relevant in vivo model to study the effects of inverse agonists at CB2 receptors because receptor constitutive activity is better assessed in animal models overexpressing the native (WT) receptor (see details in Discussion).
Brain sample preparation, immunoblot assays, and quantification of target proteins
After the various treatments, the mice were killed by decapitation without anesthesia and brain cortices were stored at –80°C until the neurochemical assays [13]. Briefly, cortical homogenates (40 µg protein) were prepared (with various protease and phosphatase inhibitors) and target proteins were resolved by electrophoresis [sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) minigels] followed by immunoblotting standard procedures [13, 14]. The primary polyclonal or monoclonal antibodies used were: rabbit anti-p-Ser217/221 MEK1/2 (~47/46 kDa isoforms sharing 78% amino acid identity, Cell Signaling, USA; the specificity of p-forms had been characterized previously [14]); rabbit anti-total MEK1/2 (Cell Signaling, USA); rabbit anti-p-Thr202/Tyr204 ERK1/2 (~44/42 kDa isoforms sharing 84% amino acid identity, Cell Signaling, USA); mouse anti-total ERK1 (clone 631122, R&D Systems, USA). Other methods [e.g., secondary antibodies and electrogenerated chemiluminescence (ECL) detection system] were reported [14]. To immunodetect total kinase content, the p-kinase blots were stripped and then reprobed with the antibody for total-kinase, which also served as the control for sample loading. The total amount of MEK1 + MEK2 or ERK1 + ERK2 was quantified as proposed [5], and MAP kinase activation was reported as the ratio of p-kinase/total-kinase. Total cortical MAP kinases were not altered by treatments with CB2 ligands (agonist JWH133, inverse agonist/antagonist AM630) or by overexpression of CB2 receptors (CB2xP, see immunoblots in Figure 1). Percent changes in immunoreactivity of a target MAP kinase (MEK or ERK) with respect to control samples (100%) were calculated for each mouse brain sample in the different gels (repeated at least three times), and the mean value was used as a final estimate.
Data and statistical analyses
Data are expressed as means standard error of mean (SEM). All series of data were analyzed with GrapPad Prism, version 6.0 (GraphPad Software, Inc; San Diego, CA, USA). Before the statistical analysis, the data were inspected for possible outliers (Grubb’s test): one outlier was detected within the group of CB2xP mice for p-MEK1/2 (very low p-content); deletion of this value did not alter the reported results. Comparison of three experimental groups (controls vs. two drug dosing procedures) was performed by one-way ANOVA followed by Bonferroni’s multiple comparison tests, see Results. Comparison of two groups (control vs. experimental procedure, see Figure 1B; or combined controls vs. combined drug doses, see Figures 1A and C) was performed with an unpaired, two-tailed t-test. The level of significance was set to P ≤ 0.05 in all statistical evaluations.
Results
CB2 receptor agonist JWH133 disrupts the sequential regulation of p-MEK1/2 to p-ERK1/2 in the mouse brain cortex
The acute treatment of mice with the CB2 agonist JWH133 (combined data for 1 mg/kg and 3 mg/kg, i.p., 1 h) modestly reduced, compared with saline controls, the basal activation of p-MEK1/2 in brain cortex (p-MEK/total-MEK: –17% ± 3.7%, n = 8; t = 1.69, P = 0.12) (Figure 1A); data for 1 mg/kg: –14% ± 5.1%, n = 4; t = 1.04, P = 0.34; data for 3 mg/kg: –19% ± 5.6%, n = 4; t = 1.42, P = 0.21. One-way ANOVA for JWH133 (MEK, three groups: F [2, 9] = 1.41, P = 0.29).
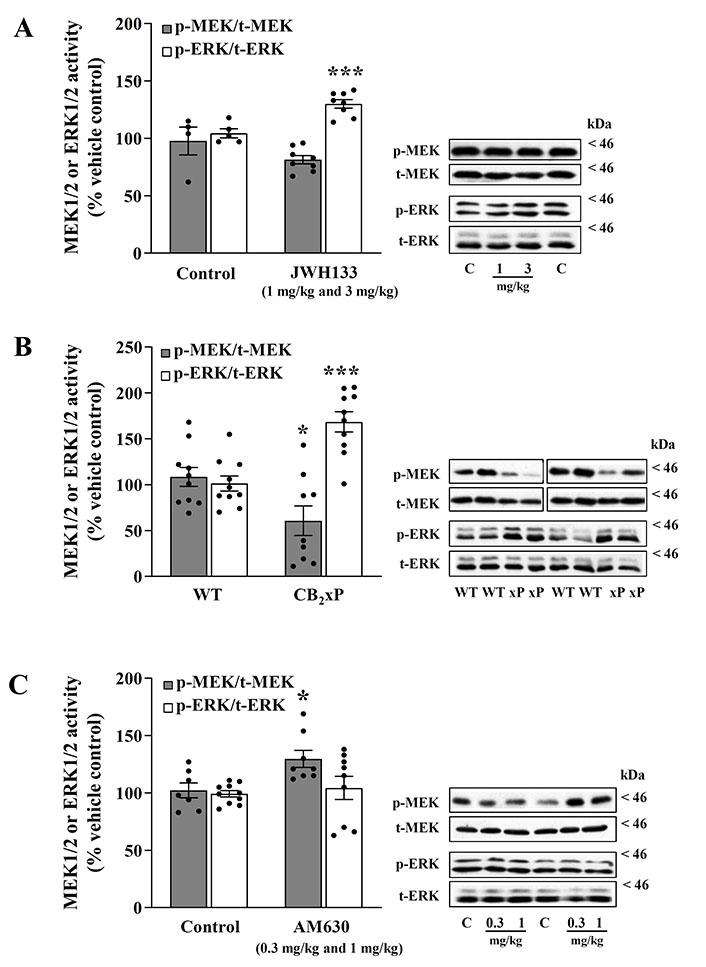
CB2 receptor ligands disrupt the sequential phosphorylation from MEK to ERK in the mouse brain cortex. A. MEK-ERK activation after acute treatment with the CB2 agonist JWH133; B. MEK-ERK activation on mice overexpressing the receptor CB2 (CB2xP); C. MEK-ERK activation after acute treatment with the CB2 inverse agonist/antagonist AM630. (A, left panel) Regulation by the CB2 receptor full agonist JWH133 (1 mg/kg and 3 mg/kg, i.p.) of p-MEK1/2 (p-Ser218/222 MEK/total-MEK ratio) and p-ERK1/2 (p-Tyr204/Thr202 ERK/total-ERK ratio) activities in the mouse brain cortex. Grey (MEK) and white (ERK) columns are means ± SEM (vertical bars) of 8 experiments (with an animal per experiment), and expressed as a percentage of saline (4–5 controls)-treated mice (100%). *** P = 0.0009 vs. the corresponding control (unpaired, two-tailed t-test). (A, right panel) Representative immunoblots of cortical MEK (p-MEK and total-MEK) and ERK (p-ERK and total-ERK) form after treatments of mice with the agonist JWH133 (1 mg/kg and 3 mg/kg) or with saline control (C). The apparent molecular masses of MEK and ERK kinase forms were determined by calibrating the blots with prestained molecular weight markers (kDa). (B, left panel) Effects of CB2 receptor overexpression (38%) on the regulation of p-MEK1/2 (p-Ser218/222 MEK/total-MEK ratio) and p-ERK1/2 (p-Tyr204/Thr202 ERK/total-ERK ratio) activities in mouse brain cortex. Grey (MEK) and white (ERK) columns are means ± SEM (vertical bars) of 9–10 experiments (with an animal per experiment), and expressed as a percentage of WT (10 controls) mice (100%). * P = 0.02, *** P = 0.0001 vs. the corresponding WT control (unpaired, two-tailed t-test). (B, right panel) Representative immunoblots of cortical MEK (p-MEK and total-MEK) and ERK (p-ERK and total-ERK) form after CB2 receptor overexpression in mice (WT, CB2 overexpression). The apparent molecular masses of MEK and ERK kinase forms were determined by calibrating the blots with prestained molecular weight markers (kDa). (C, left panel) Regulation by the CB2 receptor inverse agonist AM630 (0.3 mg/kg and 1 mg/kg, i.p.) of p-MEK1/2 (p-Ser218/222 MEK/total-MEK ratio) and p-ERK1/2 (p-Tyr204/Thr202 ERK/total-ERK ratio) activities in mouse brain cortex. Grey (MEK) and white (ERK) columns are means ± SEM (vertical bars) of 8–9 experiments (with an animal per experiment), and expressed as a percentage of saline-treated (controls 7–10) mice (100%). * P = 0.02 vs. the corresponding control (unpaired, two-tailed t-test). (C, right panel) Representative immunoblots of cortical MEK (p-MEK and total-MEK) and ERK (p-ERK and total-ERK) form after treatments of mice with the inverse agonist AM630 (0.3 mg/kg and 1 mg/kg, i.p) or with saline control (C). The apparent molecular masses of MEK and ERK kinase forms were determined by calibrating the blots with prestained molecular weight markers (kDa)
In contrast to MEK, the basal activation of p-ERK1/2 was significantly upregulated in the same cortical samples (p-ERK/total-ERK: 25% ± 3.6%, n = 8; t = 4.53, P = 0.0009) (Figure 1A); data for 1 mg/kg: 20% ± 5.7%, n = 4; t = 3.04, P = 0.02; data for 3 mg/kg: 30% ± 3.7%, n = 4; t = 5.45, P = 0.001. One-way ANOVA for JWH133 (ERK, three groups: F [2, 10] = 12.2, P = 0.002).
Therefore, the ratio of activity from MEK to ERK quantified in saline controls (ratio of kinases: 0.94) was remarkably reduced (34%) by the CB2 agonist JWH 1 mg/kg and 3 mg/kg (ratio of kinases: 0.62) (Figure 1A).
Overexpression of CB2 receptors results in marked opposite regulations of p-MEK1/2 and p-ERK1/2 in mouse brain cortex
In mice overexpressing CB2 receptors in brain cortex (CB2xP: 38%, P = 0.004; see [13]), the sustained basal activation (induced by endogenous agonists) of p-MEK1/2, compared with WT controls, was found significantly downregulated (p-MEK/total-MEK: –44% ± 16%, t = 2.56, P = 0.02) (Figure 1B).
In contrast, the sustained basal activation of p-ERK1/2 was remarkably upregulated in the same cortical samples (p-ERK/total-ERK: 67% ± 11%, t = 4.89, P = 0.0001) (Figure 1B).
Therefore, the ratio of activity from MEK to ERK quantified in WT controls (ratio of kinases: 1.07) was also strikingly reduced (66%) by overexpression of CB2 receptors (ratio of kinases: 0.36) (Figure 1B).
CB2 receptor inverse agonist/antagonist AM630 increases the basal activation of p-MEK1/2 without altering that of p-ERK1/2
The acute treatment of mice with the CB2 inverse agonist/antagonist AM630 (combined data for 0.3 mg/kg and 1 mg/kg, i.p., 1.5 h) significantly increased, compared with saline controls, the basal activation of p-MEK1/2 in brain cortex (p-MEK/total-MEK: 27% ± 7.0%, n = 8; t = 2.75, P = 0.016) (Figure 1C); data for 0.3 mg/kg: 34% ± 10.8%, n = 5; t = 2.90, P = 0.016; data for 1 mg/kg: 16% ± 2.9%, n = 3; t = 1.51, P = 0.17. One-way ANOVA for AM630 (MEK, three groups: F [2, 12] = 4.98, P = 0.027).
In contrast to MEK, the basal activation of p-ERK1/2 was found unaltered in the same cortical samples (p-ERK/total-ERK: 5% ± 7%, t = 0.60, P = 0.55) (Figure 1C); data for 0.3 mg/kg: 13% ± 8.6%, n = 5; t = 1.75, P = 0.1; data for 1 mg/kg: –3% ± 11.4%, n = 5; t = 0.38, P = 0.71. One-way ANOVA for AM630 (ERK, three groups: F [2, 17] = 1.33, P = 0.29).
Therefore, the ratio of activity from MEK to ERK quantified in saline controls (ratio of kinases: 1.01) was increased (22%) by the CB2 inverse agonist/antagonist AM630 0.3 mg/kg and 1 mg/kg (ratio of kinases: 1.23) (Figure 1C).
Discussion
The sequential in vivo activation (p) of cortical MEK1/2 (p-MEK/total-MEK ratio) to p-ERK (p-ERK/total-ERK ratio) kinases was disrupted in mouse brain cortex after the acute regulation with various, and markedly different, cannabinoid CB2 receptor ligands (i.e., direct agonist JWH133: modest decrease of MEK with ERK upregulation; endogenous agonist acting on overexpressed receptors: MEK downregulation with ERK upregulation; and direct inverse agonist/antagonist AM630: MEK upregulation with ERK unaltered) (see Figure 2). In the endocannabinoid system, these paradoxical findings for the CB2 receptor contrast with the well-known canonical activation from MEK to ERK signaling after the stimulation of CB1 receptors with the agonist WIN55212-2 (both kinases were concomitantly upregulated in rodent brain cortex [7, 8]).
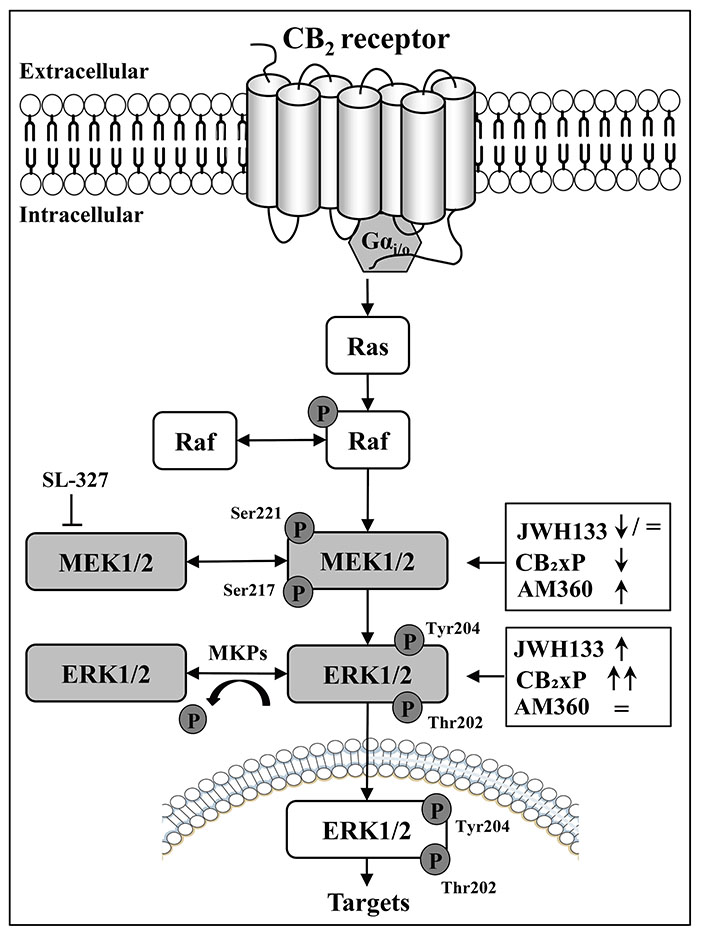
Schematic diagram depicting the disruption of the sequential regulation of MAP kinases p-MEK1/2 (ERK, MAP kinase) and p-ERK1/2 (phosphorylation and dephosphorylation processes) by CB2 receptor ligands (full agonist, JWH133; inverse agonist, AM630); and CB2 receptor overexpression (CB2xP) in mouse brain cortex. MKPs: a family of specific MAP phosphatases; SL-327: a selective MEK1/2 inhibitor; ↓: down-regulatory effects; ↑: up-regulatory effects; =: lack-regulatory effects
In the paradoxical context of MEK-ERK physiological regulation, it is important to remark that the p-MEK1/2 kinases (i.e., MAP-ERK1 and 2 kinases) are equally competent activators of their unique substrates p-ERK1 and 2, and consequently, the cellular functions of p-ERK1/2 are under the tight control of their upstream activators p-MEK1/2 [1, 5]. In fact, the acute treatment with the selective MEK1/2 inhibitor SL-327 blocked the concomitant p-ERK activity in the mouse brain [7] (Figure 2). In the endocannabinoid system, and as mentioned above, the canonical activation of p-MEK to p-ERK signaling has been well demonstrated after stimulation of CB1 receptors with selective full agonists (e.g., WIN55212-2) in rat and mouse brain cortex [7, 8]. Therefore, it was shocking to observe that treatment of mice with a selective CB2 receptor agonist (JWH133) resulted in disruption of p-MEK (decreased) to p-ERK (increased) signaling in the brain cortex (Figure 1A and Figure 2). This unexpected result prompted the in vivo assessment of other CB2 ligands for further possible divergent regulation of MEK-ERK in the brain.
Therefore, the major findings of this report demonstrate that the sequential phosphorylation of cortical MEK (ratio p-MEK1/2/total-MEK1/2) to p-ERK (ratio p-ERK1/2/total-ERK1/2) kinases are disrupted not only by the full CB2 receptor agonist JWH133 (i.e., decreased MEK with increased ERK; Figure 1A and Figure 2) but also by acute treatments with the CB2 inverse agonist/antagonist AM630, which as expected showed the opposite results to those induced by the cannabinoid agonist (i.e., increased MEK with unaltered ERK) (Figure 1C and Figure 2). In fact, an inverse agonist [10] is a ligand (e.g., AM630) that exerts the opposite pharmacological effects to a full receptor agonist (e.g., JWH133; see other relevant details in [13]). In fact, inverse agonism is better observed in those receptors that display high constitutive activity [10, 11, 13].
In this later context, and remarkably, the significant overexpression of cortical CB2 receptor density (by 38%) in CB2xP transgenic mice (see the second figure in [13]), which clearly favored the expression of CB2 receptor constitutive activity with a change in receptor function (e.g., reduced basal expression of the pro-apoptotic MAP-kinase JNK1/2; see [13]), also resulted in a striking disruption of MEK to ERK parallel activity in brain cortex (i.e., marked decrease of MEK with marked increase of ERK) (Figure 1B and Figure 2). This result is in line with the reported agonist effect of JWH133 in normal CD1 mice (with a low level of CB2 receptor expression), and suggests that after CB2 receptor overexpression (38%) the sustained basal effects induced by the cannabinoid endogenous agonists (anandamide and/or 2-arachidonoylglycerol) on MEK-ERK would be more evident as clearly it was observed (Figure 1B and Figure 2). In this context, the biochemical and functional effects of a marked receptor overexpression (up to 195 times) were first reported for the cardiac β2-adrenoceptor, which resulted in a greater myocardial function in mice (see the pioneer study by Milano et al. [15]). In the present study and in the opposite side of the above reasoning, it could have been expected that a marked decrease of cortical p-MEK activity in mice (Figure 1B and Figure 2) would be translated into a substantial reduction in the activation of its exclusive substrate p-ERK (see [3–5]). However, this anticipated theoretical result was not observed (Figure 2) further suggesting a disruption of MEK-ERK signaling in the mouse brain cortex.
The reported in vivo disruption of the sequential and parallel basal activation of p-MEK to p-ERK by CB2 receptor ligands had been observed for other Gi/o-coupled receptor and other experimental paradigms in rats and mice, including the treatment with low doses of the opioid receptor agonist fentanyl [16] as well as after treatments with structurally diverse hypnotic/anesthetic agents, which function through the allosteric activation of inhibitory gamma-aminobutyric acid subtype A (GABAA) receptors (see [14]). Interestingly, the acute treatment of mice with the irreversible (alkylating) receptor agent BU99006 (20 mg/kg, i.p., 1 h) also resulted in disruption of the expected parallel activity of MEK1/2 (p-MEK/total-MEK ratio decreased by 32%) to ERK1/2 (p-ERK/total-ERK ratio unaltered) signaling in the hippocampus [17]. In these previous studies, various molecular mechanisms were investigated in order to explain the disruption of brain MEK-ERK sequential regulation [for example, the roles of inactivated p-Thr286 MEK1 and specific MAP kinase phosphatases such as MAP kinase-1 (MPK-1), MPK-2, and MPK-3] [14–16], but the complex underlying in vivo mechanisms were not fully disclosed.
It is therefore unlikely that similar contra regulatory mechanisms could explain the remarkable disruption from MEK to ERK phosphorylation induced by the different cannabinoid CB2 ligands in the mouse brain cortex (Figure 1). It is known that the stimulation of cannabinoids CB2 receptors can engage multiple waves of different signaling pathways [12, 13], but at present the molecular signals underpinning the reported in vivo MEK-ERK dysregulation in the brain remain unknown. Moreover, some in vitro studies have reported that p-ERK1/2 signaling in rat-1 fibroblasts can be activated (with very high concentrations of peroxynitrites) via some MEK-independent pathway [18], but such a possibility contradicts the accepted postulates (mainly for in vivo experiments; e.g., see [7, 8] for specific details) of the canonical activation of MEK-ERK signaling (i.e., p-ERK1/2 are the sole cytoplasmic substrates of p-MEK1/2 kinases [3, 4], which have been validated by quantitative phosphoproteomic analysis [19]).
Further studies are needed to unravel the molecular mechanisms underpinning the striking disruption of MEK to ERK parallel regulation in the cannabinoid CB2-Gi/o receptor signaling complex in the brain. Thus, a better understanding of the complexity of MEK-ERK in vivo signaling (e.g., assessing the effects of more efficacious synthetic CB2 agonists and inverse agonists modulating neuronal death in psychiatric and neurodegenerative syndromes) would clarify the postulated roles of brain CB2 receptors in various CNS diseases and their treatments with specific drugs [20, 21].
In this context, it has recently been suggested that the chronic use (during many years) of common medications by the elderly (e.g., antihypertensive, antidepressant, hypoglycemic, and hypocholesterolemic drugs [22]), and/or the abuse of alcohol (binge drinking and alcoholism) and/or hypnotic agents among older adults (drugs which also induce disruptions of the basal activation of p-MEK to p-ERK [14] and other references therein), might have a negative impact on neuronal survival accelerating the rate of progression of apoptotic death [23] in neuropsychiatric disorders such as Alzheimer and Parkinson syndromes, mainly because the very limited capacity of adult neurons to proliferate or be replaced.
Abbreviation
| CB2: | cannabinoid 2 |
| CHO: | Chinese hamster ovary |
| Gi/o: | G-protein |
| MAP: | mitogen-activated protein |
| p-ERK1/2: | phosphorylation-ERK1/2 |
| p-MEK1/2: | phosphorylation-MEK1/2 |
| SEM: | standard error of mean |
| WT: | wild-type |
Declarations
Acknowledgement
The authors thank Dr. Jorge Manzanares (Institute of Neurosciences, Alicante, Spain) for providing WT and CB2xP mice.
Author contributions
GS: Conceptualization, Data curation, Formal analysis, Investigation, Visualization, Writing—review & editing. MÁB: Conceptualization, Data curation, Investigation, Methodology, Supervision, Visualization, Writing—review & editing. JAGS: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All animal care and experimental procedures were conducted according to standard ethical guidelines (ARRIVE guidelines and European Communities Council Directive 2010/63/EU) and approved by the Local Bioethical Committee of the University (Committee number: 64).
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Data will be made available on request (Jesús A. García-Sevilla, jesus.garcia-sevilla@uib.es).
Funding
The study was partly supported by Ministerio de Economía y Competitividad [SAF2014-55903-R] (MINECO) and
Copyright
© The Author(s) 2023.