 Review
Review

Affiliation:
Department of Pediatrics, Division of Pediatric Neurology, Dr. Behçet Uz Children’s Education and Research Hospital, Izmir Faculty of Medicine, University of Health Sciences Turkey, Izmir 35210, Türkiye
Email: drunsalyilmaz@yahoo.com
ORCID: https://orcid.org/0000-0002-7256-8557
Explor Neuroprot Ther. 2024;4:38–54 DOI: https://doi.org/10.37349/ent.2024.00069
Received: June 20, 2023 Accepted: November 10, 2023 Published: February 27, 2024
Academic Editor: Vijay K. Sharma, National University of Singapore, National University Health System, Singapore
The article belongs to the special issue The Future of Biomarkers in CNS Diseases
Over the last two decades, immunoglobulin G (IgG) antibodies against myelin oligodendrocyte glycoprotein (MOG), previously thought to be a biomarker of multiple sclerosis (MS), have been shown to cause a distinct disease called MOG antibody-associated disease (MOGAD). MOGAD accounts for approximately one-third of all demyelinating syndromes in children and is the second most common central nervous system (CNS) demyelinating disease after MS. The diagnosis is made by detecting anti-MOG IgG antibodies against the natural MOG antigen, in the presence of compatible clinical and neuroradiological features. However, due to controversies in the methodologies for detecting anti-MOG antibodies and their diagnostic cutoff values, as well as the expanding clinical spectrum, accurate diagnosis may be challenging, at least in a subset of patients. Clinical presentations of MOGAD vary by age; the highest rates are seen in acute disseminated encephalomyelitis in younger children and optic neuritis, myelitis, or brainstem symptoms in older children. Although it was previously thought to be a milder demyelinating disorder and to have a monophasic course in the majority of patients, recent studies have shown that relapses occur in about half of the patients and sequelae develop in a significant proportion of them, especially in those with persistently high antibody titers, leukodystrophy-like magnetic resonance imaging (MRI) lesions, and spinal cord involvement. However, due to the monophasic course in about half of the patients, long-term treatment is not recommended after the first clinical episode but is recommended for patients who experience relapse. Accurate and early diagnosis of MOGAD is essential for proper management and better outcome. This review covers the challenges in the diagnosis of MOGAD in children.
In recent years, immunoglobulin G (IgG) antibodies against myelin oligodendrocyte glycoprotein (MOG), previously thought to be a biomarker of multiple sclerosis (MS), have been shown to cause a distinct disease called MOG antibody-associated disease (MOGAD) [1, 2]. Although anti-MOG antibodies have been known since the 1980s, the presence of these antibodies was accepted as a finding supporting the diagnosis of MS. Later studies revealed that the anti-MOG antibodies found in MS patients were not disease-causing antibodies detected by enzyme-linked immunosorbent assay (ELISA) or Western blot methods using flattened or denatured MOG peptides as antigens, but were secondary to the demyelinating process [3]. Actually, in many patients with anti-MOG antibodies detected by ELISA or Western blot methods, no antibodies were detected with the new-generation cell-based assays (CBAs) in which full-length intact human MOG peptides were used as antigens [4]. Thereafter, numerous clinical, radiological, immunological, and pathological studies showed that the disorder in which anti-MOG-IgG antibodies detected by the new generation CBAs is a different disease from MS [1, 2, 5–7]. Diagnosis of MOGAD is mainly made by detecting anti-MOG antibodies in serum using CBAs in the presence of clinical findings compatible with MOGAD, such as multiphasic acute disseminated encephalomyelitis (M-ADEM) in younger children, recurrent optic neuritis (ON) or neuromyelitis optica spectrum disorder (NMOSD) phenotypes in older children and adolescents [3, 8–10].
MOG is a central nervous system (CNS)-specific protein found only on the myelin sheath and on the surface of oligodendrocytes. Although it constitutes only a minor component (0.5%) of myelin, it is a possible target for autoantibodies in a demyelinating disease due to its highly immunogenic position. MOG plays a role in the maturation of oligodendrocytes, as well as in myelin integrity [4]. Various environmental factors such as infections or vaccinations can trigger immune activation against MOG antigens [4].
CNS demyelinating diseases are currently classified under four groups: MS, NMOSD, MOGAD, and seronegative demyelinating syndromes [7]. These demyelinating disorders share many common clinical, radiological, and laboratory features. Since the treatment and prognosis differ, it is imperative to distinguish these four groups of diseases and to diagnose them early and accurately. In addition, early identification of MOGAD patients presenting with a first demyelinating event and at risk of relapse allows for early initiation of disease-modifying therapeutic agents, thereby improving prognosis. Since the author of the article is a child neurologist and deals only with pediatric patients, this review largely discusses the challenges in the diagnosis of MOGAD in children. Although the recommendations herein can also be applied to adult MOGAD patients, the diagnosis and clinical course of MOGAD may differ slightly in adults, especially compared to very young children whose brain development is still ongoing.
The incidence of MOGAD, the only recurrent CNS demyelinating disease that is more common in children than adults, is approximately three times higher than in adults. (0.31 vs. 0.13 per 100,000) [11]. In children, about one-third of all demyelinating syndromes are MOGAD, and it is the second most common demyelinating disease after MS [7, 12–14]. In an observational prospective multicenter hospital-based study of 210 children presented with a first acquired demyelinating syndrome, at onset, 29% were diagnosed with acute disseminated encephalomyelitis (ADEM), 18% with MS, 6% with NMOSD, and 48% with clinical isolated syndrome; after a 24-month follow-up, 46% did not relapse, 38% were diagnosed with MS, 31% with MOGAD, and 3% with aquaporin-4 (AQP4) antibody seropositive NMOSD [14]. In another study of 110 children with relapsing demyelinating syndromes, 56% of the patients were diagnosed with MS, 25% with NMOSD, 13% with M-ADEM, and 6% with recurrent ON [7]. In these studies, anti-MOG antibodies were detected in 57% of all children with ADEM, 100% of M-ADEM/ADEM-ON, 33–73% of relapsing ON (RON), 56–58% of NMOSD (83% of AQP4 negative patients), only 0–4% of MS, and none in children with the AQP4 positive patients [7, 14]. Based on these data, it can be concluded that approximately 40% of demyelinating diseases in children are MS, 30% are MOGAD, and less than 5% are AQP4 antibody seropositive NMOSD. While AQP4 antibody positivity is associated only with NMOSD phenotype; MOGAD is clinically highly heterogeneous, with nearly all M-ADEM/ADEM-ONs, three-quarters of recurrent ON, and about half of the MOG antibody-associated NMOSD and monophasic ADEM phenotypes appearing to be MOG antibody-related [7, 14, 15].
Diagnosis of MOGAD has been challenging due to controversies in antibody detection methodologies and also to the expansion of the clinical spectrum over the years. Although several diagnostic criteria have been developed over time by different groups [8–10], the International MOGAD Panel has only very recently published the formal consensus diagnostic criteria and underlined the need for validation [3]. The diagnosis basically depends on the detection of anti-MOG antibodies in the presence of clinical findings consistent with MOGAD. In this context, the spectrum of the clinical presentation compatible with MOGAD, and the methodology and sampling material for the detection of anti-MOG antibodies have been the subject of discussion. Unlike MS and AQP4 antibody seropositive NMOSD, in which the clinical course is characterized by a relapsing-remitting pattern, patients with MOGAD can have either a monophasic or relapsing course. Therefore diagnosis of MOGAD does not require clinical relapses or any radiological finding fullfilling dissemination in time.
In contrast to MS and NMOSD, the male-to-female ratio is approximately equal in MOGAD [12, 15, 16]. The median age at onset is reported as 6–7 years and differs among presenting phenotypes: 4 years in M-ADEM, 6 years in ADEM-ON, 8 years in NMOSD, and 11 years in RON [15, 17]. Unlike MS and NMOSD, which are mostly diagnosed in young adults, MOGAD is more common in children [12, 14]. This may be partly explained by the fact that ADEM is more common in children and anti-MOG antibodies are detected in a significant proportion of these patients.
Core clinical presentations suggested by the most recent International MOGAD Panel proposed criteria include ON, myelopathy, ADEM, cerebral monofocal or polyfocal deficits, brainstem or cerebellar deficits, and cortical encephalitis [3]. Pediatric demyelinating disorders can be diagnosed as ADEM, ON, transverse myelitis (TM), NMOSD, MOGAD, or MS based on clinical and paraclinical findings during the first episode. ADEM, isolated ON, and TM may remain monophasic throughout the patient’s lifetime or may evolve into diagnoses of MS, MOGAD, or NMOSD. In addition, ADEM, ON, and TM may be a component of MS, NMOSD, or MOGAD during the first episode or throughout the course of the disease. Because of this complex situation, it is difficult to determine the accurate incidence and prevalence of these syndromes and to diagnose a specific disease, especially during the first episode. The clinical phenotype and the course of MOGAD may differ according to the age of the patient. Likely due to the degree of myelin maturation, a leukodystrophy-like phenotype may be seen in very young children; ADEM in older children, and recurrent ON and NMOSD in adolescents and adults [11, 12, 16, 18]. Of all children with MOGAD, the phenotype of the first episode was reported as ADEM (46%), ON (30%), TM (11%), and NMOSD (4%) [12]. Although the NMOSD-like phenotype with concurrent ON and TM is a very rare initial presentation in children, approximately 40% of children with NMOSD-like phenotype have anti-MOG antibodies [12].
ADEM is the most common presenting phenotype of MOGAD in children (40%), whereas it accounts for only 3% of adult patients with MOGAD [2, 15]. Although it is mostly a monophasic disease, some children may have further attacks of ADEM (M-ADEM) or ON (ADEM-ON) in the following months or years. It has been reported that almost all patients diagnosed with M-ADEM and ADEM-ON have MOGAD [14, 19]. In a previous study, MOG antibodies were detected in 64% of all ADEM patients and in 96% of those with relapsing ADEM episodes, which were observed in 36% of all children with ADEM [20]. Post-ADEM epilepsy is more common in MOG-positive ADEM compared to seronegative ones [21]. Although clinical and radiological improvement is observed following high-dose steroid treatment, the prognosis is worse in this group and ongoing seizures with cognitive and behavioral problems are common. Despite chronic immunomodulatory therapy, relapses persist in about 40% of these patients [15].
ON can be limited to the optic nerve (isolated ON); or it can be the first episode of other CNS demyelinating disorders such as ADEM, MS, NMOSD, or a part of the clinical picture of these disorders; or an initial manifestation of various connective tissue or neurometabolic disorders such as systemic lupus erithematosus or mitochondrial disorders (Figure 1). Isolated ON may be in the form of a single episode (monophasic isolated ON) throughout life, as well as unilateral or bilateral relapsing ON. In particular, there is a strong association between anti-MOG antibodies and concurrent bilateral ON, unlike MS [22]. While some patients with relapsing ON have a progressive course and show steroid dependence [chronic relapsing inflammatory ON (CRION)], others do not have a progressive course or steroid dependence [relapsing inflammatory ON (RION)] [23]. In recent years, the CRION phenotype has been accepted as a MOGAD presentation since anti-MOG antibodies have been detected in almost all CRION patients [24]. In a study of four-year follow-up of children presenting with isolated ON, 45% of the patients did not relapse (55% of them were anti-MOG positive), 19% were diagnosed with MS, and 37% had non-MS relapsing demyelinating syndromes (RDS, 50% were anti-MOG positive, 8% anti-AQP4 positive NMOSD, and 42% seronegative) [20]. Prominent optic disk swelling visible both on fundoscopy and radiology, good response to treatment despite initial severe vision loss, and better prognosis distinguishes MOG-antibody-associated ON from AQP4 antibody-positive and MS-associated ON [22, 25, 26].
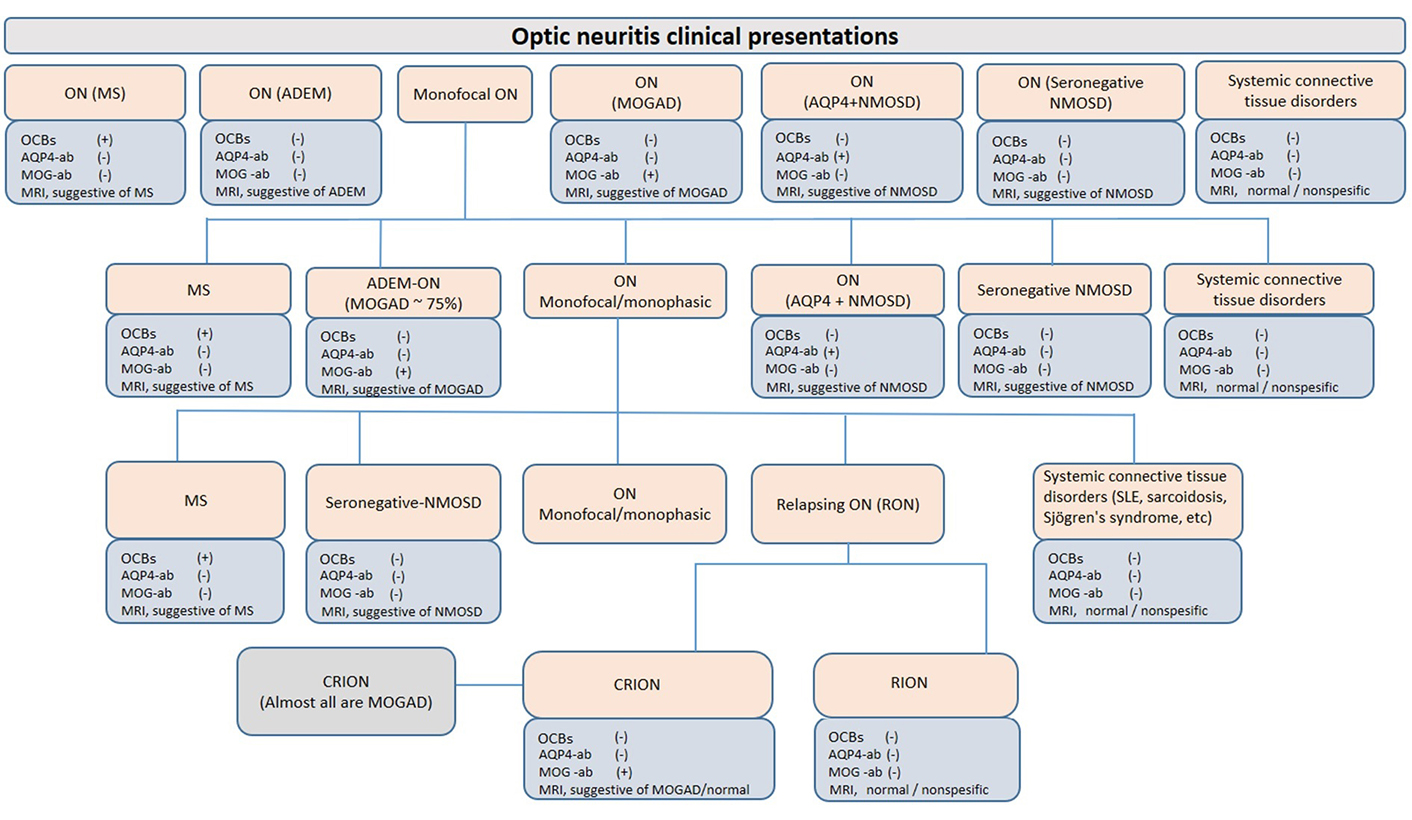
ON presentations. ON may be confined to the optic nerve and remain monophasic throughout life (isolated ON); or it can be the first episode of other CNS demyelinating disorders such as ADEM, MS, NMOSD, or a part of the clinical picture of these disorders; or a manifestation of various connective tissue or neurometabolic disorders such as systemic lupus erythematosus (SLE) or mitochondrial disorders. Paraclinical findings are presented as commonly seen. For example, cerebrospinal fluid (CSF)-restricted OCBs can be detected in up to 10% of MOGAD patients and up to 30% of NMOSD patients. OCBs: oligoclonal bands; ab: antibodies; +: positive; –: negative; MRI: magnetic resonance imaging
In a previous study, 21% of children with any demyelinating disease were found to have TM at the first clinical episode; 72% of these patients remained monophasic (8% MOG positive, the rest seronegative); 22% had non-MS RDS (73% AQP4 positive, the rest seronegative); and 6% of them have evolved to MS (all seronegative) [20]. TM in MOG antibody-positive patients can occur in isolation, as a component of ADEM, or concurrent with ON [27–29]. It has been reported that 14% of MOG-positive patients who presented with isolated TM in the first presentation later developed ON episodes and therefore evolved to NMOSD [30]. Of the patients with conus involvement, especially in males, a significant proportion of patients was reported to have persistent bladder (55%), bowel (36%), and sexual (82%) dysfunction although motor symptoms were completely resolved with steroid treatment [31].
Recently, the detection of MOG antibodies in patients who meet the diagnostic criteria for AQP4 antibody seronegative NMOSD has led to controversy over the terminology and classification of these patients. Using the diagnostic criteria for NMOSD [32] and MOGAD [3], a patient can definitely be diagnosed with both AQP4 antibody seronegative NMOSD and MOGAD, respectively. Since these patients behave as MOGAD instead of AQP4 antibody seropositive NMOSD, it seems more appropriate to classify these patients as having MOGAD. [3, 4] While anti-AQP4 antibodies are detected in more than half of adult NMOSD patients, they are found in 11–42% of pediatric NMOSDs and in only 3–6% of all demyelinating diseases in children [14, 20, 30]. On the other hand, anti-MOG antibodies were detected in about one-third of all demyelinating diseases, and 39–56% of children presented with NMOSD phenotype [14, 20, 30]. Compared to patients with AQP4 antibody-associated NMOSD, MOG antibody-associated NMOSD patients have been shown to have a younger age of onset, exhibit less of a female preponderance, slower disability accumulation, longer relapse time, more frequent cerebellar peduncle involvement, and less frequent area postrema involvement [7, 22, 33, 34]. Although brain stem involvement can also be seen in MOGAD, area postrema syndrome characterized by nausea, vomiting, and unexplained hiccups is a characteristic presentation of AQP4-positive NMOSD [35]. MOG and AQP4 antibody-associated NMOSD are also pathophysiologically distinct disorders: while the primary pathophysiology in MOGAD is myelin and oligodentrosic destruction, it is astrocytopathy in AQP4-positive NMOSD. Coexistence of anti-MOG and anti-AQP4 antibodies was very rarely reported [36]. The presence of anti-MOG antibodies is an exception in MS, and a detailed investigation should be performed before diagnosing MS in such patients [1, 2, 14, 37, 38].
Although the most recent International MOGAD Panel has defined the typical clinical presentations used in the diagnosis of MOGAD as discussed above, the clinical phenotypic spectrum of MOGAD is still expanding with the widespread availability of anti-MOG-IgG testing using fixed or live CBAs and increased awareness of MOGAD, such as leukodystrophy-like phenotype [18], isolated seizures [39], ocular flutter [40], CNS vasculitis [41], cranial neuropathies [42], or intracranial hypertension [43] (Table 1).
Unusual clinical presentations of MOGAD
| References | Clinical presentations | Number of cases |
|---|---|---|
| Hacohen et al. [18], Dev Med Child Neurol, 2018 | Leukodystrophy-like phenotype | 7 |
| Vazquez Do Campo et al. [44], Neurol Neuroimmunol Neuroinflamm, 2018 | Combined central and peripheral demyelination | 1 |
| Ramanathan et al. [39], Dev Med Child Neurol, 2019 | Isolated seizures | 4 |
| Breza et al. [40], Mult Scler, 2019 | Ocular flutter | 1 |
| Sa et al. [45], Mult Scler, 2019 | Complex movement disorder with dystonic posturing and tonic eye deviation | 1 |
| Sundaram et al. [46], Mult Scler, 2020 | Relapsing lumbosacral myeloradiculopathy | 1 |
| Macaron et al. [47], Mult Scler, 2020 | Imaging-negative myelitis | 1 |
| Patterson et al. [41], Neurol Neuroimmunol Neuroinflamm 2019 | Small vessel CNS vasculitis | 2 |
| Baba et al. [48], Mult Scler Relat Disord, 2019 | Progressive cognitive deterioration and behavioral changes with primary CNS vasculitis | 1 |
| Cobo-Calvo et al. [42], Neurol Neuroimmunol Neuroinflamm, 2019 | Cranial nerve (trigeminal, vestibulocochlear, oculomotor) involvement | 3 |
| Yılmaz et al. [49], Mult Scler Relat Disord, 2019 | An isolated seizure followed by slowly progressive behavioraland personality changes | 1 |
| Corbett et al. [50], J Clin Neurosci, 2020 | PRES-like presentation | 1 |
| Chaudhuri et al. [43], Neurol Neuroimmunol Neuroinflamm, 2022 | Intracranial hypertension and bilateral papilledema | 1 |
PRES: posterior reversible encephalopathy syndrome
A relatively rare clinical presentation of MOGAD is encephalitis, which accounts for 7–9% of all non-ADEM autoimmune encephalitis in children [15, 51]. Encephalopathy is a common characteristic of both ADEM and encephalitis. ADEM is a demyelinating disease characterized by diffuse, hazy large lesions in cerebral white matter and deep gray matter, often accompanied by ON and/or TM, whereas cortical/subcortical involvement is seen in encephalitis, in which MRI may be completely normal [52]. While encephalopathy, headache, focal neurologic deficits, and seizures were the most common symptoms in MOG antibody-associated encephalitis; pleocytosis in CSF, juxtacortical, cortical, and deep gray matter involvement in MRI are common paraclinical features [52]. The coexistence of anti-MOG and anti-N-methyl-D-aspartate receptor (anti-NMDAR) antibodies has been reported very rarely [51, 52]. Epileptic seizures may be part of the clinical presentation of cortical encephalitis or may be isolated in MOGAD [53]. Typical demyelinating episodes after 1–12 months have been reported in MOGAD patients presented with isolated seizures [49, 53]. Recently, a case of MOGAD with an ADEM episode at the age of 4 years, no other symptoms until the age of 17, and progressive spastic quadriparesis for 9 years after the age of 17 was reported [54].
An unexpected presentation of MOGAD is intracranial hypertension. A 19-year-old MOGAD patient presenting with recurrent intracranial hypertension, bilateral papilledema, only mild meningeal involvement in the left parietal region on MRI, and responding well to rituximab treatment was recently reported [43].
Very few cases of polyradiculoneuropathy with CNS demyelinating lesions associated with MOG antibodies have been reported recently [55]. However, it is not clear whether anti-MOG antibodies are the cause of the clinical findings in these cases [12]. In a recent review, a total of 11 cases of aseptic meningitis and leptomeningeal enhancement associated with MOG antibodies were identified, of which five had no demyelinating lesions [56]. A small number of unclassifiable phenotypes have also been reported. For example, anti-MOG antibodies were detected in one patient with isolated uveitis and in two patients with coexisting uveitis and ON [57]. Therefore, the clinical spectrum of MOGAD seems not limited to ADEM, ON, or NMOSD; but further expansion of the MOGAD phenotype is likely in the coming years, which may necessitate the revision of the clinical spectrum used in the diagnosis of MOGAD.
As a target autoantigen for both humoral and cell-mediated immune responses in CNS, MOG has been extensively studied for decades. Since the 1980s, experimental autoimmune encephalomyelitis (EAE) induced by immunization using MOG has been used extensively as an important animal model of MS. Earlier studies have shown that MOG autoantibodies mediate oligodendrocyte damage and demyelination [58, 59], suggesting MOG antibodies may be a diagnostic biomarker in demyelinating disorders including MS. Subsequent studies focused on the questions of whether anti-MOG antibodies play a primary role in the pathogenesis of MS or arise secondarily to myelin destruction.
MOG is expressed only on the surface of oligodendrocytes and myelin in the CNS and is found in the highest concentration in the outermost part of the myelin sheath and plasma membranes of oligodendrocytes, thereby making it a readily accessible antigenic target for the action of autoantibodies. Autoantibodies can bind to both linear and conformational regions of MOG. Traditional methods such as ELISA and Western blots detect autoantibodies developed against linear epitopes of MOG. However, animal studies have shown that pathogenic MOG antibodies recognize only conformational epitopes [60–62]. In animal studies, antibodies to native MOG were found to be associated with demyelination; however, antibodies against flattened MOG detected by ELISA were not associated with demyelination [62]. As ELISA and Western blot methods cannot distinguish between autoantibodies developed against pathogenic conformational or linear epitopes, they do not appear to be sensitive methods for detecting pathogenic anti-MOG antibodies. On the other hand, newer CBA methods can detect antibodies directed against native conformational MOG.
Earlier studies reported higher proportions of MOG positivity among patients with MS (up to 21%), other inflammatory neurological diseases, and also healthy controls [4, 63]; whereas these rates were found to be about 1% in MS and none in healthy controls in more recent studies [64, 65]. This appears to be due to the differences in antibody detection methodologies. For the detection of anti-MOG antibodies, most earlier studies used assays that utilized linearized or denatured MOG antigens, such as ELISAs and Western blots; whereas later studies mostly used CBAs for MOG antibody detection [4, 33]. Since only those anti-MOG antibodies that direct against native MOG in their conformational state, rather than linear or denatured MOG, are pathogenic [61], live CBAs have been considered the gold standard in the diagnosis of MOGAD [3]. There is an acceptable concordance between the fixed and the live CBAs, and therefore the fixed CBAs are considered to be a reasonable alternative when live CBA is not available [65]. However, the positive predictive value of live CBAs is higher when compared to that of fixed-CBAs [66, 67]. MOG antibodies may occasionally be detected, particularly at low titers, in patients with other neurologic disorders, including 0.3% to 2.5% of adults with MS; however, MOG antibody false-positivity appears to be more common in adults than children [13, 38, 64, 68].
The detection of MOG antibodies in serum appears to be more sensitive than in CSF [69–71]. In a previous study investigating anti-MOG antibodies in 33 patients with various neurological diseases and healthy controls, the corresponding CSF samples were found to be positive only in patients with serum titer values ≥ 1:640, suggesting a peripheral production of anti-MOG antibodies [72]. Similarly, other studies have reported that patients with positive anti-MOG antibodies in serum were found to be negative in CSF [58, 73]. Accordingly, the most recent International MOGAD Panel proposed criteria recommend antibody testing in serum and require an additional supporting clinical or MRI feature in order to make a definitive diagnosis in patients with CSF-restricted MOG antibodies [3], although previously proposed criteria accept positivity in CSF as a fulfillment of criteria in absence of serum positivity [8]. However, in two recent studies, 12–29% of patients with confirmed MOGAD were found to have CSF-restricted antibodies, while none of the NMOSD patients had CSF-restricted anti-AQP4 antibodies [74, 75]. In addition, it was shown that MOG antibodies are produced intrathecally in MOGAD patients, in contrast to AQP4 antibodies produced in the periphery [74]. However, since CSF positivity was detected in only 62% of seropositive patients, it was recommended that the test be performed first in serum and then in CSF if clinical suspicion persists [69].
MOG antibody testing should be performed close (< 3 months) to the onset of the acute clinical episode; a negative MOG antibody testing long after the onset of the attack does not exclude MOGAD [15, 76]. Ideally, MOG antibody testing should be performed prior to administration of corticosteroids, immunoglobulins, or apheresis because these treatments may reduce the likelihood of detecting serum MOG-IgG as observed when testing for AQP4-IgG [77].
Although a correlation between antibody titer and outcome has not been demonstrated and a monophasic/multiphasic distinction cannot be made, it is recommended to measure serum for anti-MOG antibodies every six months for up to two years or until antibodies turn negative [76]. In contrast to AQP4 antibodies, which usually remain positive at follow-up, antibodies become negative over time in a significant proportion of MOGAD patients, more often in monophasic patients [14, 15, 37]. In contrast, one study reported that 62% of AQP4-positive NMOSD patients remained positive for AQP4, while 81% of MOGAD patients remained positive for MOG antibodies (82% of monophasics, 81% of multiphasics) during a four-year follow-up [20]. Although negative conversion of MOG antibodies has a high predictive value for a lower risk of relapse, monophasic patients may also remain positive for MOG antibodies for a long time (median 12 months) [15, 76].
A clear anti-MOG antibody positivity was defined by International MOGAD Panel as at least two doubling dilutions above the assay cutoff, or above the assay-specific titer cutoff, or flow-cytometry ratio cutoff for live assays; and as titres greater than or equal to 1:100 for fixed assays [3]. MOG antibody test results are considered to be low positives if they are in the low range of the individual live CBAs, or if titres are between 1:10 and 1:100 for fixed-CBAs [3]. The positive predictive value for clinical features consistent with MOGAD increases with an increasing titre of MOG antibodies [68, 78]. While, clear positive are strongly associated with a diagnosis of MOGAD, and distinguish it from AQP4-IgG-seropositive NMOSD or MS, low titres are less discriminatory and may be seen in patients with MS, other neurological diseases, and even healthy individuals [13, 68, 78, 79].
Co-occurrence of AQP4 and MOG antibodies has rarely been reported in patients with demyelinating disorders, and almost all such patients have high AQP4 and low MOG antibody titers [36, 80]. Anti-NMDAR antibodies were found to be more commonly coexisted with MOG antibodies (12%) compared with AQP4 antibodies (0.6%) [81]. Anti-MOG antibodies may rarely coexist with anti-glial fibrillary acidic protein (anti-GFAP) in patients presenting with isolated meningitis and papillitis [82].
CSF-restricted OCBs were reported only in a minority of patients with MOGAD [22, 58, 72], supporting the hypothesis of peripheral rather than intrathecal production. Since approximately 90% of MOGAD patients do not have CSF-restricted OCBs, MS should primarily be considered in its presence [1, 22, 58, 83]. An elevated albumin CSF/serum ratio which shows blood-CSF barrier dysfunction is reported in about half of patients with MOGAD [83]. CSF pleocytosis has been reported in over 50% of patients with MOGAD [79, 83]. High CSF protein levels were reported in about 30% of patients with MOGAD and do not discriminate MOGAD from other neuroinflammatory disorders [1, 69, 79]. Intrathecal IgG synthesis was reported to be low, often transient, and mostly restricted to acute attacks in patients with MOGAD [83]. As expected, the measles, rubella and varicella zoster virus (MRZ) reaction is absent in MOGAD, the most specific biomarker of MS [83].
Visual evoked potential examination and optical coherence tomography can be used to evaluate optic pathway damage in MOG antibody-associated ON, but they are not diagnostically specific [84]. However, retinal thinning detected by optical coherence tomography in the follow-up of children with MOGAD can be used in the decision to start maintenance therapy [85].
MRI abnormalities were reported in about one-third of MOGAD patients, with a variety of findings not typical for MS [33, 34, 86]. The characteristic MRI features in MOGAD differ according to the age of patients at onset and to the presenting phenotype, and can be listed as leukodystrophy-like lesions in very young children; multiple large, poorly circumscribed supra- and infratentorial lesions with ADEM-like clinical presentation in children; longitudinally extensive spinal cord lesions that can sometimes involve the entire spinal cord, and frequently with enhancement especially on the conus in NMOSD patients; or may be completely normal or nonspecific lesions may be seen in isolated ON patients [18, 87].
Optic nerve involvement is mostly in the form of bilateral and longitudinally extensive involvement in the anterior segment with perineural enhancement [26, 86]. Optic nerve head swelling and retrobulbar optic nerve involvement are common findings in MOG antibody-associated ON [86]. Optical coherence tomography revealed more severe optic nerve swelling but less retinal neuronal loss than in patients with MOGAD compared to AQP4 antibody-positive patients [88]. Although chiasma was previously reported to be preserved in MOGAD unlike ON associated with AQP4 antibody [26], a recent study has shown that a significant proportion of MOGAD patients also showed chiasma involvement (20% in AQP4 positive ON, 16% in MOG-positive ON) [89].
Spinal MRI shows more frequent involvement of the thoracolumbar region in MOG antibody-positive patients, although up to 10% of spinal MRI scans can be normal at onset despite obvious clinical findings (MRI-negative myelitis) [90]. Although longitudinally extensive spinal cord involvement is seen in both AQP4 and MOG antibody-related myelitis, conus involvement is more common in MOGAD (37% in one study) [30, 90, 91]. Longitudinally extensive involvement is a rare finding in MS, and its presence should suggest AQP4-positive NMOSD, MOGAD, and other etiologies [92]. Contrast enhancement occasionally associated with cauda equina and pial enhancement was reported in about half of the patients with MOG antibody-associated myelitis [27, 28, 55, 93].
Rapid and complete resolution of MRI lesions has been reported in some patients, particularly after steroid therapy, which is a rare finding in MS and AQP4-IgG-NMOSD [34, 86, 94]. In a previous study, it was reported that 67% of patients had cerebral lesions at onset, and these lesions disappeared completely in approximately half of the patients at follow-up [37].
In a previous study, serial MRI examinations were performed in a five-year follow-up period of MOGAD patients, and new asymptomatic MRI lesions were detected in 14% of the patients, mostly in the first months of the disease, but relapses were seen in only 20% of these patients [95]. Based on this finding, the authors concluded that detecting an asymptomatic lesion on follow-up MRI in MOGAD patients does not require initiation of treatment [95], in contrast to the E.U. pediatric MOG consortium consensus report which suggests initiation of maintenance treatment in the presence of asymptomatic MRI progression [85].
Based on recent diagnostic criteria [3] and taking into account recent data on unusual clinical presentations of MOGAD and the possibility of the presence of CSF-restricted MOG antibodies, a diagnostic algorithm for the evaluation of patients with suspected MOGAD is presented in Figure 2.
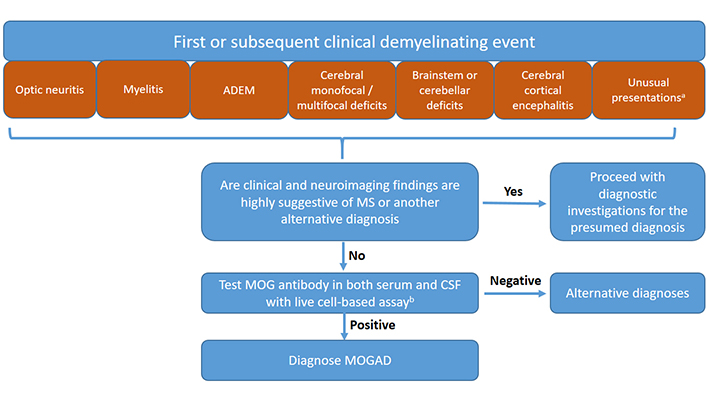
Diagnostic algorithm for the diagnosis of MOGAD in children. a Although the International MOGAD Panel has recommended MOG antibody testing in patients presented with six core clinical phenotypes [3], there is growing evidence that the clinical spectrum is not limited to these six clinical presentations, but includes and probably not limited to some other phenotypes such as intracranial hypertension, vasculitis, or ocular flutter (see Table 1). Therefore MOG antibody testing may be suggested in these patients as well. MOG antibody testing in both serum and CSF at the time of initial evaluation may also be suggested in order not to delay diagnosis. b If there is no CSF sample at the time of initial evaluation, the test can be carried out in serum. However, if the MOG antibody is not detected in the serum sample and clinical suspicion persists, the test should be repeated in the CSF sample. Fixed CBAs can be used as an alternative to live CBAs when the latter is unavailable. However, if the fixed CBA test is negative and clinical suspicion persists, the test should be repeated with live CBAs. Results of live or fixed CBA tests can be interpreted according to International MOGAD Panel criteria [3] (See also section “Interpretation of MOG antibody test results”)
Data on recurrence risk and prognosis of patients with MOGAD are highly variable and contradictory, as follow-up durations are highly heterogeneous in studies. Although previous studies with relatively short follow-up periods reported a monophasic course in most of the MOGAD patients, recent studies with longer follow-up periods demonstrated a multiphasic course in more than half of the patients [96]. However, it is not recommended to start maintenance therapy after the first clinical episode, to avoid unnecessary maintenance therapy in patients who may show a monophasic course [85]. Anti-MOG antibodies persist in more than half of the patients (50–79%), more frequently in relapsed patients [11, 14, 16, 37]. The risk of relapse is higher in those patients who remain seropositive at follow-up than in those who turn to seronegative [8, 11, 37]. In summary, more than half of pediatric MOGAD patients have antibody persistence and the risk of relapse is much higher in these patients. However, relapses may not occur in a significant proportion of patients who show antibody persistence, and relapses may occur in some patients who turn seronegative. Clinical phenotype appears to be the most important determinant of prognosis. While the outcome is worse in those patients with spinal cord involvement, it appears better in those with optic nerve involvement. However, no clinical findings were found to be associated with an increased risk of relapse [16]. In a recent study in which 61 adult and pediatric MOGAD patients were followed for at least eight years: 95% had relapses, 26% of them had relapses in the first year; 91% of all patients had at least one ON attack; the median EDSS score was 1 at the last examination, ≥ 3 in 38% of patients and ≥ 6 in 13%; bilateral blindness developed in 6%; and permanent catheterization was required in 13% of patients [97]. ADEM-like clinical presentation can cause cognitive and behavioral problems and epilepsy, especially in younger children. Cerebral involvement is more common in patients under the age of 11, while the optic nerve is more commonly affected in patients over the age of 11 [66]. In one study, moderate-to-severe deficits were reported in 15% of the patients at the end of a mean follow-up of 42 months, while in another study reporting the results of a 61-month follow-up, sequelae were reported in 58% of pediatric patients, and this rate was higher in those presenting with myelitis [15, 98]. Those patients with ADEM-like relapses and leukodystrophy-like cerebral involvement and those with diffuse cortical encephalitis were reported to have a worse outcome [15]. Although these findings show that the long-term prognosis is good in most of the patients, disability related to vision and bladder function is not uncommon. In summary, relapses occur in about half of children diagnosed with MOGAD, and permanent sequelae include blindness, loss of ambulation, genitourinary, and cognitive deficits in the long term.
The accurate and early diagnosis of CNS demyelinating diseases, which are classified under four groups MS, MOGAD, AQP4 seropositive NMOSD, and seronegative demyelinating syndromes, is essential for appropriate management and better outcomes. Although the diagnostic criteria for MS and AQP4 seropositive NMOSD have been extensively discussed and validated, the formal diagnostic criteria for MOGAD have just been established and are open to discussion [3]. Current diagnostic criteria recommend only serum MOG antibody testing in the presence of typical clinical presentations of MOGAD and suggest testing in CSF if no antibodies are detected in the serum but clinical suspicion persists [3]. However, this approach may cause a delay in the diagnosis and appropriate management in a subset of the patients. Because of these concerns, anti-MOG antibody testing in both serum and CSF may be recommended in all patients with any CNS demyelinating presentations at initial evaluation, unless clinical and neuroimaging findings are highly suggestive of MS or another alternative diagnoses.
ADEM: acute disseminated encephalomyelitis
AQP4: aquaporin-4
CBAs: cell-based assays
CNS: central nervous system
CRION: chronic relapsing inflammatory optic neuritis
CSF: cerebrospinal fluid
ELISA: enzyme-linked immunosorbent assay
IgG: immunoglobulin G
M-ADEM: multiphasic acute disseminated encephalomyelitis
MOG: myelin oligodendrocyte glycoprotein
MOGAD: myelin oligodendrocyte glycoprotein antibody-associated disease
MRI: magnetic resonance imaging
MS: multiple sclerosis
NMOSD: neuromyelitis optica spectrum disorder
OCBs: oligoclonal bands
ON: optic neuritis
TM: transverse myelitis
The author would like to thank all his colleagues for their help in reviewing the literature on follow-up of patients.
ÜY: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Validation.
The author declares that he has no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Fatemeh Afrashteh, Rayan Rajabi
Amanda C. Y. Chan ... Joy Vijayan
Grace YY Chia ... Benjamin YQ Tan
Sathish Selvam, Velpandi Ayyavoo
