
Abstract
Stroke, one of the leading causes of global morbidity and mortality, results from disrupted cerebral blood circulation, leads to cellular damage or death. Ischemic stroke, the predominant subtype, relies mainly on recombinant tissue plasminogen activator (rtPA) and endovascular thrombectomy for the treatment. Neurological impairments following ischemic stroke highlight the importance of understanding the interplay between neuroinflammation and neurogenesis in brain repair. Research reveals a complex relationship, where inflammation both promotes and hinders neurogenesis, impacting post-stroke outcomes. The subventricular zone (SVZ) of striatum and sub granular zone (SGZ) in hippocampus play pivotal roles in adult neurogenesis, with distinct characteristics and functions. SVZ neurogenesis involves neuroblast progenitors migrating to the olfactory bulb, while SGZ facilitates granule cell generation for hippocampal function. Understanding the intricate processes of neuroinflammation, neurogenesis, and angiogenesis is crucial for developing effective stroke therapeutics. Promising avenues include drug therapy, selective serotonin reuptake inhibitors, antibody therapy, angiogenesis stimulation, growth factor therapy, hormone therapy, miRNAs, extracellular vesicles, and neuroprotective agents. Stem cell therapy, exploring various cell types, holds potential for neuronal replacement and recovery. In conclusion, deciphering the roles of SVZ and SGZ in neurogenesis, unraveling the complexity of neuroinflammation’s impact on repair, and exploring diverse therapeutic approaches highlight the need for comprehensive investigations to enhance stroke outcomes. The multifaceted landscape of stroke therapeutics presents challenges, but ongoing research offers promising avenues for bridging the gap between preclinical findings and clinical treatments.
Keywords
Neurogenesis, cerebral ischemia, neuroinflammation, therapeuticsIntroduction
Stroke is among the leading causes of morbidity and the second most common cause of mortality. The treatment of ischemic stroke, the sole medication sanctioned by the US Food and Drug Administration (FDA) is recombinant tissue plasminogen activator (rtPA). Additionally, endovascular thrombectomy is employed as a mechanical intervention during acute ischemic stroke (AIS). Following an ischemic stroke, neurological symptoms may encompass challenges in maintaining balance, deficits in motor coordination, sudden onset of unilateral numbness and vision impairment, along with a decrease in reflexes and muscle strength. The cell death induced by ischemic stroke is attributed to the dysregulation of metabolic processes and the activation of glutamate receptors, primarily caused by alterations in ion distribution. This, in turn, triggers an increase in intracellular calcium concentration, ultimately leading to cellular death [1]. Research has identified targets for therapeutics to increase post-stroke neurogenesis and neuroplasticity, yet few effective strategies exist for promoting neurological recovery after a stroke. Neurogenesis in adult brains was recognized in the 1990s, with ongoing investigations focusing on various brain niches. Utilizing neurogenesis as a therapeutic target holds promise for promoting self-repair and recovery from stroke damage, but challenges remain due to intricate interactions with neuroinflammation and angiogenesis. Understanding the roles of immune cells, cytokines, and chemokines in the post-stroke environment is crucial, considering their time-dependent expression patterns and effects on brain recovery mechanisms [2]. In context of ischemic stroke, there is a significant increase in neurogenesis within two specific regions of the brain: the subventricular zone (SVZ) [3] located in the lateral ventricular wall (LV) and the sub granular zone (SGZ) situated in the dentate gyrus. The SVZ serves as a secondary proliferative area housing neural progenitor cell. Within the ventricular zone (VZ), one can find radial glial cells, which are the primary neural stem cells (NSCs) in the brain and spinal cord [4]. This area is termed the VZ due to its lining of the inner developing ventricles. During the process of adult SVZ neurogenesis, neuroblast progenitors, which eventually develop into interneurons, migrate to the olfactory bulb (OB) via the rostral migratory stream. Interestingly, the SVZ also appears to play a role in the generation of astrocytes following a brain injury [5, 6]. A research study highlights the significant contributions of environmental enrichment to adult neurogenesis, with enriched living environments doubling the total number of surviving newborn cells. Moreover, the differential effects of various behavioral protocols on neurogenesis and highlights the rapid onset of these effects. The importance of considering specific behavioral interventions in elucidating the mechanisms underlying adult hippocampal neurogenesis and its implications for brain health and cognitive function has been well studied [7]. Uncertainties exist concerning the mechanisms driving the neurological and functional recovery following an ischemic stroke. Stroke victims’ prospects have not significantly improved despite recent improvements in our understanding of the fundamental repair processes of the central nervous system (CNS). To develop novel therapeutic techniques, a deeper knowledge of the mechanisms underpinning neurological recovery following a stroke is required. The recovery from a stroke is mostly mediated by angiogenesis and neurogenesis, which may be targeted for therapeutic intervention. Concepts of brain angiogenesis and neurogenesis at the tissue and cellular levels have been developed into frameworks [8]. Post-ischemic stroke neuroinflammation, distinct from systemic inflammation, involves active resolution processes mediated by arachidonic acid derivatives. Understanding inflammation’s impact on NSCs is crucial for effective brain repair [9]. Neuroinflammation plays a role in both secondary brain injury and neurorepair post-stroke, affecting neurogenesis [10]. Inflammation’s effects on neurogenesis are complex, with both positive and negative impacts, influencing CNS injury and repair [11].
Understanding the human SVZ and the hippocampal SGZ can advance stroke research and therapies [12]. The SGZ facilitates hippocampal neurogenesis, guiding NSCs through a lineage to granule cells involved in cognitive functions [5]. Radial astrocytes in the SGZ serve as both glial cells and NSCs, while horizontal astrocytes lack radial processes and do not generate neural progenitors [13–16]. Astrocytes, with their close interactions with various cell types, play a crucial role in the process of neurogenesis within the SGZ. Neuroblasts, which originate from the neurogenic processes in the SVZ and SGZ, undergo migration to different regions of the brain by traveling through blood vessels formed because of angiogenesis. The coordination between angiogenesis and neurogenesis is highly significant. Angiogenesis is the natural process responsible for the development of new blood vessels from existing ones that were initially formed during vasculogenesis in prior stages [16–18]. This process, achieved through sprouting and division, ensures the continuous growth of the vasculature [19]. Chemical signals within the body play a crucial role in influencing angiogenesis. Some of these signals interact with receptors on the surface of regular endothelial cells, such as vascular endothelial growth factor (VEGF). When VEGF and other endothelial growth factors bind to their respective receptors on endothelial cells, it triggers messages that promote the formation and survival of new blood vessels. The intricate relationship between angiogenesis and neurogenesis is also explored in the review.
Stroke therapeutics present a diverse field with investigations exploring various approaches. Drug therapy, including agents like amphetamine and levodopa, holds potential in stroke recovery, but uncertainties persist regarding optimal dosage and timing [20]. Selective serotonin reuptake inhibitors (SSRIs), such as fluoxetine, show notable motor recovery benefits post-stroke. Antibody therapy, utilizing monoclonal antibodies like GSK249320, initially demonstrated promise in enhancing gait velocity, but subsequent trials produced mixed results. Angiogenesis stimulation, involving VEGF and induced pluripotent stem cell (iPSC)-derived cells, aims to promote new blood vessel growth [21]. Growth factor therapy, using substances like brain-derived neurotrophic factor (BDNF) and granulocyte-colony stimulating factor (G-CSF), exhibits potential in preclinical studies, despite challenges such as limited CNS penetration. The role of hormone therapy in stroke recovery remains underexplored, while miRNAs and extracellular vesicles, particularly from mesenchymal stem cells (MSCs), show promise in controlling gene expression for neuroprotection [22]. Neuroprotective agents, targeting excitotoxicity and apoptosis, are under investigation, with some demonstrating efficacy in specific subpopulations, especially those undergoing mechanical thrombectomy. Stem cell therapy explores various cell types, including marrow stromal cells and iPSCs, with potential benefits in replacing damaged neurons and promoting recovery. However, challenges like cell viability and differentiation persist. In essence, stroke therapeutics involve a complex interplay of pharmacological and biological interventions, with ongoing challenges in translating promising preclinical findings into effective clinical treatments [23].
Understanding cerebral stroke-induced neurogenesis: factors, initiation, glial response, and neural plasticity
Cerebral stroke induced neurogenesis
Ischemic strokes prompt neurogenesis, the generation of new neurons, in the infarcted area and its neighboring regions. These NSCs originate from two key locations: the SVZ within the striatum and the SGZ in the dentate gyrus. This process is vital for repairing the damaged brain area and facilitating post-stroke recovery [24, 25]. Subsequently, these freshly generated NSCs migrate to and become integrated into the infarcted and peri-infarct areas, where they later mature into functional neurons [26]. In the context of post-stroke functional recovery, the long-term survival of these newly generated neurons holds significant importance. Past research has indicated a decrease in the viability of these nascent neurons, potentially attributed to a deficiency in trophic factors and the presence of persistent inflammatory responses [27, 28]. However, it’s noteworthy that acute neuroinflammation has been observed to assist in neurogenesis and enhance the survival of neurons [29, 30]. Following an ischemic stroke, individuals frequently experience significant neurological impairments. Therapeutic interventions aim to either reduce neuronal loss in the penumbra, where cell death is delayed, or replace dead neurons in the ischemic core. Presently, stem cell therapy stands out as a leading strategy for fostering neurogenesis and facilitating neural repair post-stroke. Stem cell treatments encompass both the transplantation of external stem cells and the stimulation of endogenous NSCs to proliferate and differentiate into neural cells [31]. Moreover, age stands out as a prominent factor influencing neurogenesis, with the rate of neurogenesis gradually diminishing as individuals grow older [24, 32, 33]. Recent findings have shown that hippocampal neurogenesis is impaired in aged animals compared to their younger counterparts, whereas striatal neurogenesis after a stroke appears similar in both young and aged mice. This observation raises the intriguing possibility of distinct region-specific regulatory mechanisms and multiple potential avenues for modulation if these mechanisms can be harnessed [24].
Factors controlling neurogenesis
The modulation of neurogenesis in the post-stroke cerebral milieu is orchestrated by a myriad of factors. Typically, neurogenesis unfolds through three distinct stages: the proliferation of NSCs [22], the migration of neuroblasts and immature neurons [23], and their subsequent differentiation into mature neurons and extension of neurites [34–36], ultimately leading to the formation and stabilization of synapses. It is crucial to emphasize that the molecules governing these stages exhibit distinctions between embryonic neurogenesis and adult neurogenesis [31]. Notably, fibroblast growth factor-2 (FGF-2), insulin-like growth factor-1 (IGF-1), BDNF [25–28], and VEGF [32] are factors directly impacting the proliferation of NSCs [32]. Conversely, factors such as stromal-derived factor (SDF-1), monocyte chemoattractant protein (MCP-1), and matrix metalloproteinases (MMPs, MMP-2, 3, 9) play roles in influencing the migration of neuroblasts [33]. SDF-1 and MCP-1 serve as chemokines integral to the inflammatory response following ischemic injury, while MMPs function as MMPs involved in the extracellular matrix remodeling process [37, 38]. This process of tissue remodeling appears to be, at least in part, facilitating the migration of neuroblasts. Additionally, the degradation of matrix proteins, exemplified by perlecan, has been posited as a potential driver of neurogenesis in post-stroke cerebral contexts [39]. Within the infarct and peri-infarct regions, the C-terminus of perlecan domain V is implicated in both neurogenesis and angiogenesis [40]. In vitro studies employing neurospheres and fetal cortical neurons have elucidated the capacity of perlecan DV to foster differentiation into mature neurons and promote neurite extension. This underscores a plausible dual role for MMPs in this context [41–48].
The elevated neurogenesis observed in the dentate gyrus subsequent to a stroke is attenuated in aged rats, suggesting an age-dependent diminishment of this potential mechanism for ameliorating cognitive deficits. Conversely, striatal neurogenesis is comparably induced in both youthful and aged subjects. The functional significance of the additional striatal neurons in contributing to post-stroke behavioral recovery remains uncertain. Nevertheless, the sequential stages of striatal neurogenesis operate in animals commensurate with the temporal occurrence of strokes in humans, emphasizing the prospect of targeting endogenous brain repair mechanisms as a prospective therapeutic avenue to augment functional recuperation in stroke patients [49]. External interventions, such as rehabilitative training, introduce an additional level of intricacy by autonomously influencing the process of adult neurogenesis. Besides these findings, exercise significantly contributes to triggering neurogenesis. Additionally, research indicates that one week and four weeks of voluntary running exercise can alleviate anxiety-like behaviors in vascular dementia rats. It is hypothesized that this effect may be linked to the neuroprotective impact of exercise, which helps sustain serotonin (5-HT) levels in the hippocampal brain regions affected by vascular dementia. Studies have demonstrated that physical exercise is an effective method for decreasing damage and enhancing tissue tolerance to ischemia. Currently, two prevalent forms of exercise intervention for ischemic stroke are voluntary exercise and forced exercise [50–52].
Initiation of neurogenesis
Progenitor cells situated in the lateral ventricle and dentate gyrus of murine subjects possess the capability to induce cellular population proliferation throughout the rodent’s lifespan subsequent to ischemic events [24]. Under normal physiological conditions, approximately 10% of the cell population in the SVZ of adult mice comprises proliferating cells [50]. The enduring proliferative capacity of progenitor cells within the lateral ventricle’s SVZ and the dentate gyrus persists over the rodent’s lifespan [53, 54]. However, despite extensive research into the composition, proliferation, migration, and differentiation of cells within the SVZ and dentate gyrus, there is limited evidence concerning the proliferation of these cells in response to a stroke. Recent findings in middle cerebral artery (MCA) occlusion have revealed the following: (1) There is a noteworthy increase in the number of proliferating cells in the ipsilateral cortex, SVZ, and OB, but not in the dentate gyrus, suggesting that post-stroke cell proliferation is specific to particular areas; (2) a notable rise in bromodeoxyuridine (BrdU) immunoreactive cells in the SVZ corresponds with a substantial increase in BrdU immunoreactive cells in the OB, indicating that proliferating cells migrate from the SVZ to the OB; (3) the presence of proliferating progenitor cells in the adult cortex is suggested by clusters of BrdU-labeled cells in areas of the cortex distant from the infarction [55, 56].
The augmented expression of polysialylated-neural cell adhesion molecule (PSA-NCAM), a recognized marker denoting migrating neurons in the ipsilateral hemisphere, manifests a significant elevation, indicative of the migration of proliferating neurons toward the infarcted region [57]. Within both the cortex and subcortex, a mere 6% of BrdU-labeled cells exhibit positivity for glial fibrillary acidic protein (GFAP), while none demonstrates expression of neuronal protein markers such as NeuN and MAP2. This observation suggests the absence of regenerative processes in these regions following focal ischemia. Investigations reveal a substantial amplification in the proliferating cell population within the adult brain subsequent to focal cerebral ischemia [24, 58]. BrdU labeling data unveils a notable surge in the count of BrdU-labeled cells in the ipsilateral hemisphere subsequent to embolic blockage of the MCA, persisting for a minimum of 14 days but diminishing to 90% after a lapse of 4 weeks. The dearth of BrdU staining is attributed to cell division-induced dilution of the BrdU label below detectable thresholds and apoptosis-induced attrition of these cells, elucidating the decline in BrdU-labeled cell counts [59, 60]. These transient increments in BrdU-labeled cells align with evidence from instances of global cerebral ischemia, indicating a pronounced peak in BrdU immunoreactive cells 14 days post-ischemia, with proliferating cell numbers reverting to control levels after 35 weeks [61]. The discernment of a temporal window for endogenous brain plasticity is underscored by transient increments in proliferating progenitor cells in the injured brain. Manipulation of progenitor cell proliferation, migration, and differentiation within this timeframe holds the potential to augment brain plasticity and contribute to enhanced outcomes in stroke therapy. Experimental corroboration substantiates this proposition, demonstrating heightened functional recovery upon implantation of embryonic neocortical cells into the ischemic brain one-week post-MCA occlusion. Conversely, grafting remains efficacious three weeks post-MCA blockage, albeit with limited functional improvement at that juncture [62]. In contrast to cases of global cerebral ischemia, which primarily trigger significant increases in BrdU-labeled cells within the dentate gyrus, focal cerebral ischemia results in notable increases in BrdU-labeled cells in various regions, including the ipsilateral cortex, subcortex, SVZ, and OB, but not in the dentate gyrus [63]. These findings suggest that in response to brain damage, regional progenitor cells exhibit varying patterns of cellular proliferation and differentiation. While it’s widely recognized that focal ischemia leads to gliosis in cortical and subcortical regions, little is known about the extent of cell proliferation in the SVZ. Recent discoveries have indicated a significant increase in the number of BrdU-labeled cells in the ipsilateral SVZ following MCA blockage, implying that focal cerebral ischemia promotes the growth of progenitor cells.
During the embryonic phase, the VZ and the SVZ are responsible for generating a significant portion of the neurons and glial cells in the forebrain [64, 65]. Neuroepithelial cells within the VZ undergo differentiation into ependymal cells before birth, while the SVZ retains multipotent progenitor cells that continue to generate neurons and glial cells throughout an animal’s life. Recent research has also demonstrated that cells born in the SVZ engage in chain migration along the rostral migratory stream to the OB, where they differentiate into granule and periglomerular neurons under normal physiological conditions [66]. In a study, a notable increase in the quantity of BrdU immunoreactive cells within the SVZ aligns with a significant rise in the number of BrdU-labeled cells in the OB. This suggests that the heightened presence of BrdU-labeled cells in the SVZ, induced by focal cerebral ischemia, contributes at least in part to the increased number of BrdU-labeled cells in the OB. Although the migration of progenitors from the SVZ to the forebrain typically ceases by the second week after birth [67], there remains a possibility that some of the BrdU-labeled cells observed in the ipsilateral cortex and subcortex following ischemia originate from SVZ cells. The presence of BrdU-labeled cells within the ipsilateral ependymal layer of the brain indicates that ependymal cells undergo proliferation in response to focal cerebral ischemia. This aligns with prior findings suggesting ependymal proliferation following various brain injuries [67]. The inquiry arises as to whether these proliferating ependymal cells exhibit stem-cell-like characteristics [68]. While Johansson et al. [37] assert that the ependyma is the in vivo source of NSCs, a more recent study suggests that both the SVZ and the ependymal layer exhibit proliferative capacity, with only SVZ cells displaying NSC attributes [69]. Focal cerebral ischemia also induces gliosis [70–72]. Consistent with this, studies indicate that one week after MCA occlusion, the transplantation of embryonic neocortical cells into the ischemic brain can enhance functional recovery [73].
Glial response and changes in neural plasticity
Neuroglia manifests in three forms: astrocytes, oligodendrocytes, and microglia. Following brain injury, mature astrocytes and oligodendrocytes retain proliferative capacity [74]. Microglial activation stands as a pivotal event in the pathogenesis of various CNS disorders, notably cerebral ischemia. Activated microglia are known to elicit proinflammatory responses within the ischemic brain, presenting a potential therapeutic target. However, it is crucial to recognize that microglia also serve important anti-inflammatory functions and contribute to the synthesis of growth factors crucial for neuroprotection and post-ischemic healing. Notably, microglia can directly bolster neuroprotection by enhancing functional outcomes and curtailing the expansion of ischemic infarcts. Furthermore, in the ischemic milieu, microglia indirectly promote anti-inflammatory processes, neurogenesis, and angiogenesis—integral components of the pathophysiological cascade underlying ischemic recovery [75–77]. Cortical multipotent progenitors require specific environmental cues for differentiation into oligodendrocytes, astrocytes, and neurons in vivo. Despite the undetermined fate of these progenitors, they hold potential to generate neurons post-ischemia with appropriate signaling [78–80]. Microglia, astrocytes, and oligodendrocytes represent prominent glial cell populations prevalent in the peri-infarct milieu of the CNS, exerting regulatory roles over the immune system subsequent to stroke events. However, emerging research underscores the dualistic nature of glial cells, capable of exerting both beneficial and detrimental effects during ischemic stroke episodes. Prompt activation of microglia, responsible for monitoring CNS equilibrium and modulating innate immune responses, characterizes the initial response to ischemic insult. Yet, the release of inflammatory cytokines by activated microglia contributes to neuronal tissue damage in this context. Conversely, the reparative phase post-stroke is facilitated by alternatively activated microglia, which secrete neurotrophic factors and anti-inflammatory cytokines, aiding in tissue healing. Reactive gliosis and astrocyte activation, pivotal processes in ischemic stroke, play crucial roles in reinstating CNS homeostasis and mitigating brain injury. However, glial scarring consequent to these processes impedes neural reintegration and expansion, posing challenges to neurological recovery [81–84].
In addition to glial response, neuroinflammation, neurogenesis, and apoptosis, alterations in synaptic plasticity represent another molecular mechanism triggering the recovery mechanism post-cerebral ischemia. Post-cerebral ischemia, synaptic plasticity undergoes molecular alterations, crucial for triggering the recovery mechanism. Stroke disrupts both the structural and functional integrity of the brain, impacting various cerebral regions and causing widespread dysfunctions within neural networks. Although understanding of stroke pathophysiology has significantly advanced, effective therapy remains limited, especially in the subacute or chronic phase. Several novel therapies have emerged to enhance clinical outcomes by promoting brain plasticity. These approaches, which include targeting brain remodeling and restoration or constructing a neural bypass to circumvent brain injury, hold potential significance. Innovative concepts fostering and harnessing plasticity within the unaffected hemisphere, such as brain-computer interface (BCI) and contralateral seventh cervical nerve transfer (CC7), offer promising avenues for stroke management. Advancements and refinement of these state-of-the-art methodologies could provide valuable contributions to stroke treatment [85, 86]. Apparently there exists a plethora of research findings supporting the link between microglial activation and synaptic changes. It is still unclear, though, whether post-ischemic activation of microglial cells has positive or negative effects, especially with regard to long-term brain plasticity processes. It has been attempted to ascertain the degree of microglial cell involvement in specific neural plasticity, neurite outgrowth, and synaptogenesis processes by manipulating the post-stroke inflammatory response. Given that microglia are a source of neurotrophic factors, an attempt was also made to identify BDNF as a potential molecular player in these processes [87]. Activated microglia were traditionally viewed as detrimental after stroke due to their release of inflammatory cytokines. Modulating microglial activation to reduce neuroinflammation has been a longstanding approach for mitigating stroke injuries. However, beyond their inflammatory role, microglia also exhibit neuroprotective functions post-stroke, such as phagocytosis of cellular debris and secretion of growth factors. Understanding how these mechanisms can be effectively harnessed for stroke treatment requires further investigation.
Microglial polarization is another crucial aspect to explore. Investigating the mechanisms underlying microglial activation and polarization is essential for optimizing stroke therapy strategies. Furthermore, microglia interact with various CNS cells and modulate their function, influencing a wide array of physiological processes. Studying these intercellular communication pathways will enhance our understanding of stroke pathology and other CNS disorders. Astrocytes have emerged as potential targets for stroke treatment due to their survival and regulatory roles post-stroke. Although previous studies have explored anti-inflammatory agents and astrocyte modulators, none have shown significant advantages over placebo. The heterogeneity of reactive astrocytes post-stroke and their precise roles in neuroinflammation require further elucidation through transcriptome analysis. Oligodendrocytes play a significant role in patient recovery as major CNS glial cells. Understanding the regulation of oligodendrocyte migration, proliferation, and differentiation post-stroke, as well as their interactions with immune cells and other CNS components, is crucial for promoting survival and enhancing axonal regeneration. The immune role of oligodendrocytes post-stroke warrants further investigation, particularly regarding their potential as targets for immunotherapy [88, 89].
Interplay between neuroinflammation mediated neurogenesis and angiogenesis
Cerebral ischemia-induced neuroinflammation plays a significant role in causing damage to the brain. The effects of neuroinflammation are both beneficial and harmful, depending on the spatial-temporal context [90]. Microglia become highly activated almost immediately upon the onset of an ischemic attack, contributing to ischemia-reperfusion injury by releasing inflammatory factors. The activated microglia exhibit a diverse inflammatory phenotype based on the affected region and the severity of the ischemic attack. The M1 physiological state of microglia triggers a proinflammatory response, while the M2 state releases anti-inflammatory cytokines. Although proinflammatory factors contribute to tissue repair by enhancing phagocytic activity, excessive release of M1-mediated proinflammatory factors leads to the upregulation of inducible nitric oxide synthase (iNOS), damaging neurons and disrupting the blood-brain barrier. Studies using rodent models of ischemic stroke have shown a notable increase in the release of proinflammatory cytokines, particularly interleukin-1β (IL-1β), tumor necrosis factor alpha (TNFα), and IL-6. On the other hand, the anti-inflammatory response by M2 provides neuroprotection through tissue repair and regeneration. M2 regulates the VEGF and hypoxic-inducible factor-1α (HIF-1α) signaling pathway, promoting angiogenesis.
There exists a strong coordination between the post stroke neurogenesis and angiogenesis (Figure 1). Neuroblasts from the SVZ migrate towards the region of ischemic lesion, where post-stroke angiogenesis initiates and this movement of neuroblasts is through the cerebral vessels. The neovascularisation process requires cooperative role of angiogenic growth factors and inhibitors. The major angiogenic growth factor is VEGF-A, the level of which increases post stroke. The bioavailability of VEGF-A is further regulated by VEGF-B. The contractile elements surrounding endothelial cells are called as pericytes that integrate information from different brain cells and maintain efficient brain homeostasis. Signal transducer and activator of transcription 3 (STAT3) also functions as a transcriptional activator of VEGF under the conditions of reduced oxygen availability. It has a crucial role in regulating the post-stroke angiogenesis. The Janus kinase 3 (JAK2)/STAT3 signaling pathway has the tendency to polarize microglia towards M2 phenotype. Further the inhibition of STAT3-dependent autophagy has been shown to promote angiogenesis after ischemic attack [91]. The phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) pathway reduces the expression levels of inflammatory genes and is essential for angiogenesis since it activates HIF-1α, which in turn regulates VEGF. It has been suggested in a number of studies that plasminogen has a role in the process of post-stroke angiogenesis but the mechanistic purview is not completely understood. In a recent study, investigations were done to understand the possible mechanisms through which plasmin/plasminogen mediates the process of angiogenesis and related functional recovery post-stroke. The MCA occlusion (MCAO) mode of mice used in this study revealed that Plg−/− mice exhibited remarkably reduced recovery and contracted vessel density in the peri-infarct region of ipsilesional cortex as compared to Plg+/+ mice. Neuroblasts migrate to the infarct region from SVZ/dentate gyrus through cerebral vessels regenerated through post-stroke angiogenesis [92]. Migrating neuroblasts are localized specifically in active vascular sprouting regions of blood vessels.
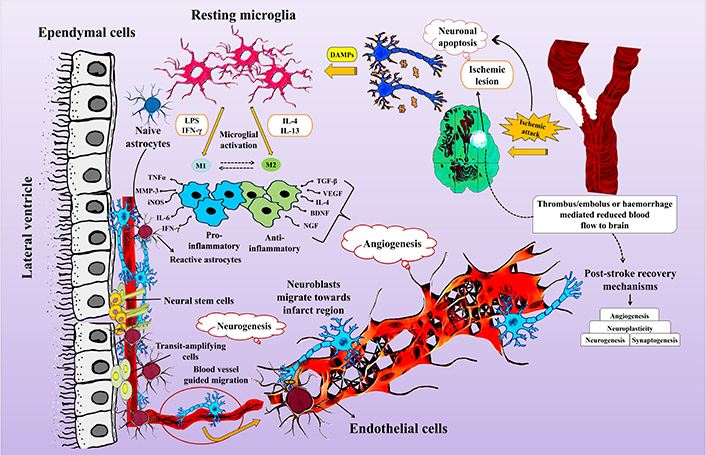
Interplay between neurogenesis and angiogenesis post stroke. BDNF: brain-derived neurotrophic factor; DAMPs: damage-associated molecular patterns; IFN-γ: interferon-gamma; IL-4: interleukin-4; iNOS: inducible nitric oxide synthase; LPS: lipopolysachharide; MMP-3: matrix metalloproteinase-3; NGF: nerve growth factor; TGF-β: transforming growth factor-β; TNFα: tumor necrosis factor alpha; VEGF: vascular endothelial growth factor
Ischemic brain damage induces inflammation, both locally and systemically, commonly referred to as sterile inflammation since it does not involve a pathogenic agent. The migration of pericytes is crucial for the branching and neogenesis of microvessels during angiogenesis. Ischemia activates astrocytes, leading to the secretion of pro-inflammatory cytokines and chemokines, triggering the release of MMPs. MMPs cause damage to the blood-brain barrier, increasing leukocyte infiltration from the blood into nervous tissue [93, 94]. Research indicates a decrease in pro-inflammatory monocytes over time, accompanied by an increase in anti-inflammatory monocytes, resulting in reduced pro-inflammatory mediators like chemokine (C-Cmotif) ligand 2 (CCL2) and an increase in anti-inflammatory factors such as transforming growth factor-β1 (TGF-β1). Assessing post-stroke angiogenesis involves measuring microvessel density in the infarct region. Both microvessel density and macrophage numbers increase over time following cerebral ischemia. Inhibition of post-ischemic angiogenesis within the first week leads to a significant decrease in neurogenesis, indicating a functional link between the two processes [95]. Studies report neuroblasts migrating toward newly formed blood vessels 16 weeks after stroke, highlighting the importance of angiogenesis in facilitating long-term neurogenesis [92, 94]. Investigations into stroke-induced rat brain endothelial cells (RBECs) show their impact on SVZ cell proliferation and differentiation, contributing to understanding the coupled regulatory mechanism of angiogenesis and neurogenesis post-stroke [96–98]. Ischemic stroke activates endothelial cells, leading to the secretion of SDF-1α and MMPs. SDF-1α facilitates neuroblast migration toward the peri-infarct region, while MMPs guide neuroblast migration to the infarct region by digesting the extracellular matrix [99]. Endothelial cells activated in the ischemic infarct region also secrete VEGF, triggering neurogenesis [100]. Overall, neurogenesis and angiogenesis are highly coordinated and mutually supportive processes that enhance brain repair after ischemic stroke injury.
Therapeutic interventions
Many post-stroke restorative therapy subcategories are currently being researched [101–105], many of which are being tested on humans. Most focus on a single agent or intervention, and combination therapies are expected to get more attention as we learn more about monotherapies. As mentioned above, some restorative therapies are started days or weeks after a stroke starts and engage with the body’s natural neurological repair mechanisms, while others are started months or years later.
Drug therapy
Numerous studies have examined the potential of small compounds to enhance stroke recovery, owing to their ability to penetrate the blood-brain barrier. Repurposed medications, particularly monoaminergic drugs like amphetamine, have received considerable attention. Investigations, including the Subacute Therapy with Amphetamine and Rehabilitation for Stroke (STARS) study, have explored their efficacy in stroke recovery through controlled experiments [106]. However, challenges persist in determining optimal dosage and timing for amphetamine administration, underscoring the necessity for further research to harness the potential of small compounds in stroke rehabilitation [106].
Dopamine, a critical neurotransmitter regulating various brain functions, particularly motor control and synaptic plasticity, plays a significant role in stroke recovery. Studies have demonstrated the effectiveness of dopaminergic medications, such as levodopa, in improving motor outcomes post-stroke. Genetic variability may influence treatment response, offering potential benefits of personalized medicine. However, trials involving other dopamine agonists like ropinirole have yielded mixed results [107–110].
Serotonin, another crucial neurotransmitter, contributes to brain regeneration and stroke recovery. SSRIs have shown promise in enhancing motor outcomes after stroke, emphasizing their potential therapeutic role in stroke rehabilitation. Further research is needed to elucidate the precise mechanisms and optimize treatment strategies utilizing these neurotransmitter systems [111–130]. The Fluoxetine for Motor Recovery after Acute Ischemic Stroke (FLAME) study provides compelling evidence for the efficacy of SSRIs in enhancing post-stroke outcomes [123]. In this double-blind [131], placebo-controlled trial, non-depressed individuals with hemiplegia/hemiparesis occurring five to ten days after an ischemic stroke were randomized to receive either oral fluoxetine (20 mg/day) or a placebo. Over three months, those assigned to fluoxetine demonstrated significantly greater improvements in the arm/leg Fugl-Meyer motor score compared to the placebo group (P = 0.003), despite a small, nonsignificant difference in baseline scores favoring the SSRI-treated group. Limited research has explored utilizing norepinephrine transmission as a therapeutic target for stroke recovery. Noradrenergic neurotransmission typically enhances overall neuronal activity, elevates general excitability, and amplifies responses to unexpected stimuli [132]. Although few studies have investigated noradrenergic medications for stroke recovery, preliminary results have been promising [133–135]. Acetylcholine plays a crucial role in facilitating plasticity by selectively augmenting expected inputs and reducing unexpected inputs [132]. While muscarinic cholinergic receptors are essential for cognitive flexibility, changes in nicotinic cholinergic neurotransmission affect attention [106]. Experimental stroke studies in rodents and primates have supported the role of acetylcholine in stroke recovery [136–142], although limited data are available from human stroke patients [143]. Nonetheless, early research findings suggest promising potential for cholinergic therapy in enhancing recovery from brain injury [139]. Limited yet encouraging data exist on the non-motor aspects of stroke recovery, with ongoing trials exploring the safety and efficacy of drugs influencing GABA or glutamate receptors, as well as phosphodiesterase type 5 inhibitors like sildenafil, in human stroke survivors [144–150].
Antibody therapy
Investigations into the potential of promoting neural repair through substantial biological molecules, such as monoclonal antibodies, have been ongoing. Monoclonal antibodies selectively bind to specific targets, such as receptors or cell surface indicators, thereby modulating the activity of particular signaling cascades. This innovative approach has significantly impacted patient care across diverse diseases, encompassing neoplastic, immunological, and other disorders. In the realm of post-stroke brain healing, monoclonal antibodies are utilized to counteract chemicals inhibiting growth in the CNS, with the goal of fostering a more conducive environment for growth. Although axonal growth is well-established in the peripheral nervous system [150], the CNS grapples with challenges posed by three major inhibitors—myelin-associated glycoprotein (MAG), oligo-myelin glycoprotein, and Nogo-A—that curtail the permissiveness of the growth environment. Further complicating matters is the upregulation of these molecules following the onset of a stroke [151].
Facilitation of axonal development can be achieved through the application of a monoclonal antibody designed to block inhibitory substances [152, 153]. In a recent trial, 42 stroke patients were randomly assigned to either a placebo or one of three doses of intravenous GSK249320, a humanized IgG1 monoclonal antibody targeting MAG with a defective Fc region. Each patient received two infusions: the first administered 24 h to 72 h after the onset of the stroke, and the second nine days later. In terms of secondary endpoints, GSK249320 exhibited a trend towards improved gait velocity compared to the placebo, and no safety concerns were identified [151]. However, a subsequent phase IIb double-blind, randomized, placebo-controlled trial involving 134 patients who had experienced an ischemic stroke 24 h to 72 h prior revealed that two doses of the antibody did not outperform the placebo in increasing gait velocity [153]. Despite this, the monoclonal antibody demonstrated good tolerance and low immunogenicity, providing valuable insights for future research aiming to utilize the antibody for targeted modulation of specific biological pathways to enhance stroke recovery.
Angiogenesis stimulation
The expression of VEGF was detected in both the blood vessel wall and astrocytes of the injured brain, with heightened immune responsiveness to VEGF observed one week after implantation. These findings suggest that VEGF secretion influences plasticity and inflammation in the early stages of recovery, with a milder reaction observed eight weeks post-transplantation in older rats. Localized implantation of iPSC-derived cells expressing the VEGF receptor fetal liver kinase-1 into the ischemic region successfully promoted new blood vessel growth. Additionally, small extracellular vesicles derived from human iPSC-derived MSCs were found to protect against brain damage post-ischemic stroke by inhibiting STAT3-dependent autophagy, enhancing angiogenesis, and preventing blood vessel growth. These findings highlight the therapeutic potential of VEGF and iPSC-derived cells in promoting neurovascular regeneration and protecting against ischemic brain injury [154, 155].
Growth factor therapy
Growth factors are crucial for healthy CNS development and are essential for spontaneous neural repair through a variety of mechanisms, including angiogenesis, cell proliferation and differentiation, migration, survival and apoptosis, synaptic plasticity, and immunomodulation [156]. As a result, growth factors have a great deal of potential as a method for repairing damaged neural tissue. Explorations in preclinical stroke research have delved into the efficacy of growth factors in facilitating stroke recovery. Consistently, preclinical investigations highlight the enduring benefits of administering exogenous growth factors 24 h or more post-stroke onset, showcasing substantial improvements in long-term behavioral outcomes. Notable examples of growth factors explored in the preclinical domain include BDNF [157], a combination of epidermal growth factor and erythropoietin [60], and human chorionic gonadotropin (hCG) in conjunction with erythropoietin. Hematopoietic growth factors, known for their established safety in human applications, have been predominantly scrutinized in trials thus far. G-CSF, one such growth factor, underwent evaluation in the AX200 for Ischemic Stroke (AXIS) study. Findings revealed that administering G-CSF within 12 h of stroke onset proved safe and well-tolerated in 44 patients. A separate trial involving 60 individuals further supported the safety of G-CSF post-stroke. However, the subsequent AXIS-2 study [67], comparing the middle G-CSF dose from AXIS (135 μg/kg) with a placebo in 328 patients within 9 h of stroke onset, indicated no significant difference between G-CSF and placebo regarding the primary endpoint, modified Rankin scale score at day 90. A meta-analysis of studies examining G-CSF administration days to years post-stroke failed to reveal favorable effects [158]. Another growth factor explored for its potential in promoting brain healing is erythropoietin. Preclinical studies suggest that when administered as a sole agent post-acute injury, such as 24 h after stroke commencement, systemically delivered erythropoietin can traverse the blood-brain barrier and enhance restorative processes. Growth factors, being large proteins with limited penetration into the CNS, have prompted various strategies to overcome this challenge. One approach involves enhancing the ability of growth factors to traverse the blood-brain barrier by conjugating them with a molecular Trojan horse [159]. Another method entails inserting a gene encoding a growth factor into exogenous stem cells, a technique explored for FGF-2, glial cell line-derived neurotrophic factor, BDNF, VEGF, placenta growth factor, and hepatocyte growth factor. It’s noteworthy that, despite the restricted access to the CNS after a stroke, growth factors can still influence brain plasticity through extraneural targets, such as the immune system.
Hormone therapy
The administration of growth hormone (GH) post-ischemic stroke holds promise for improving motor and cognitive functions. GH acts as a neurotrophic factor, promoting nerve repair, regeneration, and muscle innervation while reducing atrophy. Clinical trials have demonstrated its efficacy in enhancing motor function by stimulating cell proliferation, neurogenesis, synaptic plasticity, and angiogenesis in the peri-infarct region, along with increasing IGF-1 levels. Moreover, GH therapy has been associated with cognitive improvements post-stroke, particularly through its effects on hippocampal plasticity, synaptic transmission, and memory. Testosterone levels post-stroke exhibit complex relationships with outcomes, with reduced levels associated with larger infarct size and increased stroke risk in older men. However, testosterone’s effects on stroke appear to vary by age, exacerbating injury in young adults while potentially offering protection in middle-aged and neonatal populations. Testosterone replacement therapy (TRT) remains debated, with conflicting findings regarding its cardiovascular effects and stroke risk. Estrogen and progestin therapy have shown mixed effects on stroke risk, with alternative administration routes and hormone types potentially mitigating risks. Combined estrogen and progestin treatment have demonstrated neuroprotective effects, reducing infarct size and suppressing neuronal apoptosis in animal models. Oxytocin emerges as a potential therapeutic agent for ischemic stroke, reducing brain damage and inflammation while enhancing spatial memory. Experimental studies have shown its ability to increase neurotrophic factors and decrease cell death and apoptosis in stroke models. In summary, while sex hormones exhibit complex effects on ischemic stroke outcomes, GH therapy shows promise in enhancing motor and cognitive functions post-stroke. Further research is needed to elucidate hormone-stroke interactions and optimize hormone-based therapies for stroke prevention and treatment [160–162].
miRNAs and extravascular vesicles
Stem cell therapies hold promise for promoting regeneration in traumatic brain injury (TBI) but are accompanied by various side effects, including moral and legal concerns regarding fetal embryonic stem cell use [163]. The multipotency of stem cells increases the risk of uncontrolled development and cancer, and their administration may impede microvasculature and induce immunological reactions [164]. Interestingly, evidence suggests that MSCs may confer protective effects not only through differentiation and neuron replacement but also via paracrine release of bioactive molecules or direct cell-cell interaction, promoting resident cell survival and proliferation [165]. Isolating, preparing, and maintaining stem cell viability pose significant challenges. MSCs have emerged as a promising therapeutic avenue for TBI, showing an inclination to migrate to damaged tissue sites and differentiate into neurons and glial cells [166]. This process is linked to decreased expression of inhibitory molecules for axon outgrowth, suppression of neuroinflammation, and enhanced growth factor release, leading to notable improvements in neurological functions. Remarkably, cell-free exosomes released by MSCs have emerged as potent mediators of these beneficial effects. Systemic administration of MSC-derived exosomes in a rat TBI model resulted in neurovascular remodeling, neurogenesis in the dentate gyrus, and reduced neuroinflammation [167]. Similarly, intravenously administered MSC-derived exosomes mitigated neuroinflammation while enhancing spatial and cognitive learning in mice with TBI [168]. Exosomes, with diameters between 50 nm to 200 nm, serve as intercellular signaling molecules, facilitating the transport of proteins, RNAs, miRNAs, and lipids to recipient cells through ligand-receptor binding and internalization [169].
For instance, miR-21, a non-coding miRNA abundant in exosomes from damaged sensory neurons, stimulates pro-inflammatory responses. Antagomir treatment against miR-21 reduced neuropathic hypersensitivity and attracted fewer inflammatory macrophages to the injury site [170]. Conversely, miR-21 has been shown to inhibit neuronal autophagy in exosomes from neurons primed by damaged mouse brain extracts [171]. Furthermore, exosomes enriched with the miR-17-92 cluster promote neurogenesis, oligodendrogenesis, and axonal outgrowth in the stroke-damaged CNS. Exosome-carried miR-132 regulates brain vascular integrity as an intercellular signal [172]. Essentially, exosomes from neurons and glial cells regulate gene expression and miRNA activities, mediating neuroprotective and neurorestorative effects by fostering neurogenesis, reducing inflammation, enhancing angiogenesis, and facilitating tissue remodeling.
Neuroprotective therapy
Neuroprotectants are agents defined by their capability to reduce brain injury during ischemia by counteracting specific molecular events harmful to neurons. The principal challenge associated with the use of neuroprotectants stems from the restricted time frame for administration, given the current emphasis on promptly removing arterial blockages in acute stroke treatment. Since the 1990s, researchers have actively explored avenues for directly treating ischemic brain tissue through both pharmacologic and non-pharmacologic neuroprotective interventions. However, significant breakthroughs in this area have proven elusive. Candidate mechanisms for such agents include excitotoxicity inhibitors, apoptosis inhibitors, free radical scavengers, and anti-inflammatory medications [173, 174]. It is noteworthy that most neuroprotective clinical trials took place prior to the thrombectomy era, a period when the post or peri-reperfusion administration of neuroprotective drugs emerged as a novel and appealing treatment option. Post hoc studies, such as the uric acid trial [175], have revealed that certain neuroprotective medications show greater efficacy in subpopulations of patients who have undergone mechanical thrombectomy. In a large trial (ESCAPE NA1) [176], nerinetide, an inhibitor of the excitotoxic cell death pathway, was recently investigated in mechanical thrombectomy patients. Although the research did not indicate a difference in 90-day functional outcomes, it did reveal that nerinetide patients not receiving tissue plasminogen activator (tPA) had improved outcomes. In the current thrombectomy era, the co-administration of neuroprotective agents with recanalization could enhance the targeted delivery of these agents to specific brain regions, potentially augmenting the efficacy of stroke therapy [177].
Stem cell therapy
Various forms of cell-based therapies are under investigation (Table 1), garnering increased attention [178–180]. Examples include transformed tumor cells, adult stem cells such as marrow stromal cells, umbilical cord cells, placental cells, embryonic stem cells, and induced pluripotent cells, which can be genetically modified or cultured under specific conditions like low oxygen or exposure to neurotrophins. These cells can be administered alone or with a bioscaffold, and autologous, allogeneic, or xenograft stem cells are all viable options. The loss of neurons and glia distinguishes an intact CNS, making cell replacement a potential therapeutic strategy. Marrow stromal cells have demonstrated the ability to differentiate into various cell lineages, including glia and neurons, both in vitro and in vivo [181]. Injection of rat or human bone marrow stromal cells into the lesioned cortex following TBI resulted in migration and expression of astrocytic and neuronal markers, indicating their role in triggering neurogenesis after TBI. These histological changes were associated with sustained improvements in neurological and motor function [182, 183]. Similarly, MSCs have shown promising results in TBI research, stimulating GFAP expression and proliferation in NSC cultures.
Therapeutic interventions in stroke therapy
| Sl. No. | Stem cell types | Function | References |
|---|---|---|---|
| 1 | Embryonic stem cells | Murine embryonic stem cells (mESCs) show promise in restoring function in ischemia mouse models. However, concerns over their immunogenicity and teratoma-forming tendency hinder widespread clinical acceptance. | [190] |
| 2 | Human umbilical cord blood cells (HUCBCs) | Migrated to the injury site and differentiated into neuronal and glial cells, leading to elevated trophic factors in the brain. These factors positively correlate with decreased cerebral infarcts and improved behavioral functions. | [191] |
| 3 | Bone marrow stem cells (BMSCs) | BMSCs injection during or after cerebral infarction regenerates neurons, boosting survival rates and enhancing brain function. | [192] |
| 4 | Human fetal neural progenitors | Transplanting human fetal hippocampal cells and embryonic cortical cells into rat ischemic models, as well as human fetal striatal cells into a rodent stroke model, enhances behavior. Moreover, the transplanted human cells establish both afferent and efferent connections with host rodent cells. | [193] |
| 5 | Neural stem cells (NSCs) | NSC engraftment enhances synaptic reformation and electrophysiological properties in damaged brains by optimizing the microenvironment. It restores neuronal functions by secreting neurotrophic factors [brain-derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF)], supporting neuron health, generation, and survival, along with maintaining the extracellular matrix. | [194, 195] |
| 6 | VEGF | Facilitates angiogenesis and promotes vascular repair in ischemia-induced blood vessel damage. | [196] |
| 7 | Ciliary neurotrophic factor (CNTF), glial cell-derived neurotrophic factor (GDNF), nerve growth factor (NGF), and other such factors | Secretions from NSCs play crucial roles in safeguarding, sustaining, and fostering the proliferation of neural cells. | [197] |
| 8 | Mesenchymal stem cells (MSCs) | Neuro-restoration involves diverse mechanisms, including paracrine factor release, cell replacement, mitochondrial transfer, etc. Additionally, they induce angiogenesis through VEGF release. | [198] |
| 9 | Adipose tissue-derived MSCs | These cells efficiently aid neuro-regeneration, being easily accessible and abundant. They provide protection via extracellular vesicle release, with studies confirming the safety and efficacy of vesicles from adipose tissue-derived MSCs. | [199] |
| 10 | Human adipose stromal cells | Liposuction-derived adipose tissue stromal cells, when exposed to chemicals like 5-azacytidine, NGF, BDNF, and basic fibroblast growth factor (bFGF), can undergo differentiation into neurons. | [200] |
| 11 | Induced pluripotent stem cell (iPSC) | The occurrence of teratoma in the mouse brain underscores the imperative to address and resolve the tumorigenicity of iPSCs before clinical application. Nonetheless, iPSCs exhibit significant potential as formidable stem cells for tissue regeneration. | [201] |
| 12 | Dental pulp stem cells (DPSCs) | Neurotrophic factors, including neurotrophins, GDNF, and neurturin, are produced during dental pulp innervation initiation. Increased expression of NGF, BDNF, and GDNF mRNAs precedes dental pulp innervation onset. These factors likely support trigeminal nerve growth and tooth innervation establishment, playing crucial roles in neuronal survival, plasticity, and axon growth. | [202] |
| 13 | Skin stem cells | Murine skin-derived progenitors (mSKPs) from developmental stages exhibit osteogenic, adipogenic, smooth muscle, and neuronal differentiation potentials. They offer promise as an alternative source for addressing mesenchymal and neurodegenerative disorders. | [203] |
| 14 | Olfactory ensheathing cells (OECs) | Transplanted OECs in injured spinal cords can enhance axonal regeneration. The reconstitution of the olfactory neuroaxis is possible following central axotomy. In vivo, nasal OECs (nOECs) remain viable and respond to signals from injured neuronal cells and their processes. | [204] |
| 15 | Human fetal neural progenitors | Human fetal neuronal precursor cells exhibit ex vivo neuronal differentiation, acquiring a region-specific cholinergic phenotype. | [131] |
| 16 | Fetal kidney cells | Fetal kidney cell transplants in stroke rats express elevated GDNF and bone morphogenetic protein (BMP) levels, showing promise for neuroprotection and neuroregeneration in ischemic brain injury. | [7] |
| 17 | Haematological stem cells | Stem cell mobilization post-cerebral infarction leads to neuronal regeneration, enhancing survival rates and brain function following cerebral ischemia. | [205] |
| 18 | Adult-derived NSC | Exogenous NSC therapy in stroke reconstructs neuronal circuits and offers neuroprotection by secreting survival-promoting neurotrophic factors, either naturally or through therapeutic transgenes. | [131] |
An extensive overview is provided, highlighting a myriad of therapeutic strategies employed in the management of cerebral stroke. The table specifically delineates the utilization of different types of stem cells, showcasing the diverse approaches and regenerative interventions aimed at mitigating the impact of cerebral stroke and promoting neural recovery
Human stem cells are commonly utilized in research due to their ability to secrete neurotrophic factors like nerve growth factor (NGF) and BDNF, known for their neuroprotective effects. Transplantation of human fetal stem cells has been shown to improve cognitive and motor function, reduce lesion size and neuronal death, stimulate angiogenesis, and suppress active microglia post-injury [184]. Additionally, fetal stem cells in injured hippocampus and cortex have been found to differentiate into neurons and astrocytes due to the release of glial-derived neurotrophic factor [185]. A small-scale phase I clinical trial involving autologous marrow stromal cell transplantation in young TBI patients showed minimal neurological improvement without significant side effects [186]. Similarly, a phase I/II trial using intrathecal injections of autologous bone marrow-derived mononuclear cells in sub-acute TBI patients demonstrated no significant side effects, but only about half of the patients showed improvement in persistent vegetative state and motor dysfunction [187].
Cell replacement and neuroprotection
iPSC-derived NSCs (iNSCs) have the capability to differentiate into the three primary neural lineages: neurons, astrocytes, and oligodendrocytes, utilizing various reprogramming approaches, including an episomal plasmid-based method. The microenvironment may exert influence over the potential for iPSC differentiation. Notably, at the twelve-week mark post-transplantation, observed comparable numbers of neurons at the lesion margin in both the iNSC-treated group and normal control animals. In contrast, the non-treated group exhibited a lower number of neurons than the normal control animals [188]. Moreover, gene expression levels related to angiogenesis, neurotropism, and inflammation underwent significant alterations in the brain tissues of the iNSC-treated group. Longitudinal magnetic resonance imaging of stroke model mice revealed improvements in tissue-level parameters such as cerebral blood perfusion, white matter integrity, and brain metabolism following iNSC therapy. Another investigation reported enhanced behavioral recovery in stroke-injured rats after the transplantation of human iPSC-derived long-term expandable neuroepithelial-like stem cells (It-NESCs) into the striatum [189].
Conclusions
Stroke represents a significant health burden globally, with ischemic stroke being the most common type and often resulting in severe neurological deficits. Despite the availability of treatments like rtPA and endovascular thrombectomy, neurological symptoms persist, highlighting the urgent need for novel therapeutic approaches. One promising avenue is stroke-induced neurogenesis, particularly through stem cell therapy. However, challenges such as the limited quantity of NSCs available for transplantation and concerns regarding immunological compatibility need to be addressed. Utilizing iPSCs or MSCs derived from the patient’s own cells may mitigate immune compatibility issues due to their lack of HLA class II expression. Furthermore, optimizing cell extraction, expansion, and differentiation procedures for transplantation is crucial. Identifying factors that stimulate neurogenesis and elucidating the underlying molecular mechanisms are paramount, with inflammation post-stroke being a key area of focus. Additionally, recognizing the sex-specific pathophysiology of stroke presents opportunities for tailored pharmacological strategies, although substantial research in this area is lacking. Addressing the observed sex differences in vascular outcomes and treatment responses underscores the importance of personalized medicine in stroke management. Effective stroke therapeutics must consider the complex interactions involving neuroinflammation, angiogenesis, and neuroplasticity. While neurogenesis holds promise for neurological recovery, translating preclinical findings into clinical applications remains challenging. Understanding age-related variations in neurogenesis rates and region-specific regulatory mechanisms could inform targeted therapeutic interventions. Ultimately, developing safe and effective neuroprotective agents that account for sex-specific factors is crucial for reducing stroke-related morbidity and mortality and improving patient outcomes.
Abbreviations
| AXIS: | AX200 for Ischemic Stroke |
| BDNF: | brain-derived neurotrophic factor |
| BrdU: | bromodeoxyuridine |
| CNS: | central nervous system |
| G-CSF: | granulocyte-colony stimulating factor |
| GH: | growth hormone |
| iNSCs: | induced pluripotent stem cell-derived neural stem cells |
| iPSC: | induced pluripotent stem cell |
| MCA: | middle cerebral artery |
| MMPs: | matrix metalloproteinases |
| MSCs: | mesenchymal stem cells |
| NSCs: | neural stem cells |
| OB: | olfactory bulb |
| SDF: | stromal-derived factor |
| SGZ: | sub granular zone |
| SSRIs: | selective serotonin reuptake inhibitors |
| STAT3: | signal transducer and activator of transcription 3 |
| SVZ: | subventricular zone |
| TBI: | traumatic brain injury |
| VEGF: | vascular endothelial growth factor |
| VZ: | ventricular zone |
Declaration
Acknowledgments
Department of Knowledge and Information Management of CSIR-Indian Institute of Chemical Technology, Hyderabad, is greatly acknowledged for generating institutional publication number IICT/Pubs/2023/443.
Author contributions
MR, RK, and BSA: Conceptualization, Writing—original draft, Formal analysis. AK and SC: Conceptualization, Supervision, Funding acquisition, Validation, Writing—original draft, Writing—review & editing.
Conflicts of interest
The authors declare no conflict of interest, financial or otherwise.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
The work was funded by ICMR grant [5/4-5/3/17/Neuro/2022-NCD-1] to SC and SERB-POWER Fellowship [SPF/2021/000045]. MR and RK were supported by doctoral fellowships from CSIR India. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2024.