 Review
Review

Affiliation:
Department of Biology, Winthrop University, Rock Hill, SC 29733, USA
Email: birgbauere@winthrop.edu
ORCID: https://orcid.org/0000-0002-5555-4970
Explor Neuroprot Ther. 2024;4:349–365 DOI: https://doi.org/10.37349/ent.2024.00088
Received: April 16, 2024 Accepted: August 11, 2024 Published: August 22, 2024
Academic Editor: Dasiel O. Borroto Escuela, Karolinska Institutet, Sweden; Rafael Franco, Universidad de Barcelona, Spain
The article belongs to the special issue GPCR Heteroreceptor Complexes as Key Players in Neuroprotection
The central nervous system (CNS) is one of the most complex physiological systems, and treatment of CNS disorders represents an area of major medical need. One critical aspect of the CNS is its lack of regeneration, such that damage is often permanent. The damage often leads to neurodegeneration, and so strategies for neuroprotection could lead to major medical advances. The G protein-coupled receptor (GPCR) family is one of the major receptor classes, and they have been successfully targeted clinically. One class of GPCRs is those activated by bioactive lysophospholipids as ligands, especially sphingosine-1-phosphate (S1P) and lysophosphatidic acid (LPA). Research has been increasingly demonstrating the important roles that S1P and LPA, and their receptors, play in physiology and disease. In this review, I describe the role of S1P and LPA receptors in neurodegeneration and potential roles in neuroprotection. Much of our understanding of the role of S1P receptors has been through pharmacological tools. One such tool, fingolimod (also known as FTY720), which is a S1P receptor agonist but a functional antagonist in the immune system, is clinically efficacious in multiple sclerosis by producing a lymphopenia to reduce autoimmune attacks; however, there is evidence that fingolimod is also neuroprotective. Furthermore, fingolimod is neuroprotective in many other neuropathologies, including stroke, Parkinson’s disease, Huntington’s disease, Rett syndrome, Alzheimer’s disease, and others that are discussed here. LPA receptors also appear to be involved, being upregulated in a variety of neuropathologies. Antagonists or mutations of LPA receptors, especially LPA1, are neuroprotective in a variety of conditions, including cortical development, traumatic brain injury, spinal cord injury, stroke and others discussed here. Finally, LPA receptors may interact with other receptors, including a functional interaction with plasticity related genes.
G protein-coupled receptors (GPCRs) are one of the largest classes of receptors, with an estimated 800 in the human genome (excluding olfactory receptors). They have a large and diverse set of ligands. Furthermore, they have been the target of many pharmacological compounds in use today [1].
One class of GPCR ligands is the bioactive lysophospholipids, specifically lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P). These lysophospholipid molecules are released from cells and bind to specific receptors, whereby they mediate various cellular responses. These receptors are classic GPCRs that signal through canonical G protein signal transduction pathways. There are six known LPA receptors (LPA1 through LPA6) and five known receptors for S1P (S1P1 through S1P5), each signaling through one or more G proteins (Table 1) [2, 3]. The S1P1 receptor is unique in that all evidence to date suggests that it only signals through Gi/o, where it can decrease adenylate cyclase activity, as well as stimulate the rat sarcoma (Ras) and extracellular signal-regulated kinase (ERK) pathway to enhance cellular proliferation and activate the phosphatidylinositol-3 kinase (PI3K) pathway to inhibit apoptosis. The Gi/o pathway can also activate phospholipase C (PLC) and protein kinase C to increase intracellular calcium. The receptors S1P4 and S1P5 signal through G12/13 to activate Rho in addition to signaling through Gi/o, with some evidence that they may signal through Gs under certain circumstances. The receptors S1P2 and S1P3 signal not only through Gi/o and G12/13, but also through Gq, where they can activate PLC to increase intracellular calcium.
Lysophospholipid G protein-coupled receptors (GPCR). Nomenclature of the GPCRs and genes of S1P and LPA receptors as well as the G protein cell signaling pathways activated [2, 3]
| Ligand | GPCR (protein) | Gene nameMouse/human | Other names | Signaling pathways |
|---|---|---|---|---|
| S1P | S1P1 | S1pr1/S1PR1 | edg1, lpB1 | Gi/o |
| S1P2 | S1pr2/S1PR2 | edg5, lpB2, AGR16, H218 | Gi/o, Gq, G12/13 | |
| S1P3 | S1pr3/S1PR3 | edg3, lpB3 | Gi/o, Gq, G12/13 | |
| S1P4 | S1pr4/S1PR4 | edg6, lpC1 | Gi/o, G12/13, (Gs) | |
| S1P5 | S1pr5/S1PR5 | edg8, lpB4, Nrg-1 | Gi/o, G12/13, (Gs) | |
| LPA | LPA1 | Lpar1/LPAR1 | vzg1, edg2, lpA1 | Gi/o, Gq, G12/13 |
| LPA2 | Lpar2/LPAR2 | edg4, lpA2 | Gi/o, Gq, G12/13 | |
| LPA3 | Lpar3/LPAR3 | edg7, lpA3 | Gi/o, Gq | |
| LPA4 | Lpar4/LPAR4 | GPR23, p2y9 | Gs, Gq, G12/13 | |
| LPA5 | Lpar5/LPAR5 | GPR92 | Gq, G12/13 | |
| LPA6 | Lpar6/LPAR6 | p2y5 | Gs, G12/13 |
S1P4 and S1P5 primarily signal through Gi/o and G12/13, although there is some suggestion that they can signal through Gs (parentheses) under certain circumstances
LPA receptors, likewise, signal through various G protein pathways. The first two receptors identified, LPA1 and LPA2, signal through Gi/o, Gq, and G12/13, with the G12/13 pathway leading to activation of Rho and cytoskeletal rearrangements. The receptor LPA3 signals through Gi/o and Gq, but not G12/13. LPA4 signals through Gq and G12/13, as well as Gs (but not Gi/o). LPA5, on the other hand, only signals through Gq, and G12/13. The final validated LPA receptor, LPA6, which appears to be a lower affinity receptor [4], has been shown to signal through Gs and G12/13.
LPA and S1P receptors and their ligands have been found to be involved in a variety of physiological and pathological responses (for review, see [5]). This includes extensive roles in the nervous system and neural development (the reader is referred to other reviews [3, 6]). Activation of S1P or LPA receptors is generally considered pro-proliferative and anti-apoptotic, and thus there is considerable research on their role in the progression of cancer [7]. Furthermore, both S1P and LPA receptors are involved in inflammation, including neuroinflammation, and this is another area of active research. There is also evidence of a role of S1P receptors in the cardiovascular system and angiogenesis. In this review, I focus on summarizing evidence that LPA and S1P receptors are involved in neurodegeneration and neuroprotection.
S1P receptors are expressed throughout development and in the adult in many tissues, including the brain and spinal cord. The receptors S1P1 and S1P2 are widely expressed throughout the body [8, 9]. There is major expression in the heart for receptors S1P1, S1P3, and S1P5 [8]. The receptors S1P2 and S1P3 are expressed during development in mesenchymal tissue and somites [8]. S1P4 is highly expressed in the immune system. In the CNS, the receptors S1P1, S1P2, S1P3, and S1P5 are highly expressed [8, 9]. In the brain, expression of S1P1 and S1P4 is found in forebrain. The receptor S1P5, however, is primarily restricted to the brain and spinal cord, where it is highly expressed in oligodendrocytes [8, 10–12]. The receptors S1P1 and S1P2 are also expressed during development in neural progenitors in the ventricular zone as well as the neural tube.
Much of our understanding of the role of S1P receptors in neuropathological conditions and neuroprotection has been elucidated with various pharmacological agents (Table 2). The most widely used pharmacological tool has been the compound fingolimod (also known as FTY720, Gilenya®), which is currently in clinical use for multiple sclerosis (MS). It is an agonist for all the S1P receptors except S1P2, although it often functions as a functional antagonist (see below). However, more specific S1P receptor agonists have been developed, with the S1P1-specific agonist SEW2871 being used more recently.
Pharmacological compounds. Major pharmacological compounds used in the literature for investigation of the roles of S1P and LPA receptors
With the approval of fingolimod for the treatment of MS, MS is one of the best examples of a role for S1P receptors in neurological disease. MS is considered an autoimmune disease in which T-lymphocytes cross the blood-brain barrier and attack myelin, leading to demyelinated lesions. In the most common form, relapsing-remitting MS (RR-MS), this demyelination is repaired by oligodendrocytes, most likely newly differentiated from resident oligodendrocyte precursor cells. However, the cycle continues and leads to progressive neuronal damage, and disability, as the disease progresses.
S1P receptors, especially S1P1, are important clinical targets for MS. As mentioned previously, the pharmacological compound fingolimod has been approved by the FDA for the treatment of RR-MS after clinical trials [23], with many other compounds in clinical trials [3, 21, 24]. Fingolimod (originally called FTY720) was synthesized as a derivative of the fungal metabolite myriocin (also known as ISP-1) as an immunosuppressant for organ graft survival [25–27]. Interestingly, it led to lymphopenia with reduced circulating lymphocytes [27]. Later it was discovered that fingolimod is phosphorylated in vivo, and the phosphorylated form is the active compound [13, 14]. Phosphorylated fingolimod has structural similarities to S1P, and it was subsequently shown that fingolimod-phosphate binds to the S1P receptors S1P1, S1P4, and S1P5 with high affinity (EC50 ~0.3–0.6 nM) and to S1P3 with slightly lower affinity (EC50 ~3 nM), but not to S1P2 [13, 14]. Binding of fingolimod to the S1P receptor activates it, and fingolimod was initially characterized as an agonist [13, 14]. However, in the immune system, upon binding fingolimod, S1P receptors are internalized and degraded so that they are unresponsive to S1P, and fingolimod is now often considered a functional antagonist (see [13, 21, 22]). Recently, more specific S1P receptor agonists and antagonists have been developed, especially for S1P1, and many are in various stages of clinical trials [3]. One of the most important ones for experimental investigation has been SEW2871, which is specific for S1P1 (see Table 2).
Fingolimod’s approval for RR-MS was after extensive preclinical work where it was efficacious in the rodent model experimental autoimmune encephalomyelitis (EAE) [13, 21, 22, 28]. In MS and EAE, fingolimod works by inducing a lymphopenia of peripheral lymphocytes, including CD4+ T cells, CD8+ T cells and B cells. This lymphopenia results in sequestration of central memory T lymphocytes in lymph nodes, reducing the autoimmune activity in MS and EAE.
However, in addition to immune modulation, fingolimod has been suggested to have a neuroprotective role in EAE and potentially MS [22]. One piece of evidence for this neuroprotective role is that a higher dose of fingolimod is required for efficacy to reduce symptoms in EAE than is needed for lymphocyte sequestration [29]. Furthermore, fingolimod has been shown to restore electrophysiological function after EAE [30]. In addition, fingolimod may also work by reducing neuroinflammation through astrocytes and/or microglia [22, 31].
In addition to MS, a neuroprotective role for fingolimod has been suggested for many other neurological diseases [3, 32]. For instance, fingolimod is neuroprotective in a cerebral ischemia (stroke) model. During a stroke, a blood clot forms in the brain leading to local ischemia, and the resulting loss of oxygen results in neuronal death; it also produces a neuroinflammatory state, which, especially after reperfusion, can induce further apoptosis. S1P is released after cerebral ischemia in a rodent model, which leads to increased damage. The released S1P was shown to activate S1P receptors on microglia leading to neuroinflammation; there was also some evidence of activation of astrocytes leading to astrogliosis. This neuroinflammation led to neurodegeneration, and fingolimod was neuroprotective but not directly on neurons. Various studies suggest that the receptors S1P1, S1P2, and S1P3 are involved in mediating this response [33], although fingolimod likely acts primarily through S1P1 as its action is mimicked by SEW2871. On the other hand, in a different stroke model, it was found that neural progenitor cells migrated toward a brain infarction, and this was dependent on the receptors S1P1 and S1P2 [34], suggesting a role in neural repair processes.
There is also evidence that S1P receptor activation is neuroprotective in a Parkinson’s disease model. Parkinson’s disease is a neurodegenerative disorder characterized by the loss of dopaminergic neurons in the substantia nigra. Furthermore, in two different mouse models of Parkinson’s disease (6-OHDA and rotenone), fingolimod was neuroprotective for dopaminergic neuron loss and apoptosis, as well as reducing dopamine loss and improving motor deficits [35, 36]. This effect appeared direct, as fingolimod treatment also reduced apoptosis by 6-OHDA or rotenone in vitro in the dopaminergic SH-SY5Y cell line. In this model, fingolimod appears to act as a S1P receptor agonist, as its neuroprotective effect is blocked by a S1P1 receptor antagonist (W146). Fingolimod, as well as the S1P1 receptor agonist SEW2871, is also neuroprotective against an in vitro oxidative stress model in these SH-SY5Y cells [37], again showing a role in protecting dopaminergic neurons.
In yet another neurodegenerative disease, Huntington’s disease, fingolimod was neuroprotective, leading to reduced apoptosis as well as improvements in motor skills, electrophysiology, and survival [38]. This was also recapitulated in a striatal-derived cell line, suggesting a direct neuroprotective effect on neurons.
In amyotrophic lateral sclerosis (ALS), fingolimod slowed disease progression in a mouse model, likely moderating neuroinflammation, although there may be a direct neuroprotective effect [39]. Furthermore, in vitro, fingolimod is neuroprotective for neurons in culture as well as possibly astrocytes and oligodendrocytes [22].
A potential mechanism of neuroprotection by S1P receptors is through increasing brain derived neurotrophic factor (BDNF) expression (Figure 1). BDNF is a neurotrophin that is a survival factor for many neurons as well as their axons and synaptic connections. In a mouse model for Rett syndrome, which has low levels of BDNF, fingolimod treatment raised brain BDNF levels as well as promoted improved motor function and increased survival [40]. The authors then went on to show that in an in vitro cortical neuron culture model, either fingolimod, S1P, or the S1P1 receptor agonist SEW2871 increased excitatory neuronal activity. This was accompanied by increased phospho-ERK and phospho-CREB (cAMP-response element binding protein), resulting in increased BDNF production. This increased BDNF production was neuroprotective against excitotoxicity induced by the glutamate agonist N-methyl-D-aspartate (NMDA). This proposed neuroprotective mechanism of increasing BDNF through S1P receptor signaling is also supported in the Huntington disease model, where fingolimod treatment leads to increased BDNF levels as well as being efficacious in improving disease symptoms [38].
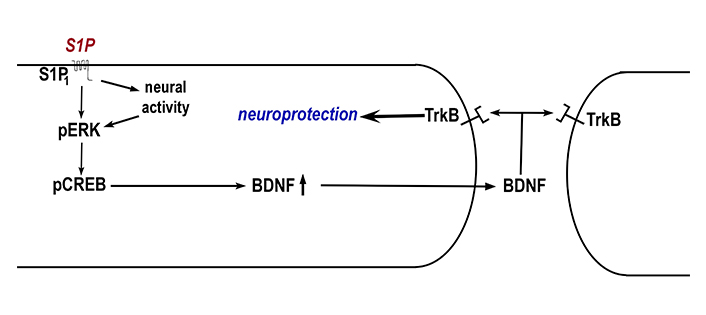
Model for neuroprotection by S1P1 receptors through BDNF. It has been found that activation of S1P1 by S1P leads to increased excitatory neural activity in cortical neurons. Activation of S1P1 as well as neural activity leads to phosphorylation of ERK (pERK). This in turn leads to phosphorylation of cAMP-response element binding protein (pCREB) that results in increased production of brain derived neurotrophic factor (BDNF). BDNF is released and binds to tyrosine receptor kinase B (TrkB) to mediate neuroprotection. BDNF could activate TrkB in the same cell (left in figure) or another cell (right in figure) (Model based upon data from [40])
Another area where S1P receptors are involved in neuroprotection is in spinal cord injury, which is a complex injury resulting in neuroinflammation, scar formation, axonal loss, and neuronal death. S1P is produced by microglia and astrocytes at the site of injury after a spinal cord lesion [41]. After a contusion spinal lesion, neural stem cells migrate to the site of injury, and this migration is blocked by a short hairpin RNA (shRNA) or inhibitor of S1P1 [41]. Furthermore, fingolimod is neuroprotective in a contusion spinal cord injury model, leading to improvements in clinical score and motor coordination [42]. Interestingly, fingolimod did not reduce inflammation, suggesting a more direct neuroprotective role, although it did reduce astrocyte accumulation. This improvement was recapitulated by the S1P1 receptor specific compound SEW2871, implicating S1P1 as the important receptor.
S1P receptors also appear to be neuroprotective in Alzheimer’s disease. S1P was found to be increased in the plasma of Alzheimer’s disease patients [43], although this is a correlation and thus may be a result, not a cause, of neuroinflammation. In rat models of Alzheimer’s disease, intracerebroventricular injection of Aβ1-42 leads to hippocampal cell apoptosis and impairments in learning and memory. In this model, treatment with fingolimod was neuroprotective for hippocampal cell loss [44, 45] as well as improving learning and memory in various paradigms [44–46]. Intriguingly, in one study, treatment with fingolimod itself impaired spatial learning and memory in control rats [44], suggesting more complex regulation, which may involve multiple S1P receptors. Furthermore, as before, BDNF may be involved. In the model of intracerebroventricular injection of oligomeric Aβ1-42, treatment with fingolimod was neuroprotective and also correlated with increased levels of BDNF [46]. The role of BDNF was corroborated in an in vitro system of oligomeric Aβ1-42 toxicity of cortical neurons, where fingolimod was shown to be neuroprotective, and this neuroprotection was shown to depend on increased BDNF production and signaling through TrkB receptors and ERK1/2 activation, as determined through the use of inhibitors [47]. A more specific role for the receptor S1P1 was seen in a hippocampal slice culture system where the specific agonist SEW2871 significantly reduced tau phosphorylation [48], another hallmark of Alzheimer’s disease.
The role of S1P receptors has been investigated in other neurological diseases as well. In a mouse model of the neurodegenerative disorder Sandhoff disease, a neuropathic lysosomal storage disorder, progression of the disease involved the receptor S1P3, as mice with null mutations in S1P3 had reduced disease progression and severity, although they were not cured [49]. This progression also required sphingosine kinase 1 (SphK1), the enzyme that catalyzes production of S1P, as a null mutation in SphK1 also reduced disease progression. In this case, the mutations were neuroprotective by reducing astrocyte proliferation and astrogliosis.
In the eye, in a rat model of retinal disease, S1P appears to be involved in retinal ganglion cell (RGC) death by glutamate excitotoxicity [50]. A sphingosine kinase inhibitor was neuroprotective and reduced the cell death, although the specific S1P receptors were not determined.
Additional human studies suggest a role for S1P receptors in a variety of neurodegenerative diseases, although this is correlative and specific receptors have not been identified. In human patients, plasma S1P levels were decreased in a variety of neurodegenerative diseases, including idiopathic Parkinson’s disease, Alzheimer’s disease (mentioned above), dementia with Lewy bodies, multiple system atrophy, and progressive supranuclear palsy [43].
Other studies have investigated the role of S1P receptors in neuroprotection in vitro. In one of the earlier studies, S1P reduced apoptosis caused by serum withdrawal in PC12 cells, a peripheral neural cell line [51], although it was not clear if this was receptor mediated. In an in vitro model of glutamate excitotoxicity in mesencephalic neurons, S1P was neuroprotective [52], but again the receptors involved were not analyzed. In astrocytes, the receptors S1P2 and S1P3 activate neuroinflammation [53].
Thus, in a variety of model systems, S1P and S1P receptors have neuroprotective roles, although whether their activation or inhibition is neuroprotective varies. Much of this hinges on whether the pharmacological compound fingolimod, as well as SEW2871, acts as an agonist or a functional antagonist, with some evidence for each role.
The roles of LPA receptors in neural development, physiology, and pathogenesis are numerous (for reviews, see [5, 6, 9, 54]). For instance, LPA receptors, especially LPA1 and LPA2, are involved in the early development of the cerebral cortex, regulating neural progenitor expansion and differentiation (see below). A significant role for LPA receptors, in this case, LPA1, LPA3, and LPA5, has been described in the initiation and maintenance of neuropathic pain (see [6, 55, 56]), whereby initiation of neuropathic pain involves a feed-forward mechanism by LPA through LPA1 and LPA3 on microglia. Subsequently, LPA5 is required for maintenance of the neuropathic pain state [57]. Additional roles for LPA receptors, especially LPA1, have been demonstrated in maturation of glutamatergic synapses in the hippocampus, with learning and memory deficits seen in Lpar1 null mice [6, 58].
LPA receptors are expressed throughout the nervous system, especially during development, with different expression patterns for the six LPA receptors. The receptor genes Lpar1, Lpar2, Lpar4, and Lpar6 are expressed during embryonic brain development in the neocortex, hippocampus, cerebellum, and olfactory bulb, as well as in adult brain in these regions. The receptor Lpar3, however, is only expressed postnatally, and Lpar5 expression may be in early brain development, although the data is less clear [59, 60].
Outside of the nervous system, LPA receptors have significant roles in cancer, the immune system, as well as other physiological processes (for reviews, see [7, 9, 54, 61–66]). Due to the importance of LPA receptors in physiology and disease, various pharmacological compounds are being developed both as experimental tools and for clinical therapeutic purposes (see [63, 67]). However, the LPA receptor pharmacological compounds have not advanced as much as for S1P receptors, and none are yet approved for clinical treatment. Nevertheless, there are a few pharmacological tools that have been used in various studies, especially that inhibit LPA1 (Table 2). The compound Ki16425 has been shown to inhibit LPA1, but also LPA3, and so is not completely specific [18]. A newer, more specific inhibitor of LPA1, AM095, is now being used [19]. In addition, a partially characterized inhibitor of LPA5, TCLPA5, has been used in some studies [20]. However, without good pharmacological agents, much of our understanding of LPA receptor function has been from mice with LPA receptor mutations.
Focusing on neuroprotection, one of the early roles of LPA receptors is in the development of the cortex. In an in vitro embryonic cortical culture system, LPA addition increases survival of neural progenitors, which is blocked by double null mutations in the receptors LPA1 and LPA2 [68]. Furthermore, in a LPA1 (receptor) null mouse (maLPA1), there was reduced neural progenitor proliferation and increased apoptosis, leading to reduced cortical layers [69]. This suggests a neuroproliferative and/or neuroprotective role of the receptor LPA1. Furthermore, in the adult hippocampus, the maLPA1 null mutant mouse had reduced hippocampal neurogenesis compared to control when exposed to an enriched environment [70], documenting a role for the receptor LPA1 in adult neurogenesis. Indeed, Lpar1 null mice have deficits in learning and memory [71, 72]; whether this was due to a neuroprotective role for LPA1 or a role in synaptic remodeling or neurophysiology is not clear.
LPA and LPA receptors have been implicated in embryonic axonal growth and, potentially, in axon guidance. LPA has been demonstrated in vitro to cause the collapse of axonal growth cones and neurite retraction in neuroblastoma and PC12 peripheral nerve cell lines [73, 74] as well as primary neurons in culture [75–79]. Although the GPCRs mediating these responses have not been identified yet (but see [78]), the response appears receptor mediated due to specificity and potency [78, 79]. This growth cone collapse and neurite retraction proceeds via the Gα12/13 signal transduction pathway, as it is blocked by inhibition of Rho and ROCK [77, 79–86]. There also appears to be a role for the receptor LPA3 in neurite branching through the novel GTPase Rnd2 [87].
In addition to roles in neural development, LPA receptors have been implicated in a variety of neurodegenerative conditions. In traumatic brain injury (TBI), LPAR2 gene expression was increased in human patients ~43 hours (range: 6 hours to 122 hours) after injury [88]. Furthermore, in human patients, levels of the ligand LPA increased significantly 24 hours after injury [89]. Using a mouse model of TBI, treatment with a monoclonal antibody against LPA (Lpathomab) was neuroprotective, leading to reduced lesion volume and decreased cytokine levels, as well as improved behavioral outcomes [89].
There has also been found a neuroprotective role of LPA receptor antagonism in spinal cord injury. In a mouse model of spinal cord injury, the receptor genes Lpar2 and Lpar3 were upregulated after injury [90]. Spinal cord injury leads to demyelination, but this demyelination was partially blocked in an Lpar1 null mutation mouse or upon treatment with the LPA1 antagonist AM095 [91]. The antagonist also led to a small, but significant, functional improvement. The receptor LPA2 also appears to be involved, as the Lpar2 null mutant mouse also showed partially reduced demyelination and some functional recovery [92]. In another study with a different model of spinal cord injury, an LPA1 antagonist led to increased corticospinal tract sprouting after injury [93], again demonstrating a neuroprotective role by blocking LPA1. Thus, in spinal cord injury, it appears that antagonism of LPA receptors is neuroprotective.
In another neurodegenerative condition, stroke and cerebral ischemia, LPA acting through LPA receptors appears to mediate neurological damage, and again antagonism is neuroprotective. In human stroke patients, plasma LPA levels increase [94, 95]; LPA levels also increase in the brain in a rodent model of stroke, a transient middle cerebral artery occlusion (tMCAO), which produces a transient focal cerebral ischemia [96, 97]. In this tMCAO model, increased LPA appears to be pathological, as addition of exogenous LPA leads to increased lesion volume [98, 99]. The receptor LPA1 appears to be involved in mediating this pathway, as the antagonist AM095 or an shRNA was neuroprotective and reduced lesion volume, neuronal apoptosis, and neurological deficits after tMCAO and reperfusion [100]. Furthermore, a role for LPA5 has also been implicated by treatment with an antagonist, TCLPA5, after tMCAO and reperfusion leading to functional recovery and reduced lesion volume [101]. Although the specificity of TCLPA5 has not been extensively tested, it inhibits LPA5 [20]. Importantly, treatment with TCLPA5 three hours after cerebral ischemia and reperfusion was neuroprotective, suggesting potential clinical benefit [101]. However, whether the neuroprotective effect is direct on neurons is not clear, as cerebral ischemia also induces neuroinflammation, and an antagonist of LPA1 or LPA5 reduces microglial activation and proinflammatory cytokines [100–103]. Interestingly, it was found that when rats are treated with a repeated hyperbaric oxygen exposure prior to ischemia, which is neuroprotective, Lpar1 gene expression was increased [104].
There is a suggestion for a role of LPA receptors in Alzheimer’s disease pathology (see [105, 106]). However, much of it is indirect, whereby characteristics of Alzheimer’s disease are mimicked by LPA or LPA receptors in other systems, sometimes with extremely high levels of LPA. For instance, LPA treatment of neuroblastoma cells leads to tau phosphorylation, which is a hallmark of Alzheimer’s disease [107]. More recently, there have been found differences in LPA receptor expression in the brain of a transgenic Alzheimer’s disease mouse model [108], but the role of these differences is not clear.
There is a potential role for receptor LPA2 in ALS. In human ALS patients, there are increased levels of Lpar2 mRNA in the spinal cord, which is also seen in the SOD1G93A mouse model of ALS [109]. Furthermore, in this mouse model, a null mutation in Lpar2 leads to reduced disease progression, suggesting LPA2 is involved in disease mediation. Interestingly, though, the Lpar2 null mutation decreases survival in this mouse model. Thus, in this ALS model, although signaling through LPA2 increases disease symptoms, it extends lifespan. Furthermore, the role of LPA2 appears to be related to inflammation and not directly on motor neurons.
There is also evidence of the role of LPA receptors in other neurodegenerative disorders. The LPA receptor LPA1 appears to be involved in posthemorrhagic hydrocephalus. In a mouse model, LPA injection into the ventricle killed ependymal cells and produced hydrocephalus, but this was partially reduced in Lpar1 null mutant mice as well as with an LPA1 antagonist [110]. In glaucoma, the receptors LPA1 and LPA2 are upregulated in a rat model of elevated ocular pressure, and an LPA receptor agonist reduced histological damage and improved retinal electrophysiology [111]. In a different oxygen-induced retinopathy model in rats, an shRNA against Lpar1 was neuroprotective for RGC loss [112].
Thus, in a variety of conditions, activation of LPA receptors appears to be pathological, while inhibition of LPA receptors appears neuroprotective.
There is also the possibility of interacting receptors that could modulate responses. For LPA receptors, one of the most intriguing possibilities is a family of receptors called the plasticity related genes (PRGs), also known as phospholipid phosphatase-related proteins (PLPPRs), which are a subfamily of the lipid phosphatase/phosphotransferase family of proteins (for review, see [113]). These are transmembrane proteins that are suggested to be receptors. Interestingly, although they are related to lipid phosphatases, they seem to have little or no lipid phosphatase activity.
The first PRG, PRG-1 (also known as PRG1, PLPPR4), was identified as a gene that was upregulated during hippocampal development as well as after lesion [114]. Interestingly, PRG-1 overexpression in neuroblastoma cells counteracted the neurite retraction activity of LPA [114], suggesting a possible interaction. In addition, other evidence showed that another family member, PRG-3, could enhance axonal outgrowth in a variety of systems [115, 116]. Interaction with LPA receptors was demonstrated in a prg-1 null mutant mouse. Deletion of prg-1 led to epileptic seizures in the mouse (with larger synaptic currents and higher mEPSC frequencies), but simultaneous deletion of Lpar2 rescued this phenotype, suggesting an interaction [117]. However, the interaction may not be direct, as PRG-1 was localized postsynaptically and LPA2 was presynaptic. In other studies, another PRG, PRG-2, was found to be important for thalamocortical axon guidance to the barrel cortex. In this case, deletion of prg-2 resulted in misrouted thalamocortical axons, and this phenotype could be rescued by inhibiting autotaxin, the enzyme responsible for LPA biosynthesis [118].
Furthermore, an interaction between LPA2 and PRG-1 has been seen in stroke. First, LPA levels increase in cerebrospinal fluid of human stroke patients. In a tMCAO mouse model of stroke, inhibition of autotaxin (which produces LPA) reduces LPA levels and improves behavioral outcomes with reduced lesion volume. Interestingly, a loss of function prg-1 mutation (R346T) resulted in increased lesion volume and worse behavioral outcomes after tMCAO [119]. An interaction with LPA2 was shown because deletion of Lpar2 along with the prg-1 mutation reversed the prg-1 mutant phenotype, resulting in lesion after tMCAO being similar to wild type injury after tMCAO. Thus, again, the Lpar2 mutation rescued the defects caused by a prg-1 mutation.
Several studies have shown a role for PRGs to be neuroprotective and counteract the inhibitory activity of LPA and LPA receptors on axonal growth. PRG-2 promoted a growth state in neurons by binding and inhibiting PTEN (phosphatase and tensin homolog) [120]. PRG-3 is strongly expressed after spinal cord injury, and PRG-3 overexpression in cortical neurons induced neurite outgrowth and overcame inhibitory LPA treatment [121, 122]. The PRG family member PRG-5 induced filopodia when overexpressed, and its overexpression attenuated LPA-induced neurite retraction [123].
A variety of experiments suggest an interaction of PRGs with LPA signaling through LPA receptors. In most cases, these receptors have not been identified, although an interaction between LPA2 and PRG-1 has been demonstrated genetically. Furthermore, whether there is direct interaction or not has yet to be established, as all the evidence so far shows only a functional interaction. Nonetheless, the potential interaction of PRGs with LPA receptors is intriguing, and much work remains to be done.
There has now accumulated substantial evidence that lysophospholipid GPCRs are involved in neurodegeneration and neuroprotection. This is especially true for S1P receptors due to the S1P receptor agonist, or functional antagonist, fingolimod, which has been clinically demonstrated to treat MS. The clinical success of fingolimod has led to the development of more specific S1P receptor pharmacological compounds as well as the investigation of S1P receptors in a variety of neurodegenerative conditions. There has been less investigation of LPA receptors in neurodegeneration, as there are fewer and less well-characterized pharmacological compounds as tools. Nonetheless, there is accumulating evidence that they, too, are involved in neuroprotection. As more LPA receptor compounds are developed and disease models examined, our understanding of the role of LPA receptors is likely to increase.
Furthermore, there exists the intriguing possibility of interactions between certain GPCRs and other receptors, including other GPCRs. There is genetic evidence of a functional interaction of PRGs with LPA receptors, although there may not be direct physical interaction. A recent report suggests a direct physical interaction between LPA1 and the GPCR C-X-C motif chemokine receptor 4 (CXCR4) [124]. Interestingly, although coexpression of LPA1 and CXCR4 did not affect LPA signaling, signaling by the ligand C-X-C motif chemokine 12 (CXCL12) through CXCR4 is reduced by LPA treatment, suggesting a functional interaction [124]. Thus, an important direction for the future is to examine this concept of GPCR heterodimerization that could influence receptor signaling.
ALS: amyotrophic lateral sclerosis
BDNF: brain derived neurotrophic factor
CNS: central nervous system
CXCR4: C-X-C motif chemokine receptor 4
EAE: experimental autoimmune encephalomyelitis
ERK: extracellular signal-regulated kinase
GPCR: G protein-coupled receptor
LPA: lysophosphatidic acid
MS: multiple sclerosis
PRGs: plasticity related genes
RR-MS: relapsing-remitting multiple sclerosis
S1P: sphingosine-1-phosphate
shRNA: short hairpin RNA
tMCAO: transient middle cerebral artery occlusion
I would like to thank Dr. Daniel Stovall, Layla Herndon and Mallika Singh for critical reading of the manuscript.
EB: Writing—original draft, Writing—review & editing.
The author declares no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
E.B. was supported by the National Institute of General Medical Sciences of the National Institutes of Health [P20GM103499]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Ahmed Hasbi, Susan R. George
Diego Guidolin ... Luigi F. Agnati
Adèle Vilette ... Peter Vanhoutte
Neelakanta Sarvashiva Kiran, Senthilkumar Rajagopal
