 Review
Review

Affiliation:
1Department of Neuroscience, University of Padova, 35121 Padova, Italy
Email: diego.guidolin@unipd.it
ORCID: https://orcid.org/0000-0003-2133-3552
Affiliation:
1Department of Neuroscience, University of Padova, 35121 Padova, Italy
ORCID: https://orcid.org/0000-0002-8860-4997
Affiliation:
2Department of Pharmacy, University of Genova, 16126 Genova, Italy
ORCID: https://orcid.org/0000-0003-3261-5264
Affiliation:
2Department of Pharmacy, University of Genova, 16126 Genova, Italy
ORCID: https://orcid.org/0000-0001-8290-3008
Affiliation:
1Department of Neuroscience, University of Padova, 35121 Padova, Italy
ORCID: https://orcid.org/0000-0002-2307-0277
Affiliation:
2Department of Pharmacy, University of Genova, 16126 Genova, Italy
ORCID: https://orcid.org/0000-0002-7909-4593
Affiliation:
3Department of Biomedical Sciences, University of Modena and Reggio Emilia, 41121 Modena, Italy
ORCID: https://orcid.org/0000-0002-8915-7295
Explor Neuroprot Ther. 2024;4:366–391 DOI: https://doi.org/10.37349/ent.2024.00089
Received: April 30, 2024 Accepted: August 14, 2024 Published: September 24, 2024
Academic Editor: Shile Huang, Louisiana State University Health Science Center, USA
The article belongs to the special issue GPCR Heteroreceptor Complexes as Key Players in Neuroprotection
Excitotoxicity represents a neuropathological process, describing the toxic actions of excitatory neurotransmitters, where the excessive or prolonged activation of glutamate receptors triggers a cascade of events leading to neuronal injury or death. Under conditions of reduced energy availability and increased oxidative stress neurons become particularly vulnerable to excitotoxicity and a large body of available evidence indicates that excitotoxicity represents a central mechanism in the pathogenesis of acute and degenerative diseases of the central nervous system. Astrocytes represent key elements in the regulation of glutamate homeostasis by their opposing functions of glutamate uptake and release, and microglial cells play an important role in the response to damage. Depending on the phenotype they assume when activated, microglial cells can trigger immune defense or neuroprotective processes. To perform their functions both glial cell populations monitor the extracellular space through a panel of receptors. Furthermore, a variety of signaling pathways also contribute to the modulation of the glutamatergic transmission, acting on specific cell receptors expressed by neurons, astrocytes, and microglia. In the last decades, evidence has been provided that receptors of almost all families can establish structural receptor-receptor interactions, leading to the formation of heteroreceptor complexes at the cell membrane of neurons and glial cells. The cooperativity that emerges in the actions of ligands of the monomers forming these assemblies provides the cell decoding apparatus with flexible dynamics in terms of recognition and signal transduction and allows an integration of the incoming signals already at the membrane level. Available data on possible modulatory roles played by heteroreceptor complexes in excitotoxic processes will be here reviewed and discussed. From the pharmacological standpoint, these findings may offer possibilities to explore novel therapeutic strategies targeting receptor complexes to address disorders of the central nervous system associated with dysregulation of glutamatergic signaling.
Functional interactions between receptors, not requiring a physical contact between receptor molecules, have been often observed [1]. They may occur by sharing signaling pathways or by mechanisms of transactivation. In the 1980s, however, in vitro and in vivo experiments by Agnati, Fuxe, and collaborators [2–4] provided indirect biochemical and functional evidence that structural interactions between G protein-coupled receptors (GPCRs) could also be established, leading to the formation of receptor complexes (dimers or high order oligomers) at the cell membrane (see [5] for historical details). The term “receptor-receptor interaction” (RRI) later was proposed to indicate such an interaction between receptor proteins involving a direct physical contact [6]. In the years that followed, several groups (see [7] for references) provided direct evidence for the existence of this molecular organization by exploiting a set of experimental techniques able to detect the spatial proximity of protein molecules [8–11].
The basic molecular mechanism characterizing the receptor assemblies are allosteric interactions [12–15], allowing the transfer of the energy associated with conformational or dynamic changes at some site of a protein to other sites, that will change their conformational or dynamical features accordingly. Thus, when protomers establish direct RRI and form a quaternary structure, energy perturbations occurring at some site of a protomer can propagate over the interface between receptors into the nearby protomers, changing their conformational and functional properties and allowing a cooperative behavior of the complex [16]. The interface between protomers and the residues involved, therefore, significantly influence the overall architecture of a receptor complex and its behavior, representing a topic of great interest in current research on receptor oligomerization. In this respect, several bioinformatics methods have been devised to predict the available interfaces (see [17–19] for specific reviews). Concerning GPCRs, experimental results indicate that the number of ways they interact in the membrane to form complexes is actually limited, the vast majority of experimentally identified receptor complexes being dimers. In addition, some interfaces have been observed to be more exploited than others for RRI [20]. Transmembrane (TM) domains TM4 and TM5 or the intracellular loop 3, for instance, have been often experimentally identified as interaction interfaces between GPCRs [19]. Nevertheless, oligomeric heteroreceptors have been detected [20–24].
The organization of receptors as receptor complexes is of particular interest under a functional perspective, since they can modulate cell activity, as shown by studies on neurons [25], where they could modulate synaptic weight [26], most likely affecting learning and memory processes [27, 28]. The collective dynamics of these supramolecular structures, indeed, allows the integration of different incoming signals reaching the plasma membrane to initiate specific patterns of signal transduction [29].
In general terms, several oligomerization-dependent signaling modifications have been identified or suggested [30], as briefly illustrated in Table 1.
Possible signaling changes following oligomerization
In addition, increasing evidence [36] shows that responses to specific ligands are also critically influenced by the environment in which receptor complexes are located, including lipid domains, scaffolding proteins, and other biochemical partners [37].
The majority of the available studies on RRI have been focused on GPCRs, with specific regard to the central nervous system (CNS) [38–40]. Oligomeric organization, however, plays an important role in the function of all receptor families, with the ion channel receptors (where multimerization is necessary) being located at one end of the spectrum and GPCRs (able to signal as monomers or stable dimers, or to give rise to transient quaternary structures [41]) at the other, indicating that it probably constitutes a general and efficient mechanism for modulating the functionality of receptor proteins [12, 30]. Furthermore, receptor multimerization was found to play a role in the physiology not only of nerve cells, but also other mammal cell populations [30, 42].
Based on these findings, a great research effort has been focused on two main pharmacological issues. On one hand, studies were directed at identifying dysfunctions or disruptions of receptor complexes that could represent a molecular basis for pathological changes associated with diseases [39, 43–47]. On the other hand, the possibility of novel therapeutic strategies based on drugs that specifically target receptor complexes has been explored [48–52].
In this context, a neuropathological process of significant interest is represented by excitotoxicity, a phenomenon describing the toxic actions of excitatory neurotransmitters (first glutamate) where the prolonged or excessive activation of glutamate receptors triggers a cascade of neurotoxic events leading to loss of neuronal function and cell death [53, 54]. A large body of evidence indicates that excitotoxicity represents a central mechanism in the pathogenesis of many diseases of the CNS [54].
In the present review article, we recapitulate mechanisms underlying excitotoxic processes, with a particular focus on the receptors involved, in order to explore possible modulatory roles played by RRI in neurons and glial cells.
The term “excitotoxicity” [55] indicates a complex process impacting on almost all subcellular compartments (namely cytosol, mitochondria, endoplasmic reticulum, and nucleus) and leading to neuronal swelling and death [53, 54]. It is triggered by a significant dysregulation of the glutamatergic system [56], characterized by a sustained overactivation of glutamate receptors, followed by a massive influx of cations. Several conditions in the neuron environment may induce excitotoxicity or contribute to its development [54]. They include physical damage of neighboring neurons with release of their glutamate content into the extracellular space [57], oxidative stress [58], oxygen deprivation (as in hypoxic/ischemic states) [59], diseases or disorders that significantly alter the CNS pH [60, 61]. Evidence has also been provided that glucocorticoids can increase neuronal vulnerability to excitotoxicity [62] by impairing energy availability and reducing the production of neurotrophic factors. Their action is mediated by the glucocorticoid receptors (GRs), belonging to a superfamily of nuclear receptors.
Glutamate homeostasis in the neuron environment is mainly controlled by astrocytes, that monitor the extracellular space through a panel of receptors to maintain the balance between their opposing functions of glutamate uptake and release [63]. Impairment of astrocytic glutamate transporters, therefore, makes neurons more susceptible to excitotoxicity. A variety of defense mechanisms, however, can be activated by neurons during excitotoxicity to decrease the damaging effects of the process [54]. Potassium channels, gamma-aminobutyric-acid (GABA) signaling, activation of adenosine A1 receptors, nitric oxide, and expression of heat-shock proteins represent reported examples [64]. Of interest in the framework of the present discussion is the available evidence that estrogens protect neurons against excitotoxic insults [65], an effect mediated by the estrogen receptors. Similar neuroprotective actions have also been documented for other hormones, such as progesterone, testosterone, and neurosteroids [65].
Some more detail on the abovementioned mechanisms and, in particular, on the role played by the receptors involved is the focus of the sections that follow.
Glutamatergic dysregulation is the key step leading to excitotoxicity [56]. Glutamate, however, does not directly damage the neurons. Indeed, the excitotoxic cascade is started by a prolonged activation of glutamate receptors [56] resulting in Na+ and Ca2+ influx and uptake by mitochondria, which may trigger the production of reactive oxygen species (ROS) and inhibition of ATP production. After glutamate receptor activation both the magnitude and duration of the increase of the intracellular Ca2+ concentration are important determinants of whether neurons degenerate. Indeed, Ca2+ concentration values much higher (low micromolar) than the typical cytoplasmic concentration in resting conditions (~100 nM) can be tolerated provided they are transient (seconds to minutes). On the other hand, even a low (~500 nM) increase of cytoplasmic Ca2+ concentration can kill the neuron if it is sustained for more than 20–30 min [53]. Glutamate receptors are classified into two main classes [66]: ionotropic glutamate receptors (iGluRs) and metabotropic glutamate receptors (mGluRs).
iGluRs are ligand-gated ion channels permeable to various cations that produce glutamate-evoked excitatory currents, while mGluRs control cellular processes.
Three types of iGluRs have been identified [67], namely N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate (KA) receptors, all involved in excitotoxic responses, the NMDA receptors being considered to play the major role due to their high permeability to calcium ions [54]. NMDA receptors are tetrameric structures composed of subunits delineating a central pore and requiring simultaneous binding of glutamate and a co-agonist (D-serine or glycine) for activation [68]. Aspartate can also activate NMDA receptors, although with a lower affinity as compared to glutamate [69]. Differences in the type of subunits and in their assembly results in different NMDA receptor subtypes with different functional features (see [54] for a discussion). In this respect, the subtype eNMDA receptor (enriched with GluN2B subunits) has been reported to be particularly involved in activating neurotoxic pathways [70]. AMPA are tetrameric receptors as well, that depending on the subunit composition exhibit different calcium permeability [71]. In particular, AMPA receptors lacking the GluA2 subunit appear to significantly contribute to excitotoxic cell death [72]. As demonstrated by structural biology studies, indeed, when GluA2 is contained within the AMPA receptor the Ca2+-permeability is profoundly decreased, due to the presence of arginine at position 607 [73]. A similar property of allowing ion influx following glutamate exposure is also exhibited by KA receptors. They, however, are permeable to sodium and potassium ions and almost impermeable to Ca2+ [74]. Furthermore, while AMPA receptors are mostly localized in the postsynaptic membrane, KA receptors are localized both pre- and post-synaptically [75, 76]. Concerning AMPA receptors, an intriguing experimental finding [77] was the observation that rat and mouse cortical neurons secrete exosomes containing two out of the four AMPA subunits which combine to form the receptor tetramer, opening the possibility of an intercellular exchange of elements of the glutamatergic decoding apparatus by microvesicles (the so called “roamer” type of volume transmission [78]) that may represent an interesting mechanism for the modulation of this transmission line.
mGluRs represent a diverse receptor family, including 8 subtypes organized into 3 groups [79]. Group 1 mGluRs (mGluR1 and mGluR5) are coupled to Gq protein. When activated, they stimulate inositol triphosphate production and the release of Ca2+ from neuronal stores, contributing to the excitotoxic process [80, 81]. On the contrary, the action of group 2 (subtypes 2 and 3) and group 3 (subtypes 4, 6, 7, and 8) mGluRs was reported to decrease NMDA receptor activity and the risk of excitotoxicity [82]. The effect appeared mediated by a regulation of voltage-gated K+ channels to induce hyperpolarization of the cell, thus reducing the opening probability of the NMDA receptors [82].
An interesting process contributing to excitotoxicity, but not related to glutamate receptors, is the sustained elevation of glucocorticoids [83]. Glucocorticoid hormones are essential for the adaptive response to stress and an important target organ is the brain [84], where the action of corticosteroids is mediated by the high-affinity mineralocorticoid receptor (MR) and the lower affinity GR. They are members of the nuclear receptor superfamily of ligand-dependent transcription factors [85]. In the absence of hormone, GR resides predominantly in the cytoplasm of cells as part of a large multi-protein complex [85]. Upon binding, GR undergoes a conformational change resulting in the dissociation of the associated proteins. This structural rearrangement allows the translocation of GR into the nucleus, where GR binds directly to target DNA sequences called glucocorticoid-responsive elements (GREs) and regulates the expression of target genes [85]. Measurements of intracellular calcium levels in hippocampal neurons exposed to glutamate and other excitatory amino acids have shown that glucocorticoids disrupt cellular calcium homeostasis and promote calcium overload [86]. The suppression of the production of neurotrophic factors and the suppression of antioxidant defense mechanisms represent other possible endangering mechanisms mediated by glucocorticoids [53]. A rapid effect of glucocorticoids on synaptic glutamate currents has also been recorded in the periventricular nucleus of the hypothalamus mediated by the endocannabinoids [87]. The rapid effects observed, however, were too fast to invoke a genomic mechanism. Thus, the possible involvement of membrane-associated GRs has been suggested [88]. In this respect, several lines of evidence are presently available supporting this glucocorticoid signaling mechanism in the brain [89, 90]. Opioid peptides are another class of stress-related hormones that can affect the excitotoxic process [53]. Dynorphin and nociceptin, for instance, can exacerbate excitotoxicity [91].
Neurons may implement a variety of mechanisms protecting against neuronal excitoxicity [54]. During action potentials, for instance, potassium channels can significantly limit neuronal excitability. A first example is provided by small-conductance calcium-dependent potassium channels [92]. Being quite sensitive to transient increases of cytosolic calcium [93] they generate a protective, hyperpolarizing signal [92, 94]. The ATP-dependent potassium channels represent a second important class of potassium channels triggering a protective mechanism. Their conductance, indeed, is enhanced by the ATP depletion that follows excitotoxic insults [95] and their activation at presynaptic sites was shown to inhibits glutamate release [96]. Further neuronal responses to cell stress include those activated by neurotrophic factors (such as BDNF) or by transcription factors (e.g., NF-κB and CREB) [53], and the expression of heat-shock proteins [97].
In the context of the present discussion, however, three more mechanisms deserve particular consideration. The first is the production of adenosine following a neural insult [98]. Adenosine, indeed, inhibits excitatory synaptic transmission, mostly through a presynaptic inhibition of glutamate release, an effect mediated by adenosine A1 receptors. In parallel, the activation of these GPCRs also modulates calcium and potassium channels at the postsynaptic level, leading to a decrease of calcium currents in response to glutamate. The second defensive mechanism involves the inhibitory neurotransmitter GABA that can decrease neural excitability by increasing Cl– influx through GABAA receptors [99]. In conditions of sustained excitation, an additional fast-acting neuroprotective mechanism has also been identified, based on the innate capacity of neurons to quench excitation by recruiting GABAB receptors [100]. Finally, of interest in the present context is emerging research demonstrating the importance of steroid hormones in the regulation of glutamatergic neurotransmission (see [65] for a specific review). The co-localization of AMPA and mGluRs with androgen or estrogen receptors in septum, amygdala, and hypothalamus of rats has been assessed by immunohistochemistry [101], and functional studies indicated a neuroprotective role played by these hormones in ischemic conditions [102]. The effect appears mediated by their receptors, as indicated by studies on estrogen receptors α and β (ERα and ERβ), showing that specific agonists of these nuclear receptors can protect hippocampal CA1 neurons during ischemia (where glutamate levels are known to be elevated) [103]. In this context, the modulating effects of progesterone, testosterone, and neurosteroids on glutamatergic neurotransmission also deserve consideration [65]. Although more specific studies on this topic would be needed, available data suggest a neuroprotective role for these hormones as well.
In addition to the well-known metabolic support given to neurons [53, 54], a key function of astrocytes in the CNS is the regulation of neurotransmitter homeostasis [104], as they can uptake synaptically released neurotransmitters, metabolize them, and release back to neurons precursors of those neurotransmitters as well as regulatory gliotransmitters. Extracellular concentrations of GABA, for instance, are under control of GABA transporters (GAT) expressed by both neurons and astrocytes. Astrocytes mainly express GAT-1 and GAT-3 subtypes and it has been estimated that about 20% of extracellular GABA is taken up by these cells [105].
Glutamatergic transmission, however, is in a special way regulated by astrocytes (see [63, 106] for specific reviews on the topic). Glutamatergic synaptic activity generates increases, that can exceed micromolar levels, of glutamate concentration in the extra synaptic space [107]. Although many CNS cells contribute to glutamate removal, astrocytes represent the most efficient elements, being responsible for about 90% of glutamate clearance [108]. The main mechanism of glutamate uptake by astrocytes is represented by two types of glutamate transporters (Na+-dependent and Na+-independent) [63]. High-affinity sodium-dependent glutamate transporters, also known as excitatory amino acid transporters (EAATs), play a major role in the removal of extracellular glutamate by astrocytes and in preventing excitotoxicity [54]. Once taken-up by astrocytes, glutamate can be amidated to glutamine, which is released to the adjacent neurons, where it is converted to glutamate (or GABA) and repackaged into vesicles for release [109]. This astrocytic glutamate-glutamine cycle [63, 110], therefore, is also involved in the maintenance of glutamate homeostasis by sustaining the synthesis of the neurotransmitter [54].
Astrocytes monitor glutamate in their environment by expressing a panel of glutamate receptors [104, 111, 112]. In these cells, the response to glutamatergic signaling is mainly mediated by mGluRs, with mGluR1, mGluR3, and mGluR5 being the most expressed [104, 111]. According to several lines of evidence [104], type 5 mGluR appears as the most relevant, mediating the response of astrocytes to glutamate in many brain areas, such as hippocampus, nucleus accumbens, and thalamus. Ionotropic receptors of the AMPA and NMDA types have also been identified in astrocytes [112, 113]. When compared to neuronal NMDA receptors, astrocytic NMDA receptors appear to exhibit some distinctive features. They, indeed, contain GluN3A receptor subunit, which lowers Ca2+ permeability [114], making astrocytes less vulnerable to glutamate-mediated excitotoxicity [113]. Moreover, astrocytic NMDA receptors lack Mg2+ block and can be activated without antecedent depolarization [115]. Glutamate receptors also play a role in modulating the activity of glutamate transporters, since group 2 mGluRs can enhance their expression, while group 1 mGluRs and iGluRs downregulate EAATs [63].
The uptake of glutamate into astrocytes can be increased by exogenously administered estradiol, the predominant estrogen in terms of activity [116]. More generally, EAATs may be affected by steroid hormones [65]. The expression of GLT-1 and EAAT3 is upregulated following ischemia, and mRNA and protein levels of these transporters resulted further increased by progesterone and estrogens as compared to ischemia alone [117]. On the contrary, glutamate uptake by astrocytes can be inhibited by adenosine signaling through the adenosine A2A receptors, which are also expressed by astrocytes, due to a physical association of A2A with Na+/K+-ATPases [118].
In addition to uptake, however, many studies indicate that astrocytes can also release glutamate to the adjacent neurons [119], and a notable subpopulation of astrocytes, selectively expressing synaptic-like glutamate-release machinery, has also been identified in defined anatomical locations [120]. Acting as a gliotransmitter [121], glutamate helps to synchronize neuron firing and modulate the excitatory or inhibitory neuronal transmission [63]. In this respect, the possible impact on synaptic transmission of a significantly increased glutamate release by astrocytes is hardly predictable [122]. Extra synaptic mGluRs should be mainly reached. If astroglial glutamate mainly involves the inhibitory mGluR2-4 (Gi/o coupled) located on the nerve terminals, a reduction of neuronal glutamate release would take place with inhibition of glutamate transmission [123] and reduced toxicity. However, if also extra synaptic and postsynaptic mGluR1 and mGluR5 (Gq coupled) are significantly activated by astroglial glutamate, an increase of glutamate synaptic strength may occur, leading to increased intracellular calcium levels [122]. Extracellular signals can modulate astrocytic glutamate release. An example is dopamine, that acting on the D2 receptors expressed by astrocytes inhibits the release of glutamate by these cells [35]. In the context of the present discussion, of interest are data showing that, in addition to glutamate, astrocytes can release D-serine (essential for NMDA receptors function), but also ATP, GABA [124], lactate [125], and citrate, chelating zinc ions inhibits NMDA receptor [126].
This dual function of astrocytes, with a balance between opposing actions of glutamate removal and release, confirms the complex role played by these cells in conditions potentially leading to excitotoxic processes [63, 113].
Another glial cell type involved in excitotoxicity is microglia, the resident immune system of the CNS. They are plastic cells, exhibiting a variety of cell morphologies and cell surface markers. They are activated by molecules from damaged neurons and, like peripheral macrophages, they are usually described as characterized by two activation states [127]. In the classical M1 phenotype microglia are cells producing proinflammatory factors. By contrast, in the M2 phenotype, they contribute to neuroprotection and injury repair. A recently characterized process of endocytosis of full-length tau protein by microglial cells [128, 129], triggered by the purinergic G protein-coupled receptor 12 (P2Y12), provides an example of protective action in Alzheimer’s disease. Increasing evidence, however, suggests that this M1/M2 dichotomy is over-simplified (see [130, 131] for detailed discussions of the topic). Different sub-types of M2-like microglia, indeed, have been identified, and simultaneous expression of classical M1 and M2 markers within individual microglial cells has been reported [132]. These findings indicate that individual microglial cells may be able to adopt complex phenotypes that exhibit both inflammatory and restorative functions. To detect molecular patterns associated with tissue damage, microglial cells exploit a large set of receptors, including all the glutamate receptor types except mGlu7 [131, 133]. Ionotropic [133, 134] and group I/II metabotropic [135] glutamate receptor activation is in general associated with the secretion of cytotoxic or inflammatory factors. Conversely, group III [131, 136] metabotropic receptors were reported to mainly trigger protective actions. Upon specific activation, microglia can also release glutamate, contributing to excitotoxicity.
A schematic view of the main mechanisms is provided in Figure 1, and examples of excitotoxic mechanisms contributing to acute and degenerative CNS diseases are reported in Table 2.
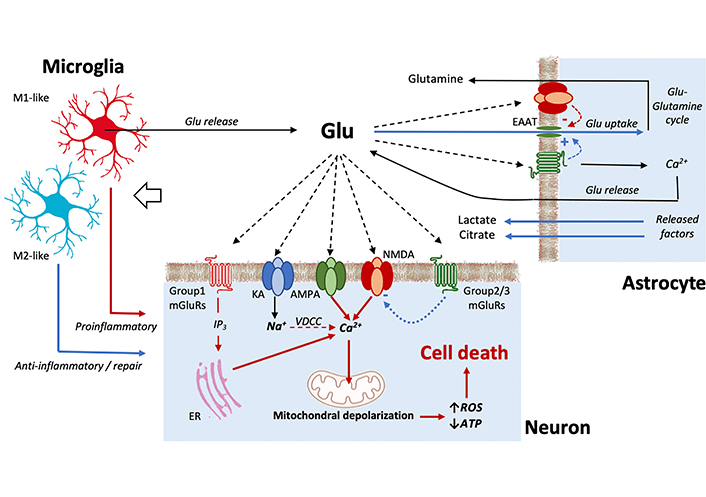
Schematic view of the main processes involved in excitotoxicity. Red lines represent processes favoring excitotoxicity; blue lines represent protective processes. In neurons [53, 54], the binding of kainate (KA) receptors results in Na+ influx and membrane depolarization, leading to the opening of voltage-dependent Ca2+ channels (VDCC). Further calcium influx is associated to the activation of some forms of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and most importantly of N-methyl-D-aspartate (NMDA) receptors. The activation of group 1 metabotropic glutamate receptors (mGluRs) also contributes to increase intracellular Ca2+ by mobilizing intracellular calcium stores. The resulting increase in cytoplasmic calcium levels may induce Ca2+ uptake into the mitochondria that, when excessive, leads to the production of reactive oxygen species (ROS) and inhibits ATP production followed by cell damage or death. The action of activated group 2/3 mGluRs, however, can decrease NMDA receptor activation and protect to some extent the cell. Concerning astrocytes [54, 63], the release of protective factors and, most importantly, their activity of glutamate uptake through excitatory amino acid transporters (EAATs) channels, represent important processes to prevent excitotoxicity. Astrocytes, however, can also release glutamate. Microglial cells monitor the extracellular environment by a large panel of receptors. Depending on the phenotype they assume when activated, microglial cells can trigger pro-inflammatory actions and release of glutamate, or neuroprotective processes [130]
Excitotoxic processes contributing to diseases of the central nervous system (CNS)
| Disease | Excitotoxic process | References |
|---|---|---|
| Ischemia |
| [137, 138] |
| Epilepsy |
| [139, 140] |
| Alzheimer’s disease |
| [141, 142] |
| [143, 144] | |
| Amyotrophic lateral sclerosis |
| [145, 146] |
| Parkinson’s disease |
| [147, 148] |
| Huntington’s disease |
| [149] |
NMDA: N-methyl-D-aspartate; AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; EAATs: excitatory amino acid transporters
In the last decades several lines of evidence demonstrated that almost all the above-mentioned receptors involved in the regulation of excitotoxicity mechanisms may establish structural, allosteric, RRI in neurons, astrocytes, and microglia [111]. These findings suggest receptor complexes as a molecular organization potentially relevant for tuning and modulating the cell processes associated with the dysregulation of the glutamatergic signaling. Thus, receptor complexes are of interest as a possible target of new pharmacological strategies for the treatment of neurodegenerative diseases associated with excitotoxic events. Examples of experimentally identified heteromers of potential interest are reported in Table 3 and will be briefly discussed in the sections that follow.
Receptor complexes containing receptors involved in excitotoxicity-associated processes
| Cell location | Receptor complex | Observed interaction | References |
|---|---|---|---|
| Neurons | AMPA-IFNγAMPA-D2NMDA-D1NMDA-D2NMDA-MORNMDA-A2ANMDA-mGluR5NMDA-D1-H3mGluR1-mGluR5mGluR2-mGluR3mGluR2-mGluR4mGluR2-mGluR7mGluR1-A1mGluR2-5HT2AmGluR5-A2AmGluR5-D2mGluR5-MORA2A-D2-mGluR5A2A-D2A1-A2AGABAA-D5A2A-GRGR-MRERα-mGluR1 | Enhanced AMPA signalingReduced AMPA signalingReduced NMDA signalingReduced NMDA signalingEnhanced NMDA signalingCross-antagonismDependent on scaffold proteinsCross-antagonismAsymmetricAsymmetricAsymmetricAsymmetricReduced mGluR1 signalingEnhanced mGluR2 signalingSynergisticReduced D2 signalingDelayed MOR desensitizationReduced D2 signalingAntagonisticReduced A1 affinitySynergisticReduced GR activitySynergisticIncreased internalization | [150][151][152][153][154][155][156][157][158][159][159][160][161][162][163, 164][165][166][23][8][167][168][169][170][171] |
| Astrocytes | A2A-D2A1-A2AA1-P2Y1A2A-OTRD2-OTR | AntagonisticAntagonisticA1 desensitizationAntagonisticSynergistic | [172][105][173][174][175] |
| Microglia | P2X4-P2X7CB1-CB2A2A-CB2GPR18-CB2 | Change of signaling pathwaysAntagonisticReduced CB2 signalingCross-antagonism | [176][177][178][179] |
AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; IFNγ: interferon-γ receptor; D1, D2, D5: type 1, 2, and 5 dopamine receptors; NMDA: N-methyl-D-aspartate glutamate receptor; MOR: μ-opiod receptor; A1, A2A: type 1 and 2A adenosine receptors; mGluR: metabotropic glutamate receptor; H3: type 3 histamine receptor; 5HT2A: type 2A serotonin receptor; GABAA: type A gamma-aminobutyric-acid receptor; GR: glucocorticoid receptor; MR: mineralocorticoid receptor; ERα: type α estrogen receptor; P2X, P2Y: purinergic ionotropic and G protein-coupled receptors; OTR: oxytocin receptor; CB1, CB2: type 1 and 2 cannabinoid receptors; GPR18: G protein-coupled receptor 18
As Table 3 illustrates, glutamate receptor subtypes were found to participate in the formation of a quite large number of receptor complexes (see [20, 180] for specific reviews).
mGluRs are class C GPCRs and dimerization is mandatory for these receptors to exert their functions. However, in addition to homodimers, this family of glutamate receptors was shown to form a number of heterodimers as well. A first set of heterodimers involves mGluRs of different types (e.g., mGluR1-mGluR2, mGluR1-mGluR5, mGluR2-mGluR3, mGluR2-mGluR4). Molecular details that govern dimerization and activation of these heterodimers have been recently explored by Wang and collaborators [159] by determining cryo-electron microscopy structures of dimers in distinct conformational states. The results showed that in the inactive states the heterodimers may assume distinct conformations at both the extracellular domains and TM domains. Upon activation, in contrast with mGluR homodimers, the heterodimers assume configurations suggesting an asymmetric mode [159, 181] of signal transduction with only one of the subunits coupling to the G protein. The choice of the subunit for G protein activation likely depends on the stability of the initial inactive conformation, highlighting the complexity of the mGluRs signaling mechanism. A second set of identified receptor complexes containing mGluRs involves receptors from a different family, such as adenosine (A1 and A2A), dopamine (D2), serotonin (5HT2A), and opioid (MOR) receptors [20], suggesting receptor complexes as a relevant molecular organization to tune glutamatergic transmission. In these heterodimers, synergistic interactions may occur. An example is the mGluR2-5HT2A receptor complex where 5HT2A-dependent phosphorylation of mGluR2 at Ser843 promotes mGluR2 signaling [182]. On the other side, antagonistic interactions were found to characterize some heterodimers containing mGluRs. An example is the mGluR1-A1 receptor complex in cerebellar Purkinje cells. In this heterodimer, the activation of adenosine A1 receptor attenuates mGluR1-mediated neuronal responses to glutamate [161]. In this context, of great interest is also obtained evidence for the existence extrasynaptically around striatal glutamate synapses of mGluR5-D2 heterodimers [165] and of D2-A2A-mGluR5 heterotrimers [23]. In these heteromers, the inhibitory action of dopamine D2 receptors on glutamate release is effectively counterbalanced by mGluR5 and adenosine A2A receptors, contributing to enhancement of the neuron excitability [20].
Heterodimers resulting from direct RRI of iGluRs (NMDA, AMPA) with other receptor types have also been identified. Both D1 and D2 dopamine receptors, for instance, may form receptor complexes with the NMDA receptor, and their activation reduces the operation of NMDA channels [152, 153]. In particular, the interaction between dopamine D1 receptor and the GluN1A subunit of NMDA appears associated with a D1-induced protection from excitotoxicity [152]. Opposingly, the interaction in the NMDA-MOR heterodimer is synergistic [154]. In this receptor complex heteromerization occurs by electrostatic interactions between the two C-termini, which dissociate when morphine activates PKC, leading to enhanced calcium currents through the NMDA channels [20]. Cross-antagonism is observed in the NMDA-A2A heterodimer [155] and in the NMDA-D1-H3 heterotrimer [157]. In both receptor complexes, indeed, the exacerbation of NMDA receptor function was reduced by antagonists of the A2A and H3 receptors respectively. Furthermore, in the heterotrimer H3 antagonists were also found to counteract the effect of an overstimulation of the D1 receptor. A more complex reciprocal interaction has been observed in the NMDA-mGluR5 heteromer. Antagonistic interactions, indeed, have been reported [156] with inhibition of the NMDA currents, but synergistic interactions have also been observed [183] and in the hippocampus, both types of actions were noted [20]. An explanation could be the dynamic association of scaffolding proteins to the heteroreceptor complex [184].
A further example of the important role played by proteins interacting with protomers is provided by the AMPA-D2 heterodimer, where D2 agonist regulation of the AMPA receptor-mediated neurotoxicity is made possible by an increased coupling of the intracellular loop 3 of the D2 receptor to NSF (ATPase N-ethylmaleimide-sensitive factor) protein [151]. An enhancement of glutamate excitotoxicity, on the contrary, characterizes the AMPA-IFNγ heterodimer [150], a neuron specific calcium-permeable complex whose formation is triggered by IFNγ.
Experimentally assessed molecular models of receptor complexes involving glutamate receptors are shown in Figure 2A.
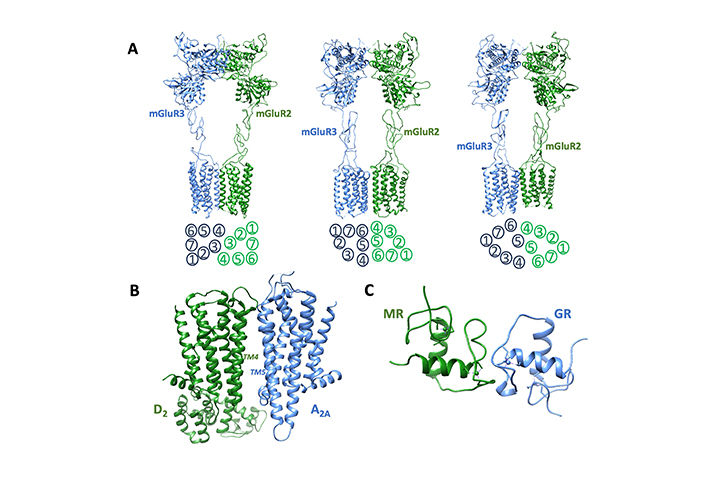
Examples of receptor complexes potentially involved in the modulation of excitotoxic processes. (A) Three inactive configurations were exhibited by the metabotropic glutamate receptor 2-3 (mGluR2-mGluR3) heterodimer according to Wang and collaborators [159]. Available data suggest that mGluR2-mGluR3 heterodimers are likely present at mossy fibers terminals in the CA3 region of the hippocampus, a region critically involved in memory processing [185]. Molecular models (Protein Data Bank codes: BJCU, BJCV, BJCZ) are shown. They differ at both the extracellular and transmembrane (TM) domains. The arrangement of TM in the three structures is schematically illustrated below each model. (B) Receptor complex formed by adenosine A2A and dopamine D2 receptors as described by Borroto-Escuela and collaborators [186]. The TM domains forming the interface are indicated. In astrocytes the A2A-D2 heterodimer controls the release of glutamate from striatal astrocytic processes and a potential role of this receptor complex in neuropsychiatric disorders has been suggested [45]. (C) Possible structure of the heterodimer between glucocorticoid and mineralocorticoid human receptors, obtained by alignment methods [187] starting from the experimentally assessed structures of the two receptor homodimers (PDB codes: 6BSF and 4TNT). It has been reported that in the hippocampus mineralocorticoid receptor (MR) and glucocorticoid receptor (GR) bind as heterodimers to the same glucocorticoid-responsive element (GRE) sites after an acute stress challenge [170]
Glutamate homeostasis and glutamatergic transmission can be regulated by a variety of extracellular signals. A first example is the typical neuromodulator adenosine [98], whose extracellular levels increase under noxious conditions. The high affinity adenosine A1 receptor plays a major role in neuroprotection, mediating a significant inhibitory action on synaptic transmission and neuronal excitability [188]. However, under noxious conditions the low affinity, facilitatory, adenosine A2A receptors are up regulated [98] and may form heteroreceptor complexes with the A1 receptors [167]. The major RRI in the heterodimer appears to be a reduction of A1 affinity by A2A agonists. Thus, at high concentrations of adenosine, able to activate A2A receptors, an increase of glutamate release was found, contributing to enhanced excitation and possible excitotoxicity [122]. This regulation, therefore, inverts the usual inhibitory action of adenosine acting on A1 receptors, with A2A actions surpassing A1-mediated effects.
As demonstrated by studies on cortico-striatal glutamate terminals, adenosine A2A receptors can also form heteromers with dopamine D2 receptors [8, 189]. Being that the D2 receptor is involved in the inhibition of glutamate release, this receptor complex significantly modulates glutamatergic transmission [190]. The A2A-D2 heterodimer (Figure 2B) is probably one of the most studied (see [191] for a recent review) and is characterized by reciprocal antagonistic interactions between the protomers. A2A agonists, indeed, lead to a reduction of the affinity of the D2 agonist-binding site [192], while D2 receptor activation inhibits the A2A-induced increase in cAMP accumulation [193] and was shown to slow and partially inhibit the binding of A2A agonists to the receptor [194]. Furthermore, new allosteric binding sites were found to emerge following the formation of the receptor complex. Homocysteine, for instance, can bind to the heterodimer without interfering with the RRI between A2A and D2 receptors, acting as an allosteric antagonist of the dopamine D2 receptor and amplifying the effect of A2A agonists [49].
Quite recent interesting findings indicate that adenosine A2A receptor is a major modulator of glucocorticoid signaling [169] as well. As mentioned before, glucocorticoids by acting on their GR receptor can disrupt cellular calcium homeostasis, promote calcium overload, and contribute to the excitotoxic process [86]. The possibility of a direct interaction between the membrane A2A receptor and the cytosolic-perimembrane GR has been suggested. It would involve direct RRI between the intracellular domains of A2A and GR, leading to a recruitment of GR and a reduction of its transcriptional activity (see [122] for a discussion). In this context, of substantial interest is a study on the hippocampus [170] showing that acute stress challenges result in an increased interaction of nuclear corticoid receptors GR and MR with their genomic recognition sites. Beyond expectancies, under the analyzed condition they interact with those sites not just as homodimers, but also as heterodimers (Figure 2C). These findings indicate an additional level of complexity in brain glucocorticoid action and may offer suggestions for future research in the almost unexplored field of RRI between nuclear receptors. In this respect, in view of the excitoprotective effects exhibited by steroid hormones (estrogens in particular) [67], of interest is evidence reporting a direct interaction between ERα and mGluR1 in rats undergoing hormonal treatment [171]. Following treatment with estradiol an increased internalization of both mGluR1 and ERα was also observed [195]. These findings support the possibility of estrogen/glutamate receptor complexes mediating an interaction between hormonal and glutamatergic signaling.
Concerning GABA transmission, a signaling pathway that can decrease neural excitability [99], heteromerization between GABAA and dopamine D5 receptors has been demonstrated by Liu and collaborators [168]. The results indicated that the formation of the complex was dependent on the co-activation of the monomers and allowed a reciprocal crosstalk, leading to a reduction in GABAA signaling and a reduced coupling of D5 to the Gs protein.
Although less investigated than in neurons, many available data indicate that RRI may play a significant role in astrocytes [111, 196].
Adenosine A2A and dopamine D2 receptors provide the first example. Both receptor types were found to form receptor heteromers in astrocyte processes [172]. From the functional standpoint, the activation of D2 receptors inhibited glutamate release, while the activation of A2A receptors, per se ineffective, abolished the D2-induced release inhibition [197]. Heterodimers between oxytocin receptor (OTR) and A2A or D2 receptors have also been recently identified [174, 175]. Functionally, both receptor complexes appear involved in the regulation of glutamatergic transmission. The interaction between OTR and D2 receptors appears facilitatory, leading to an increase of D2 receptor affinity by oxytocin. Actually, subthreshold concentrations of dopaminergic agonists (too low to activate the astrocytic D2 receptor) become effective in the presence of oxytocin [175]. On the contrary, in the A2A-OTR heterodimer the interaction was antagonistic, since the activation of the adenosine receptor abolished the oxytocin-induced inhibition of the release of glutamate by astrocytes [174].
In astrocytes a demonstrated heteromeric association involving the adenosine A1 receptor is with purinergic P2Y1 receptors [173]: As indicated by coimmunoprecipitation methods, these receptors colocalize on astroglial membranes where they organize into receptor complexes. The pharmacological results indicated that within the complex P2Y1 receptor activation induces A1 receptor desensitization. Thus, it has been suggested that this heteromer could play a significant role in the astrocytic modulation of glutamatergic neurotransmission during excitotoxic processes, when large amounts of adenosine and purines are released [173].
Adenosine receptors are also involved in the regulation of GABA uptake, which occurs via the modulation of GATs by the A1 and A2A receptors [196]. By coimmunoprecipitation and BRET assays it has been demonstrated [105] that in astrocytes these receptors can be organized as A1-A2A receptor complexes. Coupled to two different G proteins, Gs and Gi/0, both regulate GABA transport in an opposite way, with the A1 protomer mediating inhibition of GABA transport and the A2A protomer mediating facilitation of GABA transport into astrocytes. Due to difference in affinity of the two receptor types, at low levels adenosine preferentially binds the A1 protomer, while at high concentrations adenosine activates the A2A protomer. The receptor complex, therefore, was suggested to operate as a dual amplifier to control ambient GABA levels [105].
As briefly illustrated before, some receptor complexes (e.g., A2A-D2 and A1-A2A) are expressed in both neurons and astrocytes. In this respect, however, it is reasonable to assume [47] that some difference in terms of conformation and interaction of the protomers within the heteromers could occur because of differences (as, for instance, membrane potential [198] and lipid composition [199]) between the two cell types in the membrane microenvironment.
For the present discussion is also of interest the reported RRI between purinergic ionotropic receptors (P2X) (ligand-gated cationic channels) purinergic receptors expressed in microglia and controlling microglia activation [200]. P2X4 and P2X7 are the dominant forms of microglial P2X receptors [133]. Although still a matter of debate [201], the possible occurrence of P2X4-P2X7 heteromers has been reported in these cells [176], probably allowing a more sophisticated regulation of cytokine production and early inflammatory gene expression [133, 176].
As discussed before, microglia can also exert neuroprotective actions. In this respect, there is evidence that endogenous cannabinoids may favor a switch of microglial cells to an anti-inflammatory phenotype [202], and activation of cannabinoid type 2 (CB2) receptor has been proposed to be the mechanism triggering these effects [203]. A number of heteroreceptor complexes involving cannabinoid receptors in microglia have been identified. The first example is the receptor complex CB1-CB2 between the two types of cannabinoid receptors [177], where the activation of one receptor blunts the response of the partner, leading to a wide spectrum of effects when reached by endocannabinoids or by synthetic molecules acting on cannabinoid receptors [204]. CB2 receptor was also found to form heterodimers with the adenosine A2A receptor [178], and the orphan receptor G protein-coupled receptor 18 (GPR18) [179]. In the A2A-CB2 receptor complex, the blockade of the A2A receptor leads to increased CB2 signaling, while bidirectional cross-antagonism was observed in the GPR18-CB2 heteromer. Thus, both the receptor complexes are of interest from a pharmacological standpoint, since the use of antagonists targeting A2A or GPR18 receptors could be useful in the microglia-mediated protection of neuronal death in neurodegenerative diseases [178, 179].
The term excitotoxicity describes the ability of glutamate, as well as structurally related amino acids, to kill nerve cells, a process that has been proposed to take place not only in acute but also chronic diseases of the CNS [205]. Acute excitotoxic nerve cell death is thought to occur as a consequence of a variety of severe insults including cerebral ischemia, traumatic brain injury, hypoglycemia, and status epilepticus [205] and a body of evidence suggests that exposure of nerve cells to low but above normal concentrations of glutamate (or to a dysregulated glutamatergic transmission) over an extended period of time may also represent a central mechanism in the pathogenesis of many neurodegenerative diseases, including amyotrophic lateral sclerosis and Alzheimer’s disease [54].
Excitotoxicity relies on multiple cell mechanisms [53, 54]. Glutamatergic dysregulation, indeed, may occur at the receptor, transporter, or metabolic levels, leading to different types of cellular responses that ultimately culminate in neuronal death. In this respect, several lines of intercellular signaling (such as adenosine, dopamine, opioids, glucocorticoids, and steroid hormones) appear involved in the modulation of the process, acting as factors facilitating or counteracting excitotoxic mechanisms. Of particular interest is also the increasing evidence indicating the complex functional interaction between neurons, astrocytes, and microglia in influencing glutamate homeostasis [105]. The identification of this diversity of mechanisms characterizing and modulating excitotoxicity, together with the understanding that excitotoxicity is a common denominator in many CNS disorders allowed to consider a new perspective on therapy, where the targets are not specific symptoms, but the underlying processes at cell level [54]. In this regard, the quite large number of receptors expressed by both neurons and astrocytes and mediating the effect of the different signals involved in the excitotoxic machinery represent a significant pharmacological target [206].
The evidence reported and discussed in this review supports that most of these receptors can also establish direct allosteric RRI with other receptor proteins, leading to the formation of receptor complexes and allowing a modulation of signal decoding already at the membrane level, which may further expand the spectrum of available strategies. Receptor heteromers, indeed, due to the allosteric interactions between the protomers forming the complex, become endowed with a collective dynamic that significantly influences the chain of events linking ligand recognition to signal transduction [30]. In this respect, pharmacological approaches to target receptor complexes have been explored. The most followed approach has been the well-designed use of agonists/antagonists of a given protomer, since the pharmacology of some agonists/antagonists of a given protomer may show substantial differences among different receptor complexes in terms of affinity and efficacy ([51] provides the example of the A2A antagonist istradefylline, recently approved in the United States as an adjunctive treatment in Parkinson’s disease). The development of receptor-complex-specific ligands appears another very promising strategy. Indeed, the possibility to develop bivalent ligands [48] or to exploit allosteric modulators that are selective for structural domains in the heteroreceptor complexes [35, 49] has been demonstrated.
Several aspects, however, remain to be addressed to better understand the possibilities that targeting receptor complexes may offer for the modulation of excitotoxic processes. As a first point, the possibility of RRI and receptor complex formation in receptor families other than GPCRs should be considered. Some data on ion channel receptors [20] and on receptor tyrosine kinases [30] are available, while the field of cytosolic receptors is presently almost unexplored. A second point to emphasize concerns the need for a more detailed mapping of the heteromers of potential interest in order to better understand their distribution in the brain and to better characterize their location at the cellular level. In this regard, for instance, it should be noted that the research effort to identify and characterize RRI and receptor complexes has been mainly focused on neurons, while available data on astrocytes and microglia are more limited. As briefly discussed here, however, a more intense effort in pharmacological research applied to receptor complexes in glial cells may represent a topic of particular interest in the field of excitotoxic mechanisms, not only to reach a better understanding of the role of neuron-glia crosstalk but also from a therapeutical standpoint. Such a research effort, indeed, may open the possibility of exploring novel, glia-mediated strategies to address neurodegenerative disorders [196].
AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
CNS: central nervous system
EAATs: excitatory amino acid transporters
GABA: gamma-aminobutyric-acid
GAT: gamma-aminobutyric-acid transporters
GPCRs: G protein-coupled receptors
GRs: glucocorticoid receptors
iGluRs: ionotropic glutamate receptors
KA: kainate
mGluRs: metabotropic glutamate receptors
MR: mineralocorticoid receptor
NMDA: N-methyl-D-aspartate
P2X: purinergic ionotropic receptors
P2Y: purinergic G protein-coupled receptors
RRI: receptor-receptor interactions
TM: transmembrane
DG: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Supervision. CT, CC, and MM: Investigation. GM and RDC: Writing—review & editing. LFA: Conceptualization, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Ahmed Hasbi, Susan R. George
Adèle Vilette ... Peter Vanhoutte
Neelakanta Sarvashiva Kiran, Senthilkumar Rajagopal
