Review
Review

Affiliation:
1PeQuiM – Laboratory of Research in Medicinal Chemistry, Institute of Chemistry, Federal University of Alfenas, Alfenas 37133-840, Brazil
Affiliation:
1PeQuiM – Laboratory of Research in Medicinal Chemistry, Institute of Chemistry, Federal University of Alfenas, Alfenas 37133-840, Brazil
2PPGQ – Program of Post-Graduation in Chemistry, Federal University of Alfenas, Alfenas 37133-840, Brazil
ORCID: https://orcid.org/0000-0002-5524-8199
Affiliation:
1PeQuiM – Laboratory of Research in Medicinal Chemistry, Institute of Chemistry, Federal University of Alfenas, Alfenas 37133-840, Brazil
2PPGQ – Program of Post-Graduation in Chemistry, Federal University of Alfenas, Alfenas 37133-840, Brazil
ORCID: https://orcid.org/0000-0003-1674-818X
Affiliation:
1PeQuiM – Laboratory of Research in Medicinal Chemistry, Institute of Chemistry, Federal University of Alfenas, Alfenas 37133-840, Brazil
2PPGQ – Program of Post-Graduation in Chemistry, Federal University of Alfenas, Alfenas 37133-840, Brazil
Email: cvjviegas@gmail.com
ORCID: https://orcid.org/0000-0002-7799-992X
Explor Neuroprot Ther. 2024;4:411–441 DOI: https://doi.org/10.37349/ent.2024.00092
Received: April 07, 2024 Accepted: August 13, 2024 Published: October 28, 2024
Academic Editor: Nicolas Sergeant, Université de Lille, France
Alzheimer’s disease (AD) is a progressive and incurable neurodegenerative disorder, with an unknown etiology and a multifactorial pathophysiology characterized by protein misfolding, neuroinflammation, and neuronal loss. There are three well-discussed main hypotheses for the pathophysiology of AD, which are related to i) the accumulation of amyloid β (Aβ) protein aggregates in the extracellular space, ii) deposition of hyperphosphorylated tau fragments as neurofibrillary tangles, and iii) dysregulation of hemostasis of some neurotransmitters involved in the disease, such as acetylcholine (ACh) and glutamate. The association of all these factors is responsible for installing oxidative stress and neuroinflammation, which contribute to progressive neuronal death in specific brain regions. More recently, other remarkable pathological characteristics have been described, involving changes in all levels of cellular components, especially in the action and function of protein kinases. These enzymes are crucial for cellular regulation since they play a pivotal role in the phosphorylation of protein substrates by transferring a phosphate group from the ATP molecule to threonine, serine, or tyrosine residues. In more recent studies, some kinases have been especially reported by their role in inflammatory and oxidative processes associated to AD, such as cAMP-dependent protein kinase A (PKA), cyclin-dependent protein kinase 5 (CDK5), glycogen synthase kinase 3β (GSK-3β), and the microtubule affinity regulatory kinases (MARKs). Under homeostatic conditions, protein kinases act as cellular signals, directing physiological responses, but in AD pathogenesis, these enzymes have an exacerbated activity in the brain, justifying the need for a better comprehension of their function and role, and how new kinase inhibitors could lead to innovative drugs. In this context, this brief review aimed to compile the literature data related to the most recent efforts and strategies in Medicinal Chemistry in the discovery of new kinase inhibitors, opening new ways to AD therapeutics.
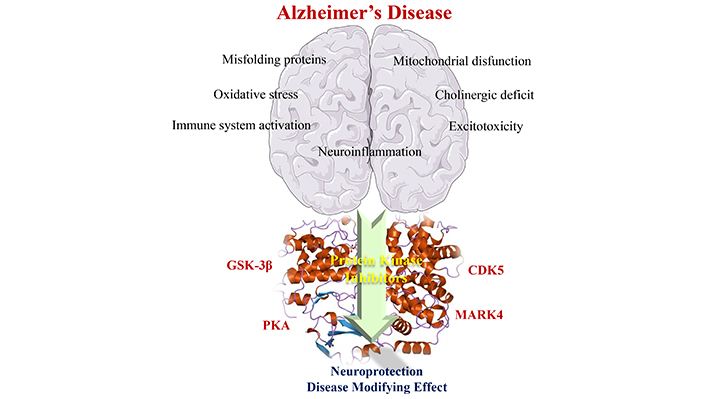
Multifactorial aspects related to AD pathophysiology have revealed the role of some protein kinases in many pathological changes in neuronal homeostasis, especially PKA, CDK5, GSK-3β, and MARKs. Part of the figure was used from pictures provided by Servier Medical Art, licensed under CC BY 3.0
Neurological disorders (NDs) are characterized by the installation of a biochemical and cellular imbalance, compromising the transmission of cellular signals and, in turn, physiological responses [1]. In this sense, Alzheimer’s disease (AD) stands out, as a pathology of multifactorial origin that alters synaptic communication and affects the cognitive and behavioral functions in patients [2–4]. Currently, AD is the most prevalent type of dementia worldwide and may affect people aged less (early AD) or more than 60 years (late AD), which is the most common risk group [2, 5]. Over the last 4 decades, different hypotheses have intensively treated the pathophysiology of AD. The cholinergic hypothesis has been the most striking and accepted one, which attributes the cholinergic deficit to a decreasing availability of neurotransmitters, especially acetylcholine (ACh). On the other hand, glutamatergic hypothesis correlates brain dysfunction and neuronal degradation to the increased glutamate levels, and finally, the amyloidogenic hypothesis attributes neuronal dysfunction to the accumulation of insoluble amyloid peptide fragments in the form of extracellular senile plaques and intracellular neurofibrils formed by hyperphosphorylated tau protein deposits [6–8].
Among the currently available treatments, we highlight the use of acetylcholinesterase (AChE) inhibitors, which aim to increase the bioavailability of ACh in the synaptic cleft, since it is the main enzyme responsible for the degradation of ACh in the neuronal environment. Based on the glutamatergic hypothesis, memantine is the most recent small molecule approved for AD therapeutics, being developed as an antagonist of N-methyl-D-aspartate (NMDA) glutamate receptors, and has been used as a single drug or in association with AChE inhibitors in most severe AD conditions [9, 10]. More recently, protein kinases (PKs) have been considered as potential therapeutic targets related to neurodegenerative and neuroinflammatory processes, which contribute to the installation and progression of neurodegeneration [11]. These enzymes are directly linked to the amyloidogenic pathway [12] and participate in the regulation of cellular activities in phosphorylation reactions, characterized by the transfer of a phosphate group from the ATP molecule to a residue of threonine, serine, or tyrosine, serving as catalysts [13]. Thus, among all known kinases, some have been highlighted in the literature for playing a pivotal role in AD pathogenesis, especially the 3’,5’-cyclic monophosphate (cAMP)-dependent protein kinase A (PKA), the cyclin-dependent protein kinase 5 (CDK5), glycogen synthase kinase 3β (GSK-3β), and microtubule affinity regulatory kinases (MARKs) [11]. These kinases are involved in the hyperphosphorylation of tau protein, which plays an essential role in the stabilization of axonal microtubules. Therefore, hyperphosphorylation of tau protein results in alteration in its functionality, leading to a collapse in the microtubule structure and, in turn, in the releasing of insoluble modified tau fragments. Once released into the neuronal environment, these protein fragments tend to aggregate and generate neurotoxic intracellular neurofibril deposits. In addition, there is also evidence of the participation of kinases in the formation of amyloid protein aggregates and deposits in the form of senile plaques in the neuronal extracellular space. Thus, considering the restricted and ineffective therapeutic arsenal available for the treatment of AD and other NDs, PKs seem to be interesting molecular targets for the development of new drugs, which may help to modulate, and even counteract the deposition of neurofibrillary tangles (NFTs) and neuroinflammatory condition [12, 14, 15].
In this context, this brief review aims to discuss the most recent contributions of Medicinal Chemistry in the discovery of small molecules acting as PK inhibitors, which may represent a genuine innovation in the development of new effective drugs, with diverse mechanisms of action, against AD. For such a goal, a literature search was carried out, covering the period from 2010 to 2023, by using PubMed, SciFinder, and Scopus as databases. The terms used as keywords in the virtual search were “neurodegenerative diseases”, “pathophysiology of Alzheimer’s disease”, and AD in association with “protein kinases” and “protein kinases inhibitors”. Thus, based on the inclusion criteria (related to the keywords used in the search and their association with AD), we have selected two papers related to PKA inhibitors, seven papers related to CDK5, seventeen to GSK-3β, and four related to MARK4. The exclusion criteria are related to non-involvement with AD, a structure previously cited in the period determined as inhibiting kinase under analysis, and availability of access.
Neurodegenerative diseases are characterized by a progressive loss of neurons in the central nervous system (CNS), culminating in loss of cognitive and motor functions of the patient [1, 10, 16–21]. As the most prevalent ND worldwide, AD was first described by the physician Alois Alzheimer in the early 20th century as a neurodegenerative pathology of multifactorial origin [2, 4, 22–24]. According to the World Health Organization (WHO), AD corresponds to 65% of the world’s dementia cases, and it is estimated that by 2030 there will be about 80 million patients affected with the disease [25]. In Brazil, approximately 50% of cases diagnosed with dementia are due to AD [26], and from 2011 to 2019 there were recorded an average of 165,000 deaths related to AD, and 65% of this population were women [27].
AD can be classified according to the age of the symptoms onset, as late, with onset after 60 years of age, and early, developed in people under 60 years old [19, 21, 28–34]. Early AD occurs in 10% of cases worldwide [28], but its symptoms usually manifest in a much faster and more aggressive way [3, 35]. One of the possible causes of the early onset has been attributed to Mendelian genetic factors, such as the autosomal dominant mutation in APP and presenilins (PSEN1 and PSEN2) genes, which are units that form the protein γ-secretase. Other possible genetic mutations are related to changes in the SORL1 receptor (an important modulator of APP traffic) and the triggering receptor expressed in myeloid cells 2 (TREM2), responsible for regulating the microglia activity, which may contribute to the amyloid plaque deposition [28]. On the other hand, late AD symptoms usually appear from the age of 65, influenced by risk factors such as age, and genetic factors, including mutations in apolipoprotein E (APOE)—protein related to increased formation of peptides Aβ [6], epigenetic factors, diet, diabetes, mitochondrial dysfunction, vascular diseases, immune system, and psychiatric disorders [36, 37]. In both forms, the progression of AD is related to an increased vulnerability to the formation and accumulation of Aβ plaques and NFTs in the brain, as well as the reduction of cholinergic neurons, which promotes the loss of brain mass leading to the development of symptoms and consequently the death of the patient [1, 21, 33, 38].
The AD’s pathophysiology can be characterized by the occurrence of distinct and interconnected physiological events, including the formation of extracellular amyloid plaques and intracellular NFTs, with reduced levels of ACh in the synaptic cleft and the increased concentration of glutamate. It is important to note although several factors contribute to trigger AD, they all favor a neuroinflammatory process, which promotes an increase in oxidative and neurodegenerative condition [32, 39–49]. The senile plaques are formed from the abnormal cleavage of APP by β- and γ-secretases, generating protein fragments prompt to oligomerization and formation of insoluble deposits of Aβ in the brain, mainly located in the amygdala regions, hippocampus, and entorhinal cortex of the temporal lobe [39–41, 50, 51]. APP is a transmembrane protein and can be cleaved by three types of secretases (α-, β-, and, γ-isoforms). The nonamyloidogenic pathway is characterized by the action of α- and γ-secretases, in which there is a first breakdown of APP by α-secretase, releasing sAPP-α in the interstice, a soluble peptide involved in the synaptic plasticity and cell survival. In the second step, sAPP-α is additionally cleaved by the action of γ-secretase, leading to the formation of soluble protein fragments known as P3 in the extracellular medium [39]. Conversely, the amyloidogenic pathway is initiated by the β-secretase-induced APP proteolysis, leading to the release of sAPP-β fragments, which exposes the C99 terminal attached to the membrane. Subsequently, γ-secretase breaks C99 structure at different sites, releasing Aβ peptides of different sizes in the medium, with Aβ1-40 fragment being originated from the cleavage of C99 between the 49 and 50 amino acids residues, and the most abundant Aβ1-42 fragment, which is resultant from C99 proteolysis between the residues 48 and 49 [6, 7, 52].
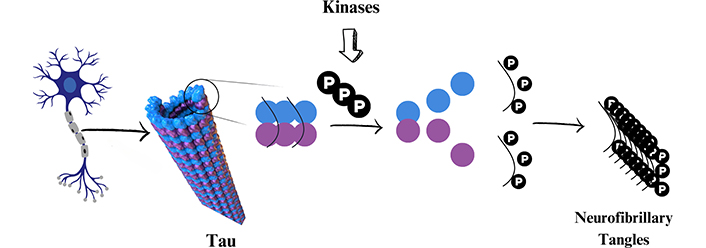
Another hallmark of AD’s pathophysiology is the massive presence of intracellular NFTs, derived from the hyperphosphorylation of tau protein, which accumulate in the entorhinal cortex and medial temporal lobes [6, 53, 54]. Tau protein is responsible for the stabilization and nutrition of axon microtubules, in addition to promoting and maintaining the link between neurons [21, 55, 56]. However, under homeostasis imbalance, tau may suffer hyperphosphorylation by PK in approximately 85 different amino acid residues, forming insoluble NFTs (Figure 1), which are redirected to the somatodendritic part of neurons and inhibiting the circulation of the glutamate receptor, culminating in the compromise of the nervous signal transmission [6, 7, 57].
Due to the formation of Aβ peptides deposits and NFTs, inflammatory and degenerative events can be observed in the brain, such as disturbance in the Ca2+ levels, impairment of mitochondrial function, oxidative stress, loss of synaptic transmission, triggering inflammatory responses, and neuronal death [39, 58–60].
In AD, a significant reduction in the concentration of ACh is often observed, whose function in the CNS is related to the maintenance of memory, learning, and attention of the individual [1, 21, 48]. The biosynthesis of ACh occurs in axon terminals and is resultant from the reaction between choline and acetyl coenzyme-A, catalyzed by the enzyme acetyltransferase (AChT). Thus, after its production, ACh is stored in vesicles and, through nerve stimuli, it is released in the synaptic cleft, and interacts with the cholinergic receptors in the postsynaptic neurons. Once in the synaptic environment, ACh is degraded by AChE into acetic acid and choline, and then part of the choline is reabsorbed into the neuronal terminals. However, in AD pathogenesis, there is a reduction in the activity of the basal nuclei responsible for synthesizing AChT. Thus, the memory decline in AD patients can be related to a significant decrease of ACh bioavailability in the synaptic clefts [48, 61].
Glutamate is another neurotransmitter with an altered activity in AD carriers, in which an exacerbated increase in synaptic communication is observed due to an intense neuronal activity caused by neuroinflammation. This occurs in response to the intense production of nitric oxide (NO) in presynaptic neurons, resulting from the exacerbated interaction of glutamate with postsynaptic receptors, especially NMDA, which stimulates the influx of Ca2+ into the cytoplasm, producing NO neuronal synthase and consequently forming NO in presynaptic neurons. Thus, once released into the synaptic clefts, free radicals stimulate greater glutamate production as a result of its interaction with specific receptors, restarting the cycle of glutamate releasing into the synaptic environment and leading to the perpetuation of the inflammatory cycle, with the consequent neuronal death [1, 20, 32, 33, 49, 62].
Currently, there are four small molecules approved by the Food and Drug Administration (FDA) as drugs against AD: donepezil (1), rivastigmine (2), galantamine (3), and memantine (4, Figure 2) [6]. In addition, some biological drugs based on monoclonal antibody therapy have been recently approved and launched on the market, such as aducanumab [63, 64] and lenanemabe [65]. The first three drugs (1–3) are selective AChE inhibitors, whereas rivastigmine is also able to inhibit butyrylcholinesterase (BuChE), and galantamine is an allosteric modulator of nicotinic receptors [66–71]. Differently, memantine is an antagonist of NMDA receptors and inhibits the action of excessive glutamate, reducing the excitotoxicity and inflammatory response observed in AD [69, 72–74].
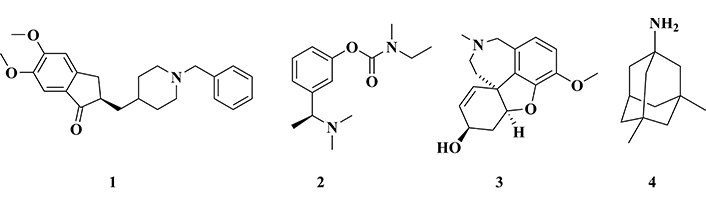
Chemical structure of the drugs used in the treatment of Alzheimer’s disease: donepezil (1), rivastigmine (2), galantamine (3), and memantine (4)
Aducanumab, a human monoclonal antibody (IgG1), acts on soluble and insoluble Aβ peptides in the medium, including the senile plaques, enabling the formation of complexes that can be metabolized [75–77]. However, its use is still contested in the literature, with claims that the results from clinical phase III are inconclusive, and that such results showed only a decrease in senile plaques after intravenous administration of high doses of the drug (10 mg/kg), without an effective and significant improvement in cognition. In addition, most patients have shown side effects, such as edema or minor bleeding in the brain, which were reversed when the treatment was discontinued. Despite its controversial effectiveness and a lack in consensus worldwide, aducanumab has been indicated for patients with mild to moderate AD [75, 76, 78, 79]. In Brazil, the Brazilian Health Regulatory Agency–ANVISA, did not approve its clinical use, and the Brazilian Society of Geriatrics and Gerontology [80] and the Brazilian Academy of Neurology [81] maintained their precautions about the use of monoclonal antibodies in AD therapy.
The most recent monoclonal antibody approved for AD treatment was lecanemab [82], a drug that acts on Aβ plaques of patients with mild AD condition and with recent diagnosis, since it selectively prevents Aβ deposition [65]. The clinical phase III study comprised 1,795 patients, where 898 received the drug. After 18 months of treatment, their clinical classification of dementia-sum of boxes (CDR-SB) decreased from 3.2 to 1.21 with the drug and 3.2 to 1.66 for placebo, showing an improvement in cognitive deficiency in approximate 25% of treated patients. The most common adverse events are related to the drug administration process (26% in the lecanemab group, and 7% in placebo), followed by reactions of amyloid-related imaging abnormalities (ARIA), with severe micro/macro brain hemorrhages (17% in the study group and 9% in placebo). Therefore, it was possible to evaluate that lecanemab effectively reduce amyloid levels and improved cognition. However, adverse reactions have been reported, showing the need of a long-term study to recommend its true clinical efficacy and safety [65].
Donamemab is another monoclonal antibody (IgG1) that targets senile amyloid plaques [83]. It is still under clinical phase I/II trials, and no conclusive results from phase III are available. In a randomized clinical trial, there was observed a 50% decrease in the amyloid plaques after 6 months of treatment with 10 mg/kg. This drug has also been well tolerated by patients, without evidence of serious adverse effects, except for few ARIA that can lead to seizures and cerebral hemorrhages (16% prevalence, 37 patients). The half-life of donanemab at the concentration of 10 mg/kg is shorter than other current monoclonal antibodies, being half metabolized in 10 days. This fact may be related to the development of antidrug antibodies emerging from treatment, present in 90% of patients in clinical phase I, after 3 months of the first dose [84]. However, some important points should be taken into account, specially associated to unchanged tau levels during the study time, showing the need for therapeutical adjustments, allied to the fact that there was no clinical distinction between the duration and severity of AD. Thus, it is necessary a more careful evaluation of the clinical phase III, and its possible application to the market [82, 85].
Thus, the currently available treatment for AD is based on drugs that act only on the mitigation of symptoms and the slowing of disease progression, which makes it necessary to explore new molecular targets for enhanced therapeutic effectiveness, enabling the discovery of new disease-modifying drugs, with diverse mechanisms of action. Therefore, PKs are targets of growing interest in the scientific scenario, since these enzymes are involved in various cellular and biochemical cascades related to AD pathogenesis, reinforcing the hypothesis that selective PK inhibitors or multifunctional PK inhibitors may represent a new frontier in AD clinics.
PKs are enzymes responsible for the phosphorylation of different cellular substrates, in various biochemical pathways, transferring a phosphate group from the ATP molecule to its specific substrate [13]. They are classified according to the type of biomolecules in which they act (substrates), and can be divided into PK, when they promote the phosphorylation of other proteins, carbohydrate-kinases protein when they act on sugars, and lipid-PK, when they exert activity on the phosphorylation of fatty acids. PKs may target the phosphorylation of sugars, constituting an important role in the maintenance of cellular energy, since they activate their metabolization. In addition, lipid kinases are enzymes responsible for the phosphorylation of lipid units, so that they can act in cell biology, promoting cell growth, intracellular movement, and signal transduction, besides other important processes in cell biochemistry [86].
PKs are responsible for the phosphorylation of different amino acid groups, being classified according to their substrate, such as serine/threonine kinases, tyrosine kinases, double specificity kinases (Ser/Thr and Tyr), and kinases capable of phosphorylating other amino acids. They play an important role in cell dynamics, being responsible for reducing activation energy and increasing the kinetics of phosphorylation reactions. They are controlled by intra- and extra-cellular stimuli, and are considered as fundamental in the orientation of cellular signals [86].
On the other hand, some PKs have been identified and specially studied for playing a pivotal role in molecular and cellular changes associated with the pathogenesis of AD. In recent decades, several kinases have been reported as potential molecular targets for AD, including GSK-3β, CDK5, MAPK, kinases c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinases 1/2 (ERK1/2), tau-tubulin kinase 1/2 (TTBK1/2), casein kinase 1α/1δ/1ε (CK1α/1δ/1ε), CK2, dual-specificity tyrosine phosphorylation and regulated kinase 1A/2 (DYRK1A/2), MARKs, phosphorylase kinase (PHK), PKA, protein kinase B (PKB/Akt), protein kinase C (PKC), protein kinase N (PKN), and calmodulin-dependent protein kinase II (CaMKII). Among all these known PKs related to neurodegeneration, PKA, CDK5, GSK-3β, and MARKs, have been especially studied for their central role in AD pathogenesis, and thus will be discussed in more detail throughout the text [11, 87, 88].
cAMP-dependent PKA was the first kinase to be discovered in the mid-1960s, with its structure determined by X-ray diffraction. PKA is particularly important in the phosphorylation of serine and threonine residues in several substrates [13, 89]. It consists of two subunits, one called as regulatory (R), where the bond with the cAMP occurs, and another one called as catalytic (C), responsible for bonding with the substrate. When R subunit is free, a tetrameric complex is formed in subunit C, leading to enzyme inactivation. Thus, when cAMP binds to PKA in R position, the release of this site occurs, and a structural change in the molecule allows the interaction of the protein to be phosphorylated in C [90]. Into the cell, PKA is responsible for phosphorylating and activating a cAMP-responsive binding protein (CREB), which modulates synaptic plasticity and is indispensable for memory maintenance (Figure 3A). However, in AD, secretases cleave the APP and are responsible for the formation of Aβ plaques, decreasing the amount of available cAMP, making it impossible to undo the tetrameric complex of PKA, and therefore, not occurring enzyme activation. As a result, there is a drastic reduction in CREB production (Figure 3B), causing the characteristic memory impairment observed in AD [11].
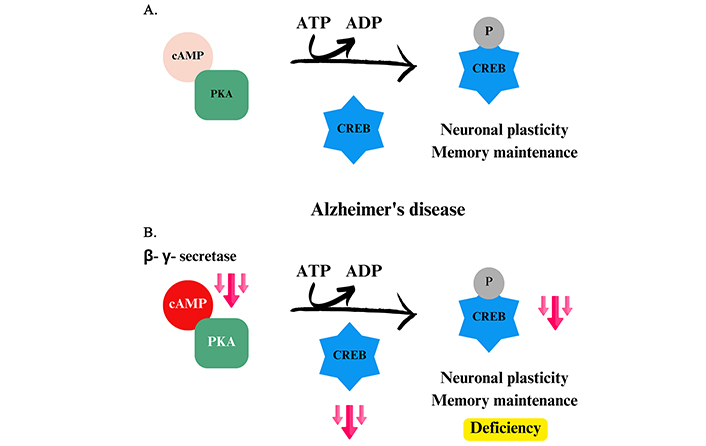
Phosphorylation of CREB by PKA. (A) Phosphorylation of CREB in a homeostatic environment, promoting neuronal plasticity and memory maintenance. (B) Imbalance caused by Alzheimer’s disease in CREB phosphorylation. cAMP: 3’,5’-cyclic monophosphate; PKA: protein kinase A; CREB: cAMP-responsive binding protein
The analysis of the PKA-CREB complex activity revealed a negative control of tau protein by reducing the gene transcription expressed in mRNA. This occurs by binding to cAMP response elements, which are linked to the expression of DNA genes, that suppress expression after binding to the complex. Thus, there is a reduction in the production of tau protein when phosphorylation of PKA-CREB occurs at its binding site (CRE). However, when a reduction in the kinase complex activity is observed, there is an exacerbation in tau levels, being more available to the action of phosphorylating enzymes, resulting in the hyperphosphorylation observed in AD patients. This reveals a mutual relationship between PKA-CREB and tau, and its maintenance is of paramount importance to ensure the stabilization of microtubules and to prevent the formation of NFTs [91, 92].
An in vitro study was carried out by Liu and co-workers [93] suggested that pre-phosphorylation of tau protein at specific serine sites by PKA (Ser214, Ser262, and Ser409) would enable subsequent tau phosphorylation by other PKs. Particularly, the activity of GSK-3β may be induced in Thr181 residues, Ser199, Ser202, Thr205, Thr217, Thr231, Ser396, and Ser422 of tau protein. In addition, experiments with monkey brains have shown that with increasing primate age, there is an increase in the rate of PKA phosphorylation of tau in the prefrontal cortex. In addition, in the communication of pyramidal neurons in dendritic spines, it was observed a decline of phosphodiesterase 4A (PDE4A), the enzyme responsible for controlling PKA signaling, and promoting its increased activity [94]. Thus, it is suggested that the maintenance of homeostasis in PKA activity is crucial, since its deregulation can promote decompensation of neuronal plasticity and its excess can provide the neurodegeneration, formation of NFTs, and Aβ plaques observed in AD.
Danshensu (5, Figure 4) is a phenolic acid found in Salvia miltiorrhiza and Prunella vulgaris var. described in the literature as a compound capable of acting in the CNS with anxiolytic and neuroprotective activities. Thus, Bae and co-workers [95] carried out an in vivo evaluation of this compound against cognitive impairment, especially the memory loss in mice. Western blotting analysis of the brains of animals submitted to classical behavioral assays, such as passive avoidance test, Y-maze, and open field tests, showed that compound 5 could promote a significant improvement in memory by the activation of dopamine receptors (D1) in the cerebral cortex. These receptors promote the phosphorylation of PKA on CREB, forming the active complex PKA-CREB, which is essential for the maintenance of cognitive ability, suggesting a possible mechanism of action to be explored in the treatment of cognitive dysfunctions related to AD [95].
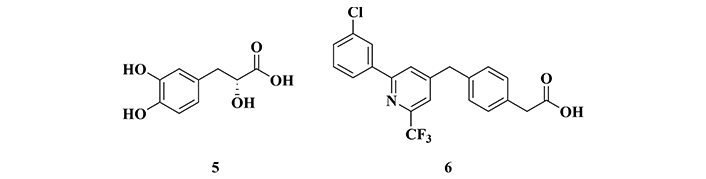
Chemical structures of danshensu (5), a phenolic acid isolated from Salvia miltiorrhiza and Prunella vulgaris, and BPN14770 (6), an allosteric inhibitor of phosphodiesterase 4 (PDE4)
In another study, Cui and co-workers [96] identified compound BPN14770 (6, Figure 4) as an allosteric PDE4 inhibitor, an enzyme responsible for maintaining the cytoplasmic levels of cAMP. Based on a set of experimental results, BPN14770 was highlighted as a new drug prototype candidate for AD, capable of promoting neuronal plasticity and memory preservation [96], possibly operating by a mechanism of action different from the other known approved drugs. Additionally, studies with animals with neuronal damage induced by Aβ1-42 deposition, revealed that BPN14770 promotes neuroprotective effects by activation of the PKA-CREB pathway, which in turn, induces structural and functional maintenance of important synaptic proteins, such as synaptophysin, and stimulated memory preservation in mice. The involvement of the PKA-CREB pathway was also evidenced by a comparative study of a control group treated with H-89, a potent PKA inhibitor, in which the beneficial effects of BPN14770 were not observed, corroborating the involvement of PKA and CREB as a probable mechanism of neuroprotection [96].
CDKs belong to a family of PK in which cyclin is the regulating molecule of phosphorylation activity. The vast majority of CDKs participate in the regulation of different stages of the cell cycle [13], and particularly CDK5 is not part of this group, being an enzyme found in post-mitotic cells, especially in the CNS [11]. The activation of CDK5 depends on two proteins located in the neuronal membranes, p35 and p39. The complex formed by this kinase, its activating proteins, and cyclin has a short half-life of around 25 minutes for p35, being very well-controlled. CDK5 has an important physiological role in the CNS, both during development and in the adult life of neurons. It is responsible for phosphorylating protein units involved in the synaptic plasticity, as well as action on the tropomyosin kinase B receptor (TrkB), and brain-derived neurotrophic factor (BDNF), the constitution of the cerebral cortex, migration of nerve cells during growth, and memory maintenance by regulating PKA signaling (Figure 5A) [11, 97].
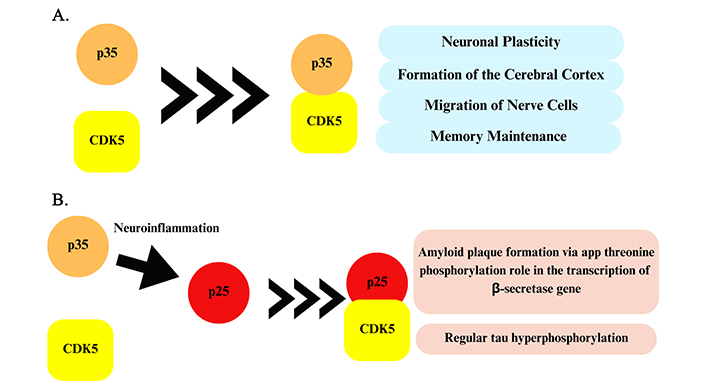
Performance of the CDK5-p35 complex and the CDK5-p25 complex. (A) Homeostatic action of CDK5 kinase and p35 protein. (B) Cleavage of p35 is caused by a neuroinflammatory environment, which culminates in the protein p25 that joins CDK5, forming a neurodegenerative complex. CDK5: cyclin-dependent protein kinase 5
Under pathological conditions of oxidative stress, overexcitation, or apoptotic signals, an excessive amount of Ca2+ enters the neurons, leading to the activation of calpains, which are calcium-dependent proteases that cleave p35 into p25 and p39 into p29 (Figure 5B). However, the CDK5-p25 complex is highly stable, which contributes to the hyperactivation of CDK5 and, consequently, to its dysregulation in the body [97]. In addition, there is an increase in the half-life of p25, which is up to 10-fold higher than p35 [98]. This factor promotes abnormal and uncontrolled hyperphosphorylation of proteins, which contributes to neurotoxicity and neurodegeneration, inducing the appearance or maintenance of diseases, such as AD, Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and encephalopathies related to prions [98, 99].
In AD, CDK5 contributes to the formation of Aβ plaques by involvement in the cleavage of APP by the phosphorylation of a threonine subunit in the APP structure and by acting on the transcription factors of genes of β-secretase, which favors the synthesis of Aβ peptides. In addition, the increase of Aβ plaques in the brain leads to an increased Ca2+ inflow to the cytoplasm, which promotes the formation of the CDK5-p25 complex, causing enzymatic hyperactivation [97]. CDK5 is also responsible for the hyperphosphorylation of numerous serine and threonine residues in tau, besides regulating other enzymes involved in the formation of NFTs, such as GSK-3β [97].
In a recent review, Maitra and Vincent [100] highlighted four possible mechanisms of action of potential CDK5 inhibitors, acting on the conversion of p35 to p25, in the stabilization of the CDK5-p25 complex, inhibition of CDK5-p25 in the catalytic site, and reduction of CDK5 and p25 expression. An in vivo study aimed at evaluating new CDK5 inhibitor candidates led to the identification of amidopyrazole derivative 7 (Figure 6), which showed a very significant effect on the reduction of enzyme activity. This derivative was able to inhibit the activity of CDK5 in a non-selective manner, acting on the CDK5-p25 complex with a nanomolar potency (IC50 = 178 nM), with good BBB permeability. Complementary studies showed a dose-dependent effect and promising druggability properties related to ADME [101].
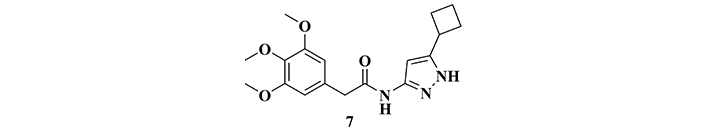
Structure of the amidopirazole derivative (7), a potent cyclin-dependent protein kinase 5 (CDK5) inhibitor
Dendrobium nobile is an Asian orchid species constituted by a high concentration of alkaloids, awakening interest in the search for anti-Alzheimer agents. A phytochemical study led to the obtaining of a mixture containing six sesquiterpene alkaloids that were identified and quantified as dendrobine (8; 92.6%), dendrobine-N-oxide (9; 3.3%), nabilonine (10; 2.0%), dendroxine (11; 0.9%), 6-hydroxy-nobilonine (12, 0.32%) and 13-hydroxy-14-oxodendrobine (13; 0.07% Figure 7). This alkaloidal extract was administered orally in mice (40 mg/kg), and submitted to an evaluation of behavioral and biological brain profiles (Western Blot), revealing a significant decrease in stress in the endoplasmic reticulum, which is an organelle responsible for protein synthesis, as well as reduction in calpain 1, CDK5 and GSK-3β levels, and a consequent decrease in the rate of tau hyperphosphorylation. Data from the behavioral evaluation were also consistent with biochemical changes, evidenced by the improvement of cognitive functions and corroborating their neuroprotective potential against AD pathogenesis [102, 103].
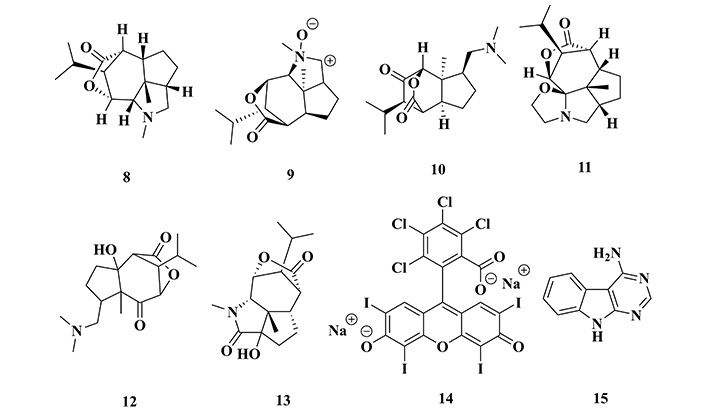
Chemical structures of the sesquiterpene alkaloids identified in Dendrobium nobile, dendrobine (8), dendrobine-N-oxide (9), nabilonine (10), dendroxine (11), 6-hydroxy-nobilonine (12) and 13-hydroxy-14-oxodendrobine (13), the synthetic rose cane (14), and pyrimido[4,5-b]indole derivative (15) with neuroprotective effects
The derivative 4,5,6,7-tetrachloro-2’,4’,5’,7’-tetraiodofluorescein (14, Figure 7), a dye also known as rose bengal, is a synthetic compound capable of promoting the stabilization of the cell cytoskeleton and preventing microtubule disorganization. Recently, in vivo studies have shown that this compound attenuates the cognitive deficit caused by the deposition of NFTs in Drosophila [104]. Based on this evidence, Mou’s group [105] evaluated a possible mechanism underlying this neuroprotective effect, which could be related to the inhibition of kinases, such as CDK5 and GSK-3β. In vivo assays revealed a decrease in CDK5 activity in the hippocampus, which was corroborated by in silico results, suggesting that compound 14 interacts with CDK5 through the ATP binding site. Thus, it was evidenced that the neuroprotective effect observed for compound 14 could probably be due to a reduced rate of tau hyperphosphorylation by CDK5 inhibition [105].
To study the neuroprotective effects of pyrimido[4,5-b]indole derivatives, Loidreau and co-workers [106] synthesized a series of eighteen 4-pyrimidoindole derivatives, which were evaluated for their inhibition profile over the CDK5-p25 complex. In vitro, results revealed derivative 15 (Figure 7) as the most promising CDK5 inhibitor (IC50 = 6 µM), being also capable to inhibit casein kinases CK1δ and CK1ε (IC50 = 0.7 µM for both), two important kinases involved in cardiac signaling [107], and 1A kinase, that is regulated by double specificity tyrosine phosphorylation (DYRK1A, IC50 = 3.1 µM) [106], mainly associated with diabetes pathogenesis [108].
The protein glycogen synthase 3 was discovered in the mid-1980s [109] and is one of the most studied PKs, capable of phosphorylating serine and threonine groups in their specific substrates. Its importance was soon recognized, since its substrates are involved in various cellular signaling processes, including the regulation of cell life, cytoskeleton organization, gene transcription, and energetic metabolization, promoting the consolidation of memory, neuron formation processing, and synaptic plasticity [110]. In addition, recent studies have shown the participation of GSK-3β in the Wingless pathways (Wnt), with a capital role during embryonic development [111]. Its name derives from its ability to phosphorylate the enzyme responsible for converting glucose into glycogen, causing its inactivation [112]. GSK can occur in two isoforms, GSK-3α originated from the gene gsk3α, and GSK-3β, transcribed from the gene GSK-3β [113, 114]. Both forms have a 98% structural homology, and their differentiation occurs in the catalytic site. GSK-3β is found in higher a concentration in the CNS [110], and is capable of autophosphorylation in the tyrosine 216 residue, which promotes its activation. The inactivation process is mediated by other PKs such as PKA, PKB, and ribosomal S6 kinases of 90 kDa (p90RSK), in the serine 9 residue and phosphorylation of p38 from MAPK in the serine 389 residue [110, 114].
Recent studies have shown that the relationship between kinase activity and AD can be associated with PSEN1, a gene responsible for the action of γ-secretase enzymes and, consequently, in the APP cleavage and formation of Aβ plaques. Another relevant fact is that GSK-3β can regulate the expression of the BACE1 gene, an enzyme responsible for the synthesis of β-secretase by the NF-κβ signaling pathway, whose function is related to the triggering of inflammatory processes [115]. As a result, GSK-3β influences the increase of the abnormal cleavage of APP by β-secretase, contributing to the formation of Aβ plaques and memory impairment observed in AD (Figure 8) [116]. Finally, the association between GSK-3β and PSEN1 also promotes tau hyperphosphorylation, contributing to the formation of NFTs [113]. Studies conducted by Ly and co-workers [116] revealed that the specific GSK-3β inhibition can induce a significant reduction in the production of β-secretase, resulting in a decreased Aβ aggregation and, therefore, being a potential target for AD treatment.
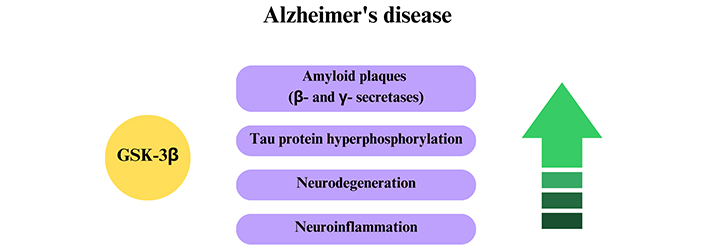
The action of GSK-3β on Alzheimer’s disease. The increase of GSK-3β activity culminates in increased pathological factors of Alzheimer’s disease (in purple)
One of the most cited GSK-3β inhibitors is lithium. The mechanism of action associated with this effect is not fully elucidated, but it is believed that Li+ can compete with Mg2+, preventing its binding to GSK-3β, causing a destabilization of ATP conformation in the enzyme and, consequently, inhibiting its catalytic activity [117]. Some marine-origin bioactive compounds have shown promising results as allosteric inhibitors of GSK-3β, such as palinurine (16, IC50 = 1.9 μM, Figure 9) and the quinone derivative VPO7 (17, IC50 = 2.8 µM, Figure 9) [117]. Based on experimental results, it has been well accepted that selective inhibition of GSK-3β could be a promising strategy for AD treatment, aiming to reduce tau hyperphosphorylation and other pathogenic factors related to neurotoxicity, protein misfolding, cognitive impairment, and motor functions, contributing to the discovery of innovative and more effective drugs.
Chemical structures of palinurine (16) and VPO7 (17), two allosteric inhibitors of GSK-3β, meridianin (18) and lignarenone (19), two bioactive natural products of marine origin, and regorafenib (20), a multi-kinase inhibitor
Other studies with marine natural products led to the identification of meridianin (18, Figure 9), an indole-pyrimidine metabolite obtained from the tunicate Aplidium Savigny, and lignarenone (19, Figure 9), obtained from mollusks of the Scaphanderlignarius genus. Both compounds showed inhibitory activities of GSK-3β, promoting the increase in the phosphorylation of the Ser9 residue, which leads to a reduction in the enzymatic activity [118].
Regorafenib (20, Figure 9) is a multi-kinase inhibitor, which targets tumor cells and tumor microenvironment, approved for the treatment of bowel cancer. Further studies aimed at expanding its applications and drug repositioning, led to the evaluation of its potential neuro-anti-inflammatory properties in AD models. In vivo studies with regorafenib being administered orally to mice for two weeks, followed by immunohistochemical analysis, showed a significant decrease in Aβ plaque deposition and tau hyperphosphorylation as a result of a marked effect on the inhibition of GSK-3β in the cortex and hippocampus. In addition, regorafenib reduced cytokine-induced neuroinflammation in in vitro experiments with microglial cells [119].
A study carried out with thieno[3,2-c]pyrazole-3-amine derivatives revealed a new chemical architecture with promising inhibitory activity of GSK-3β. Particularly, derivative 21 (Figure 10) showed a surprising in vitro nanomolar inhibitory potency (IC50 = 3.1 nM), reducing tau phosphorylation in Ser396 residue, without significant toxicity at concentrations lower than 50 μM in SH-SY5Y neuroblastoma cells type [120].
Chemical structures of pyrazole derivative (21), α-carboline derivative (22), and harmine (23), a known inhibitor of GSK-3β and its potent bioactive analogue (24), with significant GSK-3β inhibitory activities
A group of Chinese researchers studied a new series of α-carboline derivatives, that was previously proven to act as antimicrobial, anti-inflammatory, and anxiolytic, as well as showing CNS excitatory properties. A series of eighteen new compounds were synthesized and evaluated for their effects on the GSK-3β inhibition, revealing derivative (22, IC50 of 1.71 µM, Figure 10), as the most promising inhibitor, with low cytotoxicity. Additional western Blot experiments indicated that compound 22 was capable of reducing okadaic acid-induced tau hyperphosphorylation in SHSY5Y neuronal cells, which was corroborated by in silico studies. Computational data suggested that this compound could interact by H bonding involving its different nitrogen functional groups, as well as hydrophobic interactions between the carbonyl group and the catalytic site of GSK-3β [121].
The molecular framework of harmine (23, Figure 10), a known GSK-3β inhibitor, was used as a structural model in the design of a novel series of β-carboline analogues with potential neuroprotective effects. Pharmacological evaluation stood out compound 24 with the best inhibitory profile for GSK-3β (IC50 = 71 nM), being 450-fold more potent than the prototype harmine (IC50 = 32.1 μM). In addition, this compound strongly inhibited DYRK1A (IC50 = 103 nM), apparently competing with ATP, and a Western Blot analysis confirmed the inhibition of tau hyperphosphorylation in SH-SY5Y cells treated with okadaic acid. Further animal studies with APP/PS1/tau transgenic mice in the classic Morris aquatic labyrinth (MWM) behavioral model showed that administration of compound 24 led to a significant improvement in cognitive and memory capacity, with good BBB permeability. In silico studies evidenced the interaction of compound 24 at the ATP-binding site of GSK-3β, revealing some key structural subunits for structural recognition and affinity, such as the carbonyl ester function that acts as an H-binding acceptor group. Despite its moderate cytotoxicity, compound 24 was considered an interesting drug candidate prototype and a useful model for the development of new drug candidates against AD [122].
Quinoxalines have also been explored for their potential as GSK-3β inhibitors, leading to the design of different architectures of structural analogues, preserving the quinoxaline nucleus as a central pharmacophore. In a recent example, Kumar Jain and co-workers [123], searching for novel quinoxaline-based GSK-3β inhibitors, synthesized a series of compounds, in which the quinoxaline nucleus is connected to an aromatic system through a hydrazone spacer subunit. Diverse substituents on the phenyl moiety were used to furnish structure-activity relationships and their pharmacophoric insights. As a result, derivatives 25 and 26 (Figure 11) were highlighted for their best results in the selective inhibition of GSK-3β, with IC50 values of 0.27 and 0.39 μM, respectively. Molecular docking studies suggested their good affinity for the enzyme due to a H-bond interaction with a Val135 residue, and hydrophobic interactions with Arg141 and Thr138 residues, which would be the main recognition sites in GSK-3β [123].
Molecular architecture of new quinoxaline analogues (25 and 26), chemical structures of B3C (27), a bioactive dimmer of tacrine (28), and glycosylamide flavonoid derivative (30), designed by molecular simplification of flavonoid (29), with good GSK-3β inhibitory activities
Bis(propyl)-cognitin (B3C, 27, Figure 11) is a dimer derived from tacrine (28), with the presence of three methylene groups intercalating two 4-aminoquinoline fragments, with improved AChE inhibitory activity. To evaluate its potential therapeutic effects against AD, Hu and co-workers [124] investigated possible mechanisms of action of B3C in the attenuation of Aβ-induced neurotoxicity. In an in vitro model for neuroprotection, using PC12 cells treated with Aβ1-42 fibrils, it was observed a significant reduction of 81% in the GSK-3β activity. This result was then confirmed in vivo, in which a western blot analysis confirmed that the treatment of mice with compound 27 resulted in a significant reduction of phosphorylation of Ser9 residue due to GSK-3β inactivation [124].
Liang and co-workers [125] proposed a molecular simplification of iso-orientin 6-C-glycosyl compound 29 (Figure 11), a flavonoid with neuroprotective properties against NFTs and selective and reversible GSK-3β inhibition, in the structural design of a new series of glucosyl-flavonoid analogues. Thirty new compounds were synthesized and biologically evaluated, revealing compound 30 as the most potent analogue (IC50 = 0.59 μM) in the inhibition of GSK-3β, being 300-fold more potent than the original prototype 29 (IC50 = 184.9 μM). In addition, molecular modeling studies revealed important hydrophobic interactions that may explain its selectivity for the GSK-3β in detriment to the GSK-3α isoform [125].
Searching in the literature for small molecules capable of inhibiting GSK-3β, Zhang’s group [126] carried out a virtual screening approach, leading to the selection of twenty new potential bioactive ligands. Further biological studies, highlighted compounds 31 and 32 (Figure 12) for their strong in vitro inhibitory activity of GSK-3β (> 70%) at a concentration of 100 µM, and IC50 values of 8.48 and 2.19 µM, respectively. In addition, the amino acid residues Ile62, Val70, Tyr134, and Leu18 were identified as the main interaction sites of both compounds, being described as promising prototypes for the development of novel GSK-3β inhibitors with therapeutic potential against AD, especially for compound 32 which showed a 4-fold higher potency than 31 [126].
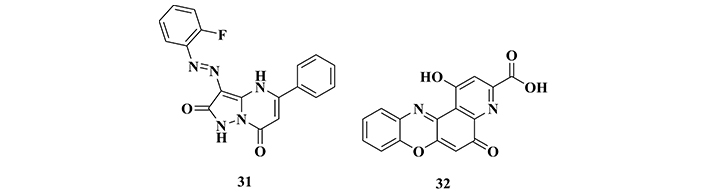
Chemical structures of compounds 31 and 32, two new selective inhibitors of GSK-3β selected from a virtual screening strategy
One of the most used and versatile strategies for the rational design of new bioactive molecules is molecular hybridization (MH). This approach is based on the combination of two or more pharmacophoric or auxophoric subunits from different bioactive compounds, originating a new single hybrid molecular architecture, that preserves or reproduces the pharmacological properties of the original prototypes [127–132]. By this approach, medicinal chemists have used MH for planning new bioactive chemical entities with innovative structural features, as well as improved potency and/or affinity, reduced potential drug-drug interactions and side effects, and especially for the design of multifunctional or multi-target directed ligands (MTDLs) [133]. Based on these principles and considering that AD is known for its multifactorial pathophysiology, with many concomitant changes in biochemical pathways, cellular dysfunctions, and imbalanced protein processing, the combination of different pharmacophores in a single molecule seems to be a promising approach to access bioligands capable to modulate multiple molecular targets concomitantly, including inhibition of PK, in addition to inhibition of AChE, monoamine oxidase B (MAO-B), BACE1, metal chelation, antioxidant, and anti-inflammatory properties [129, 130, 134–147].
Tacrine (28) and pyrimidone 33 (Figure 13) were used as prototypes to generate a new series of 4-aminoquinoline-piridyn-pirimidinone molecular hybrids. The biological evaluation addressed different targets related to AD pathogenesis led to the identification of compound 34 (Figure 13), showing a potent dual pharmacological profile by inhibiting both AChE (IC50 = 51.1 nM) and GSK-3β (IC50 = 89.3 nM). In addition, compound 34 reduced tau phosphorylation in Ser199 and Ser396 residues in SH-SY5Y cells, improving memory skills and spatial orientation in mice behavioral models, without significant cytotoxicity [148].
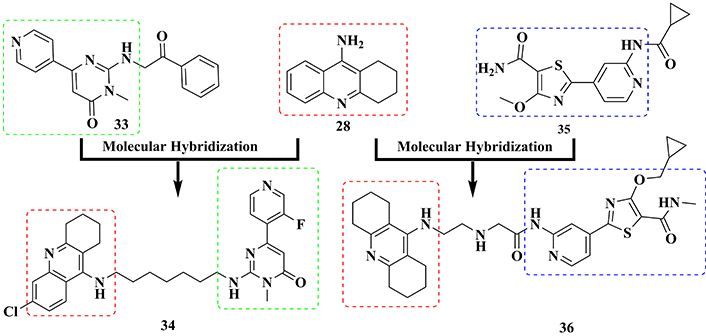
Molecular hybridization of tacrine (28) and pyrimidone (33), a known inhibitor of tau hyperphosphorylation, and with compound G1 (35), an ATP competitive inhibitor, in the molecular design of the multifunctional AChE-GSK-3β inhibitors (34) and (36), respectively
In another approach, Jiang and co-workers [149] also used tacrine as an AChE inhibitor prototype in the MH with G1 (35, Figure 13), an ATP competitive inhibitor with high affinity and ability to reduce in vivo tau hyperphosphorylation, to generate a new scaffold of thiazolopyridyl tetrahydroacridines, designed as potential dual cholinesterase-GSK-3 inhibitors. Pharmacological evaluation of 28 synthetic compounds revealed derivative 36, with a nanomolar inhibitory potency of both GSK-3β and AChE, with IC50 values of 22.2 and 1.2 nM, respectively. Further studies confirmed that this compound was effective in the inhibition of tau phosphorylation, contributing to the decrease in neurofibril deposits in the intraneuronal environment. In vivo experiments based on a behavioral scopolamine-induced amnesia assay evidenced that compound 36 was also able to restore memory and cognition in mice, suggesting it could be a promising multitarget ligand for further development addressed to AD therapy [149].
Oukoloff’s group [150] explored the combination of the structures of tacrine and valmarin (37, Figure 14), connected by an alkyltriazole fragment, to generate a new hybrid molecular architecture represented by compound 38 (Figure 14). This compound was highlighted in a series of eight tacrine-valmarin hybrids, which was evaluated in vitro against AChE and GSK-3β, exhibiting a highly potent dual inhibitory effect on human AChE (IC50 = 9.5 nM) and GSK-3β (IC50 = 7 nM). Further in silico studies suggested that the triazole subunit in the spacer moiety plays an important role in the molecular recognition, acting as an auxophore in the inhibitory activity of both enzymes. Moreover, compound 38 showed low toxicity in both human neuroblastoma (SH-SY5Y) and HuH7 liver cell cultures [150], representing another important contribution to the development of novel multitarget-directed drug candidates against AD.
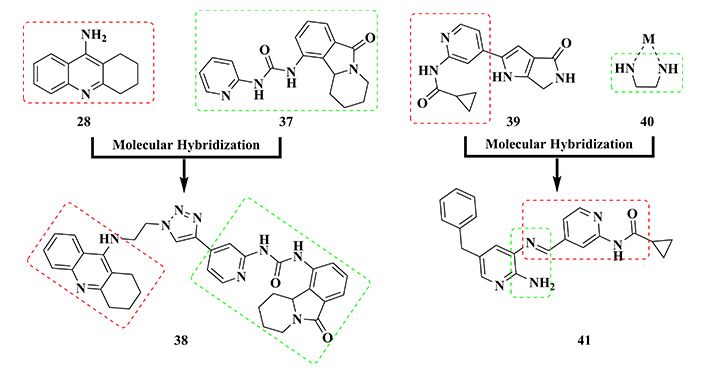
The use of molecular hybridization strategy between tacrine (28) and valmarin (37), and pyrrolopiridinone core (39) and 1,2-diamine metal chelator (40) in the design of the bioactive multifunctional GSK-3β inhibitors (38) and (41), respectively
In 2015, Sivaprakasam and co-workers [151] reported pyrrolopiridinone derivative 39 (Figure 14) as a novel orally in vivo potent GSK-3β inhibitor. Based on these findings, the structure of compound 39 was used as a bioactive model for combination with the metal chelator 1,2-diamine fragment 40 (Figure 14), aimed to generate a new molecular pattern with multifunctional properties against AD. The rationale of this designing approach took into account that imbalanced concentration of biometals, such as Cu2+, Fe3+, and Zn2+, and consequent oxidative stress and exacerbation in the ROS production are intimately associated with neuronal dysfunction and AD progress. As a result, a new series of acylamidopyridine hybrids was synthesized and biologically evaluated, leading to the identification of the imine derivative 41 (Figure 14) as the most potent GSK-3β inhibitor (IC50 = 49 nM), reducing Aβ aggregation and tau hyperphosphorylation, besides a metal chelating ability for Cu2+ and Al3+. In addition, compound 41 was able to reduce ROS formation, exhibiting a significant antioxidant activity (IC50 = 7.3 μM) and neuroprotective effect in SH-SY5Y and PC12 cell cultures, without considerable cytotoxicity. Altogether, these results suggest derivative 41 as a new multifunctional ligand to be developed as a promising drug candidate prototype for AD therapy [152].
In another approach for searching new dual GSK-3β/AChE inhibitors, Jiang and co-investigators [153] used donepezil and pyridylthiazole derivatives 42 and 43 (Figure 15), which had been already described as GSK-3β inhibitors, as molecular prototypes in the design of a new series of N-benzyl-pyridylamide hybrids. As a result, 60 new compounds were synthesized and evaluated in vitro, resulting in the identification of the 4-fluorbenzyl derivative 44, which exhibited the best dual inhibitory profile on AChE (IC50 = 0.1 nM) and GSK-3β (IC50 = 31 nM), also inhibiting BuChE (IC50 = 5.3 μM). Molecular docking studies indicated that the isonicotinamide fragment of 44 was able to interact at the ATP catalytic site through a hydrogen bond, reinforcing the affinity for GSK-3β. In addition, this compound showed adequate BBB permeability and neuroprotective properties, reducing glutamate toxicity in HT-22 cells and tau hyperphosphorylation in a dose-dependent manner in neuroblastoma N2a cells. Thus, hybrid compound 44 was featured as a promising drug candidate prototype for AD, with an innovative neuroprotective mechanism of action [153].
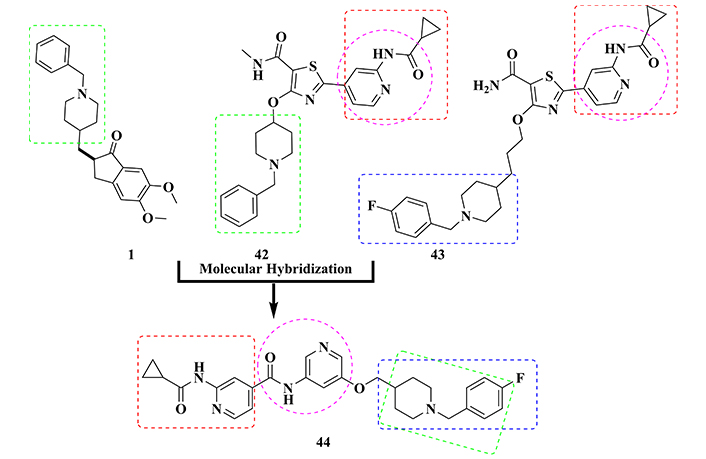
Structural design of the dual cholinesterase/GSK-3β inhibitor 44, based on the molecular hybridization between the structures of donepezil (1) and GSK-3β inhibitors 42 and 43
Based on the previous results obtained for compound 44 and still exploring the pyridylthiazole core as a pharmacophore for selective kinase inhibition, Jiang’s group [154] proposed a second generation of dual cholinesterase/GSK-3β inhibitors, based on the MH of the selective GSK-3β inhibitor 45 (Figure 16) and the N-benzyl-piperidine subunit form donepezil. The biological evaluation of 29 synthetic N-benzyl-piperidine-pyridylthiazolediamide hybrids, highlighted compound 46 as the most promising multifunctional ligand, exhibiting an improved dual inhibitory action of GSK-3β (IC50 = 0.3 nM) and AChE (IC50 = 0.3 μM). Moreover, in vivo studies evidenced its anti-inflammatory properties and good bioavailability in brain tissue after oral administration, as well as its ability to reduce LPS-induced ROS production and neuroprotective activity in neuronal cells [154]. Overall, these results not only revealed compound 46 as a promising innovative disease-modifying drug candidate prototype, but also highlighted the advantage of exploring the pyridylthiazole core in the search for multifunctional ligands suitable for further drug development against AD.
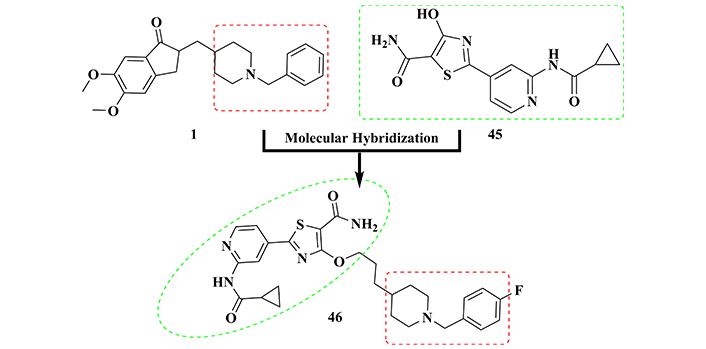
Molecular hybridization of donepezil (1) and GSK-3β inhibitor 45 in the structural design of 46, with dual AChE/GSK-3β inhibitory activity, in addition to antioxidant, anti-inflammatory, and neuroprotective properties
Donepezil (1) was also used as a structural model in the design of a novel molecular architecture of a series of potential multitarget-directed ligand candidates, in combination with chromone subunit 47, which has been described as a pharmacophore related to MAO inhibition and antioxidant activity, and with the dihydropyrimidine subunit present in SQ 32926 (48) and its triazine bioisoster (49) (Figure 17), described as acting on Ca2+ receptors and inhibiting GSK-3β, respectively. As a result, fourteen new hybrids were synthesized, and their biological evaluation revealed the racemic derivative 50, as the best multifunctional ligand, inhibiting both GSK-3β (IC50 = 29.1 μM) and AChE (IC50 = 0.06 µM). Further evaluation of the isolated enantiomers of 50 revealed (+)-50 as the eutomer, exhibiting multiple and selective inhibitory properties of GSK-3β (IC50 = 10.6 μM), MAO-B (IC50 = 11.2 μM) and AChE (IC50 = 0.09 µM), whereas the distomer (–)-50 also exhibited multifunctional effects, but with a 2-fold lower potency on GSK-3β (IC50 = 24.3 μM), being equipotent against MAO-B (IC50 = 12.3 μM) and AChE (IC50 = 0.07 μM). In addition, (+)-50 exhibited significant antioxidant and neuroprotective activities, affecting Ca2+ channel functionality, reducing Aβ and tau aggregation, and the consequent senile plaque formation and NFTs deposits, respectively, as well as good BBB permeability. Altogether, these biological data highlight compound 50, and particularly the (+)-50 isomer, for its potential innovative contribution to the development of new drugs with improved pharmacological mechanisms of action against AD [155].
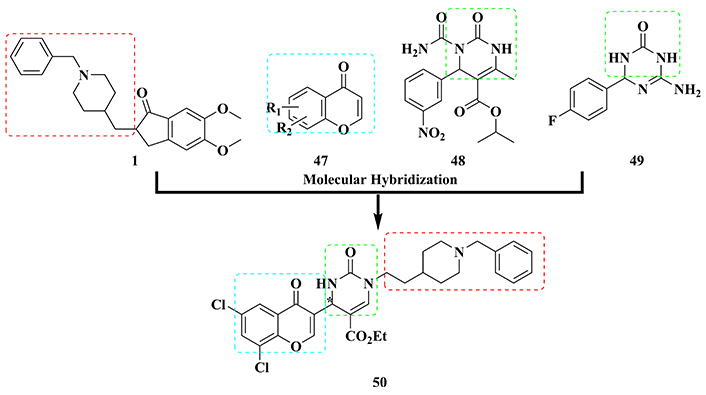
Molecular hybridization of donepezil (1), chromone core (47), SQ 32926 (48), and triazinone (49) to generate a new series of hybrid multi-target directed ligands represented by compound (+)-50
MARKs are a group of calcium/calmodulin-dependent proteins, consisting of four enzymes involved in several cellular processes, and associated with the regulation of the microtubule dynamics by phosphorylation of tau protein in the cell cycle of CNS cells, transduction of intracellular nerve signals, maturation of axons, and in conservation of synaptic plasticity [11, 156]. MARK1 is involved in the microtubule stabilization and signaling processes by the Wnt pathway. The MARK2, in turn, is present in several cellular processes, involved in the regulation of the cell cycle by Cdc25 phosphorylation, neuronal migration, signaling processes of the Wnt pathway, microtubule stabilization, determination of cell polarity and gene transcription by HDAC phosphorylation (histone deacetylases, responsible for inhibiting gene transcription in the nucleus). Similarly, MARK3 is also present in the Wnt signaling pathways, being involved in microtubule stabilization, cellular signaling pathways linked to Ras/Raf, and gene transcription by HDAC phosphorylation. In addition, MARK3 was identified as being associated with the phosphorylation of PTPH1, an important protein in cell signaling, and PKP2, the protein involved in the cellular junction. Finally, like the other three MARK isoforms, MARK4 plays a crucial role in the microtubule dynamics. MARK4 is also involved in programmed cell death, since it is upregulated in the early stages of ischemic events. However, experimental data from different studies have also suggested that the upregulation of MARK4 increases the likelihood of neurons to die, exerting a central role in neurodegeneration [156, 157]. In AD, it was evidenced that MARK2 and MARK4 have an increased enzymatic capacity, being able to hyperphosphorylate tau in the Ser262 residue. Furthermore, the genes encoding MARK4, which is more active than MARK2, are highly expressed in AD [11, 156]. Thus, after tau phosphorylation, MARK4 seems to be more likely to suffer hyperphosphorylation by other kinases, such as GSK-3β, CDK5, MAPK, and even by MARK itself, which leads to exacerbation in the microtubule destabilization and the consequent formation of NFTs, a well-known hallmark in the AD’s pathophysiology [156].
Studies on the development of selective MARK4 inhibitors as drug candidates have gained growing attention in recent years. In 2020, Turab Naqvi and co-workers [12] published a very interesting review highlighting the most recent insights related to the state-of-art of MARKs functions and their relevance as targets to address innovative and improved disease-modifying drug candidates for AD.
Particularly interested in the modulation of MARK4 activity related to AD pathogenesis, Voura and co-workers [158] explored a series of acridone derivatives as potential MARK4 inhibitors. Biological studies led to the identification of derivatives 51, 52, and 53 (Figure 18), as the most potent and selective MARK4 inhibitors, being capable of inhibiting this isoform in a dose-dependent manner with IC50 values of 1.80, 2.20, and 4.5 μM, respectively. Further in vitro experiments evidenced that these three compounds were capable of significantly reducing tau hyperphosphorylation in SH-SY5Y cells (at 10 μM concentration), but all promoted an increase in ROS production [158].
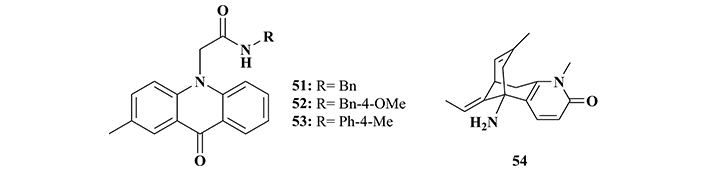
Chemical structures of acridone derivatives 51–53 and huperzine A (54), identified as MARK4 inhibitors
Shamsi and co-workers [159] carried out in silico studies to evaluate the activity of donepezil and rivastigmine, two well-known selective AChE inhibitor drugs used in AD clinics, regarding their inhibitory properties on MARK4. The results showed that both compounds were able to significantly inhibit this kinase in a very similar potency, expressed by their IC50 values of 5.3 and 6.74 μM, respectively. Apparently, the MARK4 inhibition profile is due to the possible interaction of both compounds with the ATP binding site, blocking its action in MARK4. Thus, these results were considered a relevant starting point in the design of drug candidates, with an innovative dual-target-based mechanism of action, targeting the concomitant inhibition of AChE/MARK4, and expanding the therapeutic application of the parent-approved drugs [159].
In 2023, Alrouji and co-workers [160] reported that huperzine A (54, Figure 18), a known natural product with a remarkable AChE inhibitory activity, was also able to inhibit MARK. In vitro studies showed that huperzine A significantly inhibits MARK4 (IC50 = 4.91 μM), and subsequent computational studies suggested that its affinity for this kinase could be a result of H-bond interactions in the active site. Given these experimental data, huperzine A, which has been widely reported in the literature as a prototype of AChE inhibitors, has its importance expanded as a structural model for the design of new multifunctional inhibitors for the development of new drugs against diseases related to MARK4 [160].
In another computational approach, a 1,280 structurally diverse synthetic bioactive library was studied, leading to the identification of 6 new potential MARK4 inhibitors. Further in vitro studies revealed compounds PD-173952 (55) and PD-166285 (56, Figure 19) as the most prominent nanomolar inhibitors with IC50 values of 3.3 and 3.5 nM, followed by PF-431396 (57, IC50 = 11 nM), sunitinib malate (58, IC50 = 38 nM), DMH-4 (59, IC50 = 0.27 µM), and PHA 767491 (60, IC50 = 0.8 µM). Interestingly, the most potent compound 55 showed a nanomolar inhibitory range on the other MARKs (IC50 MARK1 = 10 nM, IC50 MARK2 = 52 nM, and IC50 MARK3 = 100 nM), showing a 3.3, 15.7, and 30.3-fold higher selectivity for MARK4, respectively. On the other hand, compound 56, which showed to be equipotent to 55 in the inhibition of MARK4, was not significantly selectively related to other MARK isoforms. Despite their different inhibitory potencies, compounds 57 and 59 showed similar selectivities for MARK4, while sunitinib malate exhibited a non-selective nanomolar inhibition of MARK2 (IC50 = 13 nM) and MARK3 (IC50 = 2.7 nM). Conversely, compound 60, identified as the weaker MARK4 inhibitor, showed a 2.4 and 5-fold higher selectivity for MARK1 (IC50 = 0.33 μM) and MARK2 (IC50 = 0.16 μM), respectively. Importantly, compound 60 has been already described as a nanomolar GSK-3β inhibitor (IC50 = 22 nM), reinforcing its multi-target pharmacological profile and its importance for further development as a drug candidate with an innovative mechanism of action against AD [161].
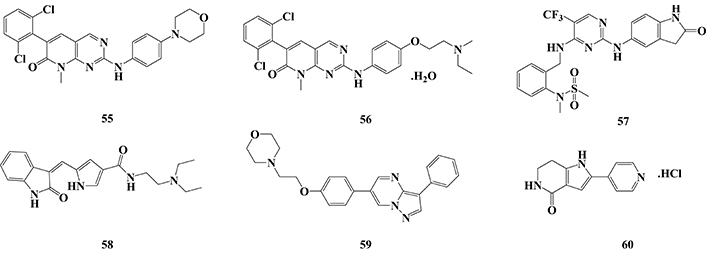
Chemical structures of MARK inhibitors PD-173952 (55), PD-166285 (56), PF-431396 (57), sunitinib malate (58), DMH-4 (59) and PHA 767491 (60)
To date, it is a consensus that pathophysiology of NDs is particularly complex and with several known biochemical and cellular changes related to the disease installation, progress, and severity, but maybe a number even higher of factors to be clarified. In AD, there is a sum of interconnected factors that lead to homeostasis impairment, either in the intracellular or extracellular environment, which promotes oxidative stress, protein misfolding, immune system activation, neuroinflammation, and neuronal death. In the last twenty years, only five small molecules were launched in the market for AD therapy, and most of them had their conception based on the cholinergic hypothesis, acting as AChE inhibitors. However, considering the lack of therapeutical effectiveness, other pharmacological hypotheses have emerged, such as the glutamatergic one, that resulted in the development of memantine. More recently, the amyloid hypothesis has received special attention, resulting in several drug candidates in clinical trials, and some innovative biological drugs, which were developed to target the clearance of Aβ plaques from the neuronal environment.
In the most recent years, more detailed studies on the multifactorial aspects related to AD pathophysiology have revealed the role of some PKs in many pathological changes in neuronal homeostasis, especially PKA, CDK5, GSK-3β, and MARKs. Thus, the modulation of such specific kinases, which are associated with microtubule stability, secretase activation, and APP cleavage, has emerged as a new paradigm in the search for innovative and more effective drug candidates for AD therapeutics. In this new pharmacological approach, the selective kinases inhibition could open new windows to counteract protein misfolding and the resultant formation of NFTs, with benefits in the reduction of neuroinflammation, oxidative stress, and neuronal injury. Therefore, a better understanding of the physiological functions of kinases in AD is an emerging issue in the literature and the first studies on the design of new small molecules addressed to selective modulation of kinases, acting as single inhibitors or MTDLs have been recently reported. In this scenario, the available data from literature show the inhibition of GSK-3β as the most common strategy adopted among medicinal chemists in the proposal of new drug candidate prototypes, but there is still no clear evidence for proof-of-concepts and druggability that could attest to the effectiveness of this approach. Until now, despite the few small synthetic molecules that have been reported as single or multifunctional kinase inhibitors focused on AD protocols, the early available results encourage these continuing efforts in the discovery of more effective disease-modifying drug candidates in the near future.
ACh: acetylcholine
AChE: acetylcholinesterase
AD: Alzheimer’s disease
Aβ: amyloid β
cAMP: 3’,5’-cyclic monophosphate
CDK5: cyclin-dependent protein kinase 5
CK: casein kinase
CNS: central nervous system
CREB: cAMP-responsive binding protein
DYRK1A/2: dual-specificity tyrosine phosphorylation and regulated kinase 1A/2
GSK-3β: glycogen synthase kinase 3β
MAO-B: monoamine oxidase B
MARKs: microtubule affinity regulatory kinases
MH: molecular hybridization
NDs: neurological disorders
NFTs: neurofibrillary tangles
NMDA: N-methyl-D-aspartate
NO: nitric oxide
PKA: protein kinase A
PKs: protein kinases
Wnt: Wingless pathways
IMFS: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. GdRRF: Conceptualization, Writing—review & editing. VSG: Writing—review & editing, Supervision. CVJ: Conceptualization, Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
Claudio Viegas Jr. and Vanessa Silva Gontijo are the Guest Editors of Exploration of Neuroprotective Therapy, but they had no involvement in the journal review process of this manuscript. The other authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The authors are grateful to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil, [#406739-2018-8], [#303804-2020-3]), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG, Brazil, [#APQ-CEX 00518-17]), Instituo nacional de Ciência e Tecnologia em Fármacos e Medicamentos (INCT-INOFAR, CNPq, Brazil, [#465249/2014-0]) for grant support and fellowships, as well as to the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Finance Code [001]) for fellowships to GdRRF. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.