 Review
Review

Affiliation:
1Instituto de Farmacología Experimental de Córdoba (IFEC-CONICET), Departamento de Farmacología, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Córdoba CP5000, Argentina
2Instituto de Investigaciones Bioquímicas de La Plata (INIBIOLP-CONICET), Facultad de Ciencias Médicas, Universidad Nacional de La Plata, Buenos Aires CP1900, Argentina
ORCID: https://orcid.org/0000-0003-1387-4922
Affiliation:
1Instituto de Farmacología Experimental de Córdoba (IFEC-CONICET), Departamento de Farmacología, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Córdoba CP5000, Argentina
ORCID: https://orcid.org/0000-0002-4030-4725
Affiliation:
1Instituto de Farmacología Experimental de Córdoba (IFEC-CONICET), Departamento de Farmacología, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Córdoba CP5000, Argentina
ORCID: https://orcid.org/0009-0004-9442-5567
Affiliation:
2Instituto de Investigaciones Bioquímicas de La Plata (INIBIOLP-CONICET), Facultad de Ciencias Médicas, Universidad Nacional de La Plata, Buenos Aires CP1900, Argentina
†These authors contributed equally to this work.
Email: mariajosebellini@med.unlp.edu.ar
ORCID: https://orcid.org/0000-0001-5737-9101
Affiliation:
1Instituto de Farmacología Experimental de Córdoba (IFEC-CONICET), Departamento de Farmacología, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Córdoba CP5000, Argentina
†These authors contributed equally to this work.
Email: claudia.herenu@unc.edu.ar
ORCID: https://orcid.org/0000-0002-3135-7024
Explor Neuroprot Ther. 2024;4:442–458 DOI: https://doi.org/10.37349/ent.2024.00093
Received: July 17, 2024 Accepted: September 24, 2024 Published: October 31, 2024
Academic Editor: Hyun-Ock Pae, Wonkwang University, Republic of Korea
Insulin-like growth factor-1 (IGF-1) elicits a variety of effects on the regulation of oxidative stress, a topic that remains shrouded in controversy. This intricate regulation plays a pivotal role in the aging process and its associated diseases. Notably, it centers around the challenge posed by endogenous antioxidant defenses, which often struggle to counteract free radicals-induced damage to various neural cell macromolecules. The interplay between IGF-1 and oxidative stress holds significant implications. Both factors are intertwined in the context of degenerative and inflammatory disruptions within the central nervous system (CNS), giving rise to dysfunctions in neurons and glial cells. These dysfunctions encompass detrimental outcomes such as excitotoxicity, neuronal attrition, and axonal impairment, all of which are closely related to behavioral irregularities. However, the complexities of IGF-1’s impact remain a topic of debate. Divergent research findings present IGF-1 as both an antioxidative agent and a catalyst to produce reactive oxygen species (ROS) in various neuropathologies. This diversity of outcomes has contributed to the ongoing controversy in the field. The present theoretical review undertakes a comprehensive vision, shedding light on the role of IGF-1 as a regulator within the mechanistic framework of oxidative stress responses. This regulatory role serves as the basis for the emergence of progressive neurodegenerative and neuroinflammatory conditions. Particularly compelling is the exploration of IGF-1 as a potential target for promising therapeutic interventions in this domain. However, the review also highlights significant limitations, including the considerations to work with this factor and the need for further research to clarify IGF-1’s role. Future perspectives should focus on refining our understanding of IGF-1’s mechanisms and exploring its therapeutic potential in more detail.
Insulin-like growth factor-1 (IGF-1) is widely recognized as a multifaceted peptide crucial for fundamental growth and developmental processes. Its structural resemblance to insulin further underscores its significance, playing a key role in growth and development by activating both the MAP kinase and PI3K signaling pathways across nearly all tissues [1, 2]. Through its transmembrane ligand-activated tyrosine kinase receptor (IGF-1R), similar to the insulin receptor, IGF-1 drives anabolic processes that promote growth and maturation with widespread biological effects [3].
In early life, IGF-1 supports brain development by facilitating neurogenesis, synaptogenesis, neurite growth, myelination, and cellular survival [4]. Along with growth hormone (GH), it forms the somatotropic axis, essential for regulating aging [5]. However, GH/IGF-1 signaling diminishes significantly by the third decade of life, leading to a complex role for IGF-1 in the aging brain due to its mixed beneficial and detrimental effects [5, 6]. Consequently, the role of IGF-1 in the aging brain is complex and debated, due to its conflicting positive and negative effects.
IGF-1R signaling in neurons triggers intracellular processes vital for synaptic function and neuronal health. It enhances calcium influx via phosphorylation of voltage-gated calcium channels, inhibits tau hyperphosphorylation, and facilitates glutamate receptor trafficking into dendritic spines, contributing to synaptic remodeling [7].
IGF-1 also influences microglial and astrocytic activity. In microglia, it promotes an anti-inflammatory phenotype via the TLR4/NF-kB pathway [8]. In astrocytes, IGF-1 signaling is essential for maintaining neurovascular unit integrity, with its disruption impairing neurovascular coupling [9] a key process in cerebral blood flow regulation. Additionally, IGF-1 modulates mitochondrial metabolism, enhances glucose uptake, and increases the availability of glutamate transporters, preventing excitotoxicity [10–12]. It also regulates astrocytic phagocytosis and inflammation through PI3K signaling [13].
IGF-1 is known to modulate the expression and activity of several key antioxidant enzymes, contributing to the defense against oxidative stress (OS). Enzymes such as superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), glutathione reductase (GR), and thioredoxin reductase play essential roles in neutralizing reactive oxygen species (ROS), which are harmful byproducts of cellular metabolism. IGF-1 enhances the activity of SOD, which catalyzes the conversion of superoxide radicals into less harmful molecules such as hydrogen peroxide, while catalase and GPx further reduce hydrogen peroxide to water, preventing oxidative damage and consequently, ROS production [14]. Similarly, thioredoxin reductase helps maintain the reduced state of thioredoxin, a crucial molecule in redox regulation [15]. By enhancing both the expression and the activity of these enzymes, IGF-1 contributes significantly to cellular antioxidative capacity, reinforcing its role in protecting cells from oxidative damage.
In certain contexts, reduced IGF-1 signaling can enhance survival in various experimental models by limiting the production of ROS, reducing autophagy and stress responses, and thereby decreasing the likelihood of age-related neurodegenerative disorders (NDs) [16–18]. Conversely, a deficiency in IGF-1 has been linked to cognitive impairments and a higher risk of poor cognitive performance in humans. Increasing IGF-1 levels in the bloodstream can mitigate these problems, aiding in neural tissue repair, enhancing cellular resilience, and reducing the buildup of age-related cellular debris [17, 19–21]. IGF-1’s multifaceted effects on neurodegenerative and neuroinflammatory disorders may lie in its role in addressing OS.
NDs encompass a cluster of incapacitating diseases characterized by progressive neuronal loss. Prominent examples include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and multiple sclerosis (MS). While the precise etiology of these largely protein-misfolding diseases remains elusive, they often arise from a complex interplay of genetic predisposition, environmental triggers, aging, and neuroinflammation. The latter pertains to the immune response within nervous tissue, involving neurons, astrocytes, oligodendrocytes, and microglia [22]. Nonetheless, a skewed redox system equilibrium may be a common denominator in the progression of these acute and chronic disorders, regulating their advancement rather than being the primary cause [23]. Biomarkers—naturally occurring molecules indicative of distinct physiological or pathological processes—play a critical role in this context [24]. Within this realm, an intricate interplay between elevated OS biomarker concentrations and diminished antioxidative biomarker levels, along with the interaction of ROS and biomolecules such as proteins and neurotrophic factors, emerges as a heterogeneous modulator. The literature suggests that both diminished and elevated IGF-1 signaling might significantly influence OS regulation in these diseases—contributing to the deposition of abnormal proteins or metabolites and cell death via ROS. Acknowledging these dualistic attributes, the aim of this study is to delve into the existing literature regarding the interplay between the IGF-1 signaling pathway and its implications for OS regulation in the context of NDs (Figure 1).
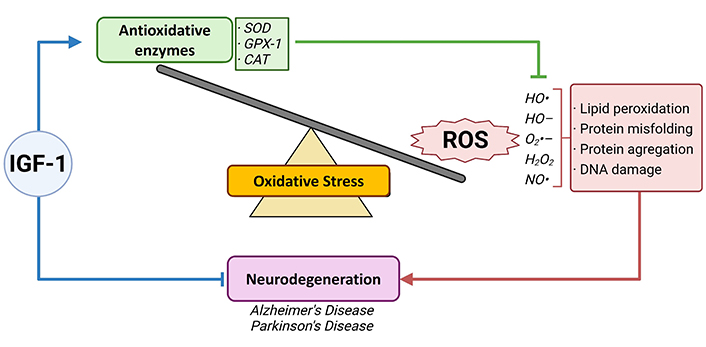
IGF-1, oxidative stress and neurodegenerative disorders. OS is characterized by either an imbalance between reactive oxygen species (ROS) production, an antioxidative system malfunctioning, or both. ROS and its detrimental effects on cell functioning are associated with several events leading to neurodegenerative disorders, such as Alzheimer’s and Parkinson’s disease, among others. Insulin-like growth factor-1 (IGF-1) is known as a neurotrophic factor and is studied as a potential therapeutic approach in different disorders of the brain. Therefore, IGF-1 arises as a formidable neurotrophic factor that could contribute to improving OS adverse effects in the neurodegenerative brain
AD stands as an age-associated neurodegenerative ailment and holds the grim distinction of being the primary cause of mortality and debilitation among the elderly across the globe [25]. Central to the identification of this neuropathology is the detection of misfolded proteins: Aβ peptides that aggregate into amyloid plaques and the tau protein that assembles into neurofibrillary tangles (NFTs) [26, 27]. The “amyloid cascade hypothesis” posits that Aβ peptides are produced in normally aging brains when the amyloid precursor protein (APP) undergoes proteolytic cleavage by β- and γ-secretases [28, 29]. Dysfunctional autophagy leading to impaired control over APP metabolism results in the accumulation of Aβ protein 42 (Aβ42) and Aβ protein 40 (Aβ40) as senile plaques (SPs) [30]. Numerous therapeutic avenues are now focusing on targetable factors that could alleviate the so-called ‘amyloid burden’ characteristic of AD brains and their distinct tissue attributes. Given that cognitive impairment is the predominant clinical hallmark of AD, several investigations propose a connection between IGF-1 and memory processes, with this neurotrophic factor playing a crucial role in specific forms of associative learning within neural circuits [31–33]. Altered levels of circulating IGF-1 have been identified as prognostic indicators in AD patients [34, 35] and animal models involving rodents [36, 37]. Significantly enriched expression of IGF-1 is particularly observed in the hippocampus, contributing to the regulation of the excitatory/inhibitory equilibrium pivotal for cognition and synaptic plasticity processes [7].
Among the central hypotheses underpinning Aβ neurotoxicity in AD etiology is OS [38]. SPs contribute to the peroxidation of lipid membranes and modify products involved in protein transformation, such as 4-hydroxynonenal (4-HNE). Additionally, they impede the dephosphorylation of the microtubule-associated protein tau. Beyond this, oxidative modification of proteins leads to a surge in carbonyl group numbers due to the oxidation of vulnerable amino acids in neurons and glia of individuals with AD. The interaction between superoxide radical (O2•−) and nitric oxide (NO) with proteins yields a potent oxidizing agent called peroxynitrite, the damaging effects of which extend beyond the conventional brain regions affected by AD, implying widespread oxidative damage in the brain [39]. In addition to protein oxidative damage, cytoplasmic RNA, as well as nuclear and mitochondrial DNA, are susceptible to oxidative modification, a consequence of hydroxylated products impacting their bases and the disruption of repair mechanisms [40].
Numerous molecular and animal studies are diligently unraveling the intricate interplay between OS and its modulation through IGF-1 signaling. Within the confines of the established selection criteria, the comprehensive assessment of Table 1 centers on the prevailing body of evidence scrutinizing the nexus of IGF-1, OS, and AD. A series of rodent investigations have indicated that a relative dearth or perturbation within the IGF-1/IGF-1R signaling pathway results in an escalation of OS in hippocampal neurons of AD transgenic mice, particularly those bearing the APPswe/PS1ΔE9 mutations (APP/PS1). Additionally, this trend was observed in mice subjected to intracerebroventricular infusion of amyloid-β oligomers (AβOs) [33] or streptozotocin (STZ) administration [41], as well as in AD-associated astrocytes [12].
IGF-1 regulation of oxidative stress in Alzheimer’s disease (AD) and related dementias
| Reference | Author (publication year) | AD model | IGF-1 model | Oxidative stress markers | Effects of IGF-1 |
|---|---|---|---|---|---|
| [33] | Selles et al., 2020 | Primary hippocampal neuronal cultures.16–19-month-old transgenic APPswe/PS1ΔE9 (APP/PS1) mice 3-month-old Swiss mice that received a single intracerebroventricular infusion of amyloid-β oligomers (AβOs). | Recombinant adenoviral vector, RAd-IGF-1 | Various types of (reactive oxygen species) ROS formation, including hydrogen peroxide, hydroxyl radical, peroxyl radicals, and peroxynitrite | IGF-1/IGF-1R pathway is affected in the AD hippocampus as well as in the hippocampus of mouse models of AD.Adenoviral-mediated expression of IGF1 protects hippocampal neurons from AβO toxicity and prevents memory impairment induced by AβOs in mice. |
| [41] | Liu et al., 2020 | Adult male Sprague–Dawley rats, intracerebroventricular injections of streptozotocin (STZ) | Physiological IGF-1 measures | Silibinin significantly decreased STZ-induced tau hyperphosphorylation (ser404) in hippocampus and cerebral cortex, markedly inhibited apoptosis of neurons induced by STZ | Silibinin increased the expression of insulin receptor and IGF-1R protein in the hippocampus. |
| [12] | Logan et al., 2018 | Mice GFAP-CreERT2/Igfrf/fYoung and old C57Bl/6Astrocyte primary cultures | Reduction in circulating levels of IGF-1 | Mitochondrial electron transport chain function and oxidative phosphorylation mitochondrial respiration, ROS generation, and OXPHOS coupling efficiency | IGFR deficient astrocytes also displayed altered mitochondrial structure and function and increased mitochondrial ROS production associated with the induction of an antioxidant response. |
| [42] | Liou et al., 2019 | Human brain tissuePostmortem frontal (Brodmann’s area [BA]8/9 and BA24) and temporal (BA38) lobe homogenates | Physiological IGF-1 measures | HNE | The brains with FTLD had significantly elevated mean levels of pTau, ubiquitin, TGF-b1, and HNE, reflecting known neuropathologic processes including aberrantly increased Tau phosphorylation, protein ubiquitination, and neuroinflammation.Oxidative stress with lipid peroxidation product accumulations, which overlap with altered expression of insulin/IGF signaling ligands and receptors.IGF-1 was increased in BA24, and IGF-1R was upregulated in all three brain regions. |
| [43] | Kuang et al., 2014 | Male 10-month-old SAMP8 and SAMR1 mice293T cells | Physiological IGF-1R measures | The lipid peroxidation product MDA, carbonylated proteins, and the DNA oxidation product 8-hydroxy-desoxyguanosine | Ligustilide induced the IGF-1 signaling inhibition and FoxO1 activation through Klotho upregulation.Klotho might alleviate oxidative stress by activating FoxO transcription factors that are negatively regulated by the insulin/IGF-1 signaling pathway, inducing the transcription of antioxidative enzymes, such as manganese superoxide dismutase and catalase, and removing ROS. |
| [44] | Cheng et al., 2005 | Transgenic mice | IGF-I gene-targeted mice (Igf1−/−) | Deposition of NT residues and enzymes involved in the production of nitric oxide, nitric oxide synthetases (NOS) | IGF-I regulates tau phosphorylation in the aging brain. |
Moreover, intriguing parallels have emerged between OS and the accumulation of lipid peroxidation products, along with altered expression patterns of insulin/IGF signaling and receptors in postmortem human brain tissue from cases of frontotemporal lobe dementia [42]. Partial amelioration of this condition was achieved through recombinant adenoviral IGF-1 gene therapy [33], which impacted various OS markers. Cell culture studies have demonstrated that IGF-1-enriched serum protects cerebellar neurons against OS induced by H2O2 [45]. Therapeutically, inhibiting IGF-1 signaling with ligustilide activates FoxO1 through the upregulation of Klotho, potentially reducing OS [43]. Conversely, silibinin administration increases the expression of insulin receptors and IGF-1R proteins in the hippocampus, thereby reducing tau hyperphosphorylation and its associated neurotoxicity [41]. However, Cheng et al. [44] reported a limited impact of IGF-1 on oxidative damage in a transgenic mice model.
In synthesis, the preponderance of experimental evidence leans towards the notion that IGF-1 downregulation exacerbates OS in the progression of AD. Simultaneously, IGF-1 is regarded as a neuroprotective and antioxidative molecule. While the dualistic attributes of IGF-1 in the aging brain generate a backdrop of controversy and contrasting assertions, these empirical inquiries are poised to furnish a deeper comprehension of the potential druggable targets within IGF-1 signaling and its role preceding the onset of disease—a face that still remains to be definitively ascertained.
PD is the second most common ND after AD, with an unclear etiology [39]. The hallmark of PD is the progressive loss and dysfunction of dopaminergic (DA) neurons in the nigrostriatal pathway [46]. Clinical manifestations of PD include motor symptoms such as rigidity, bradykinesia, and tremor, as well as a wide range of non-motor symptoms that appear in the premotor stage and progress throughout the disease [47]. The primary underlying cause of cellular dysfunction and death in PD is believed to be OS [48]. DA neurons are particularly susceptible to OS and mitochondrial dysfunction due to the presence of ROS-generating enzymes like tyrosine hydroxylase (TH) and monoamine oxidase (MAO) [39]. These neurons, located in the substantia nigra (SN), contain iron, which catalyzes the Fenton reaction and exacerbates OS [49]. This OS can initiate a cascade of events leading to protein aggregation, apoptosis, and neuroinflammation. Castilla-Cortázar et al. [50] identified parallels between the cellular and molecular mechanisms in the pathogenesis of PD and IGF-1 deficiency. Consequently, the age-related decline in serum IGF-1 levels may be a risk factor for PD development, where partial IGF-1 deficiency exacerbates brain oxidative damage, inflammation, blood-brain barrier (BBB) integrity issues, and behavioral impairments. Replacement therapy could potentially mitigate these alterations [32, 50–53]. Moreover, Bhalla et al. [54], review the pharmacological actions of IGF-1 and GLP-1 receptor activators indicating that IGF-1R analogues bind to their receptor, restoring the PI3K/AKT/mTOR signaling pathway, inhibiting neuronal apoptosis and ROS production, and thus ameliorating the pathological condition of neurodegenerative processes among them, PD.
In Table 2, we summarize the most relevant studies of IGF-1 regulation of OS in PD. Experimental exposures to pesticides are an appropriate approach to reproducing and understanding the functional effects of severe parkinsonian syndrome [55]. Among these, MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) is a prodrug to the neurotoxin MPP, lipid-soluble that penetrates the BBB and has a selective damage for SN pars compacta DA neurons as seen in PD [56]. Nadjar et al. [57], found that a reduced signaling of IGF-1 provoked directly a downregulation of OS genes and anti-inflammatory pathways in neurons under MPTP toxicity conditions. Moreover, IGF-1 acts as an indirect mediatory regulator of neuroprotective molecules, like loganin, against MPP+-induced OS in neuronal cultures [58]. Another pesticide widely used in experimental studies of parkinsonism is rotenone. Rotenone exerts toxicity by decreasing the mitochondrial membrane potential and increasing ROS production [59]. In this regard, Avila-Gomez et al. [60], reported a cellular rescue of IGF-1 to rotenone-induced ROS. Besides pesticides-derived toxins, there are other neurotoxins to induce parkinsonism, one of which is the most used: 6-hydroxydopamine (6-OHDA). This drug has affinity for catecholamines and is taken up by the dopamine transporter, which allows selective damage to DA and noradrenergic neurons of different brain areas [59]. The main molecular mechanism of 6-OHDA is auto-oxidation, leading to the formation of ROS and direct inhibition of the mitochondrial respiratory chain complex I [61]. In rat models, the supplementation with IGF-1 activates different pro-survival cascades that protects DA neurons [62] via PI3K/AKT signaling pathway [63]. All these studies suggest the pro-oxidative character of IGF-1 downregulation, but mostly its rescue and neuroprotective strategies in different antioxidant systems.
IGF-1 regulation of oxidative stress in Parkinson’s disease (PD)
| Reference | Author (publication year) | PD model | IGF-1 model | Oxidative stress markers | Effects |
|---|---|---|---|---|---|
| [57] | Nadjar et al., 2009 | MPTP | Heterozygous IGF-1R+/– mice | Oxidative stress-associated genes. | Downregulation of oxidative stress-associated genes, but also several anti-inflammatory signaling pathways.IGF-1 signaling can reduce the neuro-inflammation-dependent sensitivity of neurons to MPTP. |
| [58] | Tseng et al., 2019 | Primary mesencephalic neuronal cultures treated with MPTP | Physiological IGF-1 measures | The production of reactive oxygen species (ROS) was determined by H2DCF-DA staining, and the fluorescence of the product dichlorofluorescein (DCF) was analyzed by the Coulter CyFlow Cytometer. | Loganin protects neurons against MPP+-induced apoptotic death, neurite damage, and oxidative stress, and activations of IGF-1R and GLP-1R play important roles in the neuroprotective effects of loganin. |
| [60] | Avila-Gomez et al., 2010 | Rotenone human lymphocyte cells | Physiological IGF-1 measures | Assessment of superoxide anion radical generation and hydrogen peroxide. | IGF-1 was able to rescue lymphocytes pre-incubated with rotenone by reducing rotenone-induced ROS. |
| [62] | Ayadi et al., 2016 | 6-OHDA Sprague Dawley rats | IGF-1 mediums and serum | Changes in phosphokinases associated. | IGF-1 protects dopamine neurons against oxidative stress: association with changes in phosphokinases.IGF-1 can protect the nigrostriatal pathway in a progressive PD model and that protection is preceded by activation of key pro-survival signaling cascades. |
| [63] | Qin et al., 2021 | 6-OHDA male Wistar rats | IGF-1-related sequences | SOD activity by nitroblue tetrazolium method, MDA content by thiobarbituric acid method, GSH-Px activity by colorimetry and CAT content by double antibody sandwich method. | miR-185 targeted IGF-1 to activate the PI3K/AKT signalling pathway. Up-regulation of IGF-1 mitigated restored miR-185-mediated effects on PD rats. |
Among protein misfolding diseases we find HD, which results from the nucleotide sequence CAG repeat expansion in the HD gene, increasing the polyglutamine tail at the N-terminal of huntingtin (Htt). Mutant Htt causes the neurodegeneration of striatal and cortical pathway and leads to the characteristic cascade of events described earlier: excitotoxicity, proteasomal dysfunction, transcriptional deregulation, mitochondrial dysfunction, impaired energy metabolism, and OS [64]. Furthermore, amyotrophic lateral sclerosis (ALS) is another ND characterized by the presence of protein misfolding inclusions and, as a consequence, the loss of motor neurons in the motor cortex, brainstem, and spinal cord [65, 66]. In these inclusions, the main protein detected is TAR DNA binding protein of 43 kDa (TDP-43) and its insoluble, ubiquitinated, and cleaved C-terminal fragments. One of these latter is the ∼25 kDa C-terminal fragment (TDP-25), identified as a key player in the pathogenesis of this disease, which interacts with multiple factors—ubiquitinated aggregates, OS, mitochondria dysfunction, apoptosis-that contribute to the injury of motoneurons [67].
In addition to these ND, a group of animal and human brain diseases, characterized by neuronal loss, has been named prion diseases. Prion diseases are peculiar since they are caused by an infectious agent, prion, whose main component is an abnormal isoform (PrPSc) of prion protein (PrP) [68]. A specific sequence of PrP (106–126) has been targeted as the underlying cause of apoptosis, mitochondrial dysfunction and associated OS in the neurodegenerative mechanisms of prion disorders [69]. About cell loss and degeneration, we found that OS along with the inflammatory response constitute the primary event leading to the loss of functional neurons and demyelination in spinal cord injury (SCI) [70], axonal degeneration [71] and in aging progression [72]. Moreover, Siddiqui et al. [73], summarizes the potential role of IGF-1/GLP-1 signaling activation in intracerebral hemorrhage, concluding that the disease could be aggravated by the downregulation of IGF-1 and GLP-1 receptors and can lead related neurodegeneration disorders.
As we mentioned above, IGF-1 is well-known growth factor to exert trophic effects on neuronal regeneration and stimulate neurons and glia protein synthesis, and favor neuronal survival, inhibiting apoptosis key events of OS response [74] and being considering as a mitochondrial protector in several experimental models [75].
Table 3 includes a variety of neurodegenerative studies underlying IGF-1 regulation of OS, with the exception of AD and PD mentioned above. In protein misfolding diseases experimental models we found that increased OS, elicited by ligases of the ubiquitin-proteasome system (UPS), degrades IGF-1R protein in a model of neurodegeneration induced by zinc [76]. Thus, IGF-1 deficiency increases OS response and frailty in aging mice [77]. Furthermore, exogenous treatment with this neurotrophic factor serum reduces mitochondrial ROS production in models of HD [78, 79]. The same phenomenon was observed with the delivery of adenoviral IGF-1 in a model of ALS [68]. Regarding prion disorders, Park et al. [69], confirmed that replacement therapy of IGF-1 prevents ROS induction.
IGF-1 regulation of oxidative stress in other neurodegenerative disorders (NDs)
| Reference | Author (publication year) | ND model | IGF-1 model | Oxidative stress markers | Effects |
|---|---|---|---|---|---|
| [67] | Li et al., 2017 | Amyotrophic lateral sclerosisTDP-25 cells | AAV9-IGF-1 | Nuclear factor erythroid 2-related factor 2 (Nrf2), as quinone oxidoreductase 1 (NQO1), and the regulatory subunit of glutamate cysteine ligase (GCLM). MitoTracker® Red CM-H2XRos | Delivery of AAV9-IGF-1 to TDP25 cells protected mitochondria, such as improved mitochondrial swelling, cristae dilation, and reduced the level of reactive oxygen species (ROS). |
| [69] | Park et al., 2012 | Prion diseases PrP (106–126) treatmentSH-SY5Y human neuroblastoma cell line | IGF-1 reagent | DCFH-DA assay was carried out to ascertain ROS generationAntioxidant agents (GSH and NAC) | IGF-1 prevents PrP (106–126)-induced ROS generation.IGF-1 treatment prevents prion-mediated mitochondrial dysfunction and neurotoxicity in neuronal cells. |
| [71] | Calixto et al., 2012 | C. elegans axonal degenerationMice C57BL/6J | Physiological IGF-1 measures | ROS analyzed with the fluorogenic probe CellROX Deep Red Reagent | Insulin/IGF-1-like signaling is essential in maintaining neuronal homeostasis under pro-degenerative stimuli and identifies ROS as a key intermediate of neuronal degeneration in vivo. |
| [77] | Kwak et al., 2012 | In vitro neurodegeneration induced by zinc (AD, ALS, PD)Primary cultured cortical neurons | Physiological IGF-1 measures | H2O2 and zinc | NEDD4-1 is upregulated by a variety of neurotoxins that elicit oxidative stress in neurons, leading to IGF-1R protein degradation via ubiquitin-proteasome system (UPS). |
| [78] | Tarantini et al., 2017 | Male mice homozygous for a floxed exon 4 of the Igf1 gene | IGF-1 deficiency | Oxidative stress-mediated MMP | IGF-1 deficiency impaired hypertension-induced adaptive media hypertrophy and extracellular matrix remodeling, which together with the increased MMP activation likely also contributes to increased fragility of intracerebral arterioles. |
| [79] | Ribeiro et al., 2014 | Huntington’s diseaseKnock-in striatal cells | IGF-1 mediums and serum | Mitochondrial-driven ROS generation | Insulin and IGF-1 treatment further reduced mitochondrial.ROS production and normalized mitochondrial SOD activity. |
| [80] | Sadagurski et al., 2011 | R6/2 and R6/2•Irs2ntg miceHuntington’s disease | Role of insulin/IGF-IRS2 signaling | [NAD+]/[NADH] ratioATP concentrationROS concentration in brain extracts was carried out using the cell-permeable dye 2′,7′-dichlorofluorescein diacetate (H2DCFDA) | Increasing Irs2 levels in the brains of R6/2 mice significantly reduced life span and increased neuronal oxidative stress and mitochondrial dysfunction. |
| [81] | Allahdadi et al., 2019 | Spinal cord injurySCI contusion model in C57Bl/6 mice | Bone marrow-derived mesenchymal stem cells, overexpressing IGF-1 | NOS2 (type I activation marker, Inos), nitrite concentration | Nitrite concentration was significantly reduced in the BMMSC-IGF-1-treated group when compared to saline. |
| [82] | Gustafsson et al., 2004 | Diabetic neurite degeneration induced by hyperglycemia SH-SY5Y cells | Human recombinant IGF-1 in insulin-free N2 medium | UCP3 protein expressionMMP, intracellular levels of ROS, and total glutathione | IGF-1 prevented a decline in UCP3 protein expression following glucose treatment. Simultaneously, glucose-induced increases in MMP and mitochondrial ROS production and reduction in the number of neurites per cell were abolished or significantly reduced. |
Moreover, IGF-1 signaling is implicated in the maintenance of neuronal homeostasis under ROS conditions in axonal degeneration [71, 80] and its overexpression through cell delivery reduces the amount of nitrite concentration in a model of SCI [81]. Another mechanism of IGF-1 to reduce ROS production is through the modulation of uncoupling proteins (UCPs) which lower the mitochondrial inner membrane potential (MMP) in a culture model of neurite degeneration [82]. In accordance with the studies mentioned before, we found the same pattern of IGF-1 regulation in different models of neurodegeneration: a correlation between IGF-1 deficiency and increased OS response and IGF-1 overexpression and reduced OS response.
There is a direct and consequent relationship between neurodegeneration and neuroinflammation, with OS as an intrinsic mechanism of these pathological processes. In this context, glia and immune cells are crucial players in regulating inflammatory responses in the nervous system. The cascade of events, already mentioned, of neurodegeneration may facilitate immunocyte infiltrations and mediate neuroinflammation [83] or vice versa playing synergistic roles. Moreover, it is settling a self-toxic feedback loop between inflammatory cytokines, mitochondrial dysfunction and the production of ROS [84], since under pathological conditions mitochondrial membranes and DNA are damaged, and the release of mitochondrial components is induced. These mitochondrial components are recognized by pattern recognition receptors (PRRs) as DAMPs, indicating that cells are damaged, which trigger the innate immune response [85, 86]. This situation activates the NLRP3 inflammasome complex acting as a sensor of mitochondrial dysfunction, and activation of this complex leads to the production of IL-1β [84]. Among therapeutic approaches to modulate this loop, IGF-1 properties allow it to exert anti-inflammatory actions by the reduction of brain cytokine expression via the downregulating of glia activation [52]. Moreover, our group recently demonstrated that IGF-1 gene therapy resulted in significant changes in several parameters related to microglia and astrocyte phenotypes (particularly in the CPu and dorsal hippocampal areas), supporting the use of IGF-1 as a therapeutic molecule for neuroinflammatory processes [87].
According to our selection criteria, Table 4 lists the studies related to IGF-1 regulation of OS in neuroinflammatory diseases. Astrocytes protect neurons against oxidative injury and IGF-1 is implicated in this astrocyte neuroprotection in vitro and in vivo experimental models of stroke [88] and oxidative damage [89], elucidating astrocytes rescuing effect on neurons. Regarding microglia, IGF-1 reduces microglia activation, associated OS and proinflammatory molecules in models of spread depression [90] and in a LPS model of neuroinflammation [91]. Moreover, exogenous administration of IGF-1 regulation reduces inflammatory cytokines [92]and the levels of reactive species [93]. In agreement with the analyses of the previous tables, there is a positive modulation of IGF-1 to prevent or reduce distinct inflammatory cells and molecules release.
IGF-1 regulation of oxidative stress in neuroinflammation
| Reference | Author (publication year) | Neuroinflammatory model | IGF-1 model | Oxidative stress markers | Effects |
|---|---|---|---|---|---|
| [88] | Hayes et al., 2021 | StrokeE17-19 rat neuronal, astrocytes, and endothelial cell culturesGlutamate-induced toxicity | Exogenous IGF-1IGF-1R inhibition with PPP | Dichlorodihydrofluorescein diacetate (DCFDA) | IGF-1 can protect neurons from glutamate.Inhibition of IGF-1R impairs the neuroprotective capabilities of astrocytes.Inhibition of IGF-1R significantly increases ROS production in both astrocytes and endothelial cells. |
| [89] | Genis et al., 2014 | Postnatal rats and mice for in vitro cultures (P0–3 days for astrocytes and P7 for neurons) and 3-month-old mice for in vivo experiments | Physiological IGF-1 measuresIGF-1 deficiency and blockade | H2O2 | IGF-1 signaling participates in astrocyte neuroprotection against oxidative injury.IGF-1 decreases the pro-oxidant protein thioredoxin-interacting protein 1 and normalizes the levels of reactive oxygen species. |
| [90] | Grinberg et al., 2013 | Rat hippocampal slice culturesSpreading depression | IGF-1 mediums | CellRox™ | IGF-1 mitigated microglial but not astrocytic oxidative stress from SD. |
| [91] | Tien et al., 2017 | Intracerebral injection of LPS to 5-day-old Sprague-Dawley rat pups | Intranasal administration of IGF-1 | COX-2 expression | The protective effects of IGF-1 are associated with the decrease in microglia activation and reduction of neuronal oxidative stress by suppressing neuronal COX-2 expression in the rat SN, VTA, OB, and LC. |
| [92] | Bake et al., 2014 | StrokeMiddle-aged female Sprague Dawley rats | Intracerebroventricular infusion of IGF-1 (human recombinant) | Levels of thiobarbituric reactive species (TBARS) | IGF-1 reduces infarct volume in the acute phase of ischemia-reperfusion injury.IGF-1 did not attenuate TBAR levels in ischemic tissue.IGF-1 mediated regulation of inflammatory cytokines in the postischemic brain. |
| [93] | Montivero et al., 2021 | Traumatic brain injuryMale Wistar rats | Adenoviral IGF-1 gene therapy | Protein (AOPP) and lipids (MDA) peroxidation | Locally administered IGF-1 gene therapy a few minutes after TBI, significantly reduces recognized oxidative stress markers, such as the protein and lipid (AOPP and MDA concentrations respectively) peroxidation increments observed at 24 h and impairs its long-term maintenance, also by reducing their levels 7 days after TBI. |
Throughout this review, it is evident that inflammation is a common factor in various central nervous system (CNS) neurodegenerative pathologies, often exacerbated by a decrease in IGF-1 pathway activity.
The brain produces natural antioxidants, including growth factors like IGF-1, which may have regenerative and neuroprotective actions partly through antioxidant mechanisms. Alterations in IGF-1 signaling may lead to free radical generation and adverse outcomes in both animal models and humans. Consequently, modulating the IGF-1 signaling pathway presents a promising therapeutic strategy to alleviate the harmful effects of OS, aging, and neuroinflammation.
Neurotrophic peptides, while promising for promoting neuronal survival and regeneration in the CNS, are constrained by several limitations. A major challenge is their poor ability to cross the BBB, which hinders their delivery to the CNS in therapeutically relevant concentrations. As other insulin-like peptides, IGF-1 is described to cross through the BBB by transcytosis [94]. In any case, as some therapeutic approaches may involve delivery systems such as nanoparticles or viral vectors, these should also be able to cross the BBB.
Another concern is the potential for off-target effects, as trophic peptides can act on multiple receptor types, leading to unintended biological responses. Thus, it is important to restrict IGF-1 overexpression to the SNC cells, since IGF-1 could have negative effects such as tumorigenic effects in obesity and endocrine-related cancer [95]. Moreover, the risk of inducing aberrant neuronal growth or hyperactivation further complicates their therapeutic use, necessitating precise control over their activity. Consequently, a deeper understanding of IGF-1’s role in the CNS is required. By characterizing the interaction between OS and IGF-1, we can identify specific downstream molecules in the IGF-1 signaling pathway that may serve as novel and effective therapeutic targets.
This review consolidates evidence showing that IGF-1 has antioxidant and anti-inflammatory effects that can improve and reduce alterations associated with NDs. Thus, we suggest that disruptions in IGF-1 signaling could induce free radical production and cellular damage. Therefore, modulating IGF-1 signaling offers a new therapeutic avenue in neurodegeneration, potentially mitigating the detrimental effects of OS, aging, and inflammation in the brain. However, this neurotrophic factor exhibits pleiotropic activity that varies depending on the specific cell type it acts upon. Even within the same cell type, its effects can differ based on peptide levels. Therefore, further research is essential to obtain more conclusive and promising results.
6-OHDA: 6-hydroxydopamine
AD: Alzheimer’s disease
ALS: amyotrophic lateral sclerosis
APP: amyloid precursor protein
BBB: blood-brain barrier
CNS: central nervous system
DA: dopaminergic
GH: growth hormone
GPx: glutathione peroxidase
HD: Huntington’s disease
Htt: huntingtin
IGF-1: insulin-like growth factor-1
MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
NDs: neurodegenerative disorders
OS: oxidative stress
PD: Parkinson’s disease
ROS: reactive oxygen species
SCI: spinal cord injury
SN: substantia nigra
SOD: superoxide dismutase
SPs: senile plaques
MLH: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. LGC: Methodology, Investigation, Writing—review & editing. ALO: Conceptualization, Visualization. MJB and CBH: Supervision, Writing—review & editing.
All the authors are members of the National Council on Scientific and Technical Research (CONICET).
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

