 Mini Review
Mini Review

Affiliation:
Department of Genetic Engineering and Biotechnology, East West University, Dhaka 1212, Bangladesh
Email: afsana.b.toma@gmail.com
ORCID: https://orcid.org/0000-0003-2120-5948
Explor Neuroprot Ther. 2024;4:485–496 DOI: https://doi.org/10.37349/ent.2024.00095
Received: September 28, 2024 Accepted: December 18, 2024 Published: December 25, 2024
Academic Editor: Lucio Marinelli, University of Genoa, IRCCS Ospedale Policlinico San Martino, Italy
Repeat expansion diseases (REDs) are genetic disorders caused by unusual expansions of DNA sequences within certain genes. They cause several neurodegenerative diseases including Huntington’s disease (HD), myotonic dystrophy (DM), spinal and bulbar muscular atrophy (SBMA), fragile X syndrome (FXS), and others. The pathogenic repeat expansions disrupt normal cellular processes by producing aberrant RNA repeat sequences, leading to toxic protein aggregation, RNA foci, and altered gene expression. Although they belong to the rare disease group, such diseases must be investigated to understand integral mechanisms and prevention. Current methods for alleviating these diseases involve—gene silencing therapies by antisense oligonucleotides (ASOs) and RNA interference (RNAi), CRISPR/Cas9 gene editing, small molecule therapies, etc. ASOs and RNAi reduce toxic protein production genes while CRISPR/Cas9 excise or alter expanded repeats. Small molecule therapies targeting RNA repeat-binding or proteostasis regulation are being developed to alleviate toxic protein accumulation, prevent RNA toxic foci formation, and promote the degradation of misfolded proteins. Additionally, gene replacement and regulatory element modification restore normal gene function. Some researchers tried to modulate toxic protein aggregation using heat shock proteins and chemical chaperones. This is a comprehensive review on the available research on RED treatment and their ongoing challenges, such as efficient delivery of therapies to the central nervous system, minimizing off-target effects in gene editing, sustaining therapeutic efficacy, and addressing toxicity and scalability in large-scale applications.
Repeat expansion diseases (REDs), a group of rare disorders due to the abnormal expansion of nucleotide sequences mostly occur in noncoding regions. Nearly 50 neurological diseases have been identified so far, of which 26 diseases are related to repeat expansion in coding, non-coding, and 5’ and 3’ UTR regions. These expansions result in a wide variety of neurodegenerative and neuromuscular diseases, including myotonic dystrophy type 1 (DM1), amyotrophic lateral sclerosis/frontotemporal dementia (C9ORF72 mediated ALS/FTD), fragile X syndrome (FXS), Huntington’s disease (HD), spinal and bulbar muscular atrophy (SBMA) and different types of spinocerebellar ataxia (SCA) like spinocerebellar ataxia type 3 (SCA3) [1]. They cause neurological disorders through the production of RNA with aberrant repeat expansion sequences. Both somatic and intergenerational instabilities were observed in affected individuals with large repeat expansion. These repeat expansion disorders are characterized by progressive decline in motor, cognitive, and other bodily functions, sensory loss with devastating impacts on patients’ quality of life [2]. Since the discovery of the first RED FXS in 1991, research on therapeutic strategies to mitigate the effects of expanded repeats has been continued [3]. This review outlines some of the most prominent therapeutic approaches currently being explored to treat REDs (Table 1).
Current approaches for repeat expansion diseases
| Therapeutic approach | Description | Diseases targeted | References |
|---|---|---|---|
| Gene silencing therapies | |||
| Antisense oligonucleotides (ASOs) | Synthetic strands of nucleic acids targeting specific RNA to degrade or prevent translation. | HD, SCA3, C9ORF72 mediated ALS/FTD, SBMA, DM1 | [4–20] |
| RNA interference by siRNA | Uses siRNA to degrade mRNA encoding toxic proteins. | DM1, HD, C9ORF72 mediated ALS/FTD | [21–29] |
| RNA interference by miRNA | Uses miRNA to degrade mRNA encoding toxic proteins. | HD, SBMA | [15, 30–34] |
| CRISPR/Cas9 gene editing | |||
| Direct repeat excision | Excises expanded repeat sequences using CRISPR/Cas9. | HD, SCA, DM1 | [35–40] |
| Base editing | Alters specific nucleotides in expanded repeats without creating double-stranded breaks. | HD | [41–43] |
| Small molecule therapies | |||
| RNA repeats or transcription factor binding molecules | Small molecules bind expanded RNA repeats or transcription factors to prevent toxic RNA foci formation. | DM, HD | [44–46] |
| Proteostasis regulators | Enhance degradation of toxic proteins via the proteasome or autophagy pathways. | HD | [47–53] |
| Gene therapy | |||
| Gene replacement | Introduces functional copies of affected genes. | SBMA, DM | [54–56] |
| Regulatory element modification | Modifies regulatory elements to upregulate normal alleles or downregulate mutant ones. | FXS | [57] |
| Modulating toxic protein aggregation | |||
| Heat shock proteins (HSPs) | Enhance protein folding and clearance of misfolded proteins. | HD | [58] |
| Chemical chaperones | Stabilize protein structures to prevent toxic aggregation. | HD | [59–63] |
HD: Huntington’s disease; SCA3: spinocerebellar ataxia type 3; ALS/FTD: amyotrophic lateral sclerosis/frontotemporal dementia; SBMA: spinal and bulbar muscular atrophy; DM1: myotonic dystrophy type 1; FXS: fragile X syndrome
Gene silencing is one of the primary therapeutic strategies aimed at reducing or eliminating the production of toxic proteins encoded by expanded repeats. This approach holds promise for REDs, where mutant protein accumulation is toxic for patients.
Antisense oligonucleotides (ASOs) are short, synthetic strands of nucleic acids aimed to bind to specific RNA sequences, leading to their degradation or preventing their translation into protein [4, 5]. ASOs have been developed for HD, targeting the mutant huntingtin (HTT) transcript to reduce its production. Studies have shown that reducing HTT levels in animal models leads to improvement in motor function and reduced neurodegeneration [6]. Some ASOs have been developed as splice-modulator like branaplam and PTC518 or as knockdown ASOs like tominersen, WVE-120101, and WVE-120102. In 2015, the first continuous clinical trial (NCT02519036) aimed at reducing HTT levels was launched. The trial utilized ASOs administered intrathecally to individuals in the early stages of HD, with the goal of lowering HTT alleles. Some ASOs, such as tominersen (Figure 1) entered clinical trials, showing encouraging initial results in HD patients. However, phase 3 clinical trials (NCT03761849) were halted due to off-target inflammatory effects. New phase 2 clinical trial is under process to mitigate these effects [7].
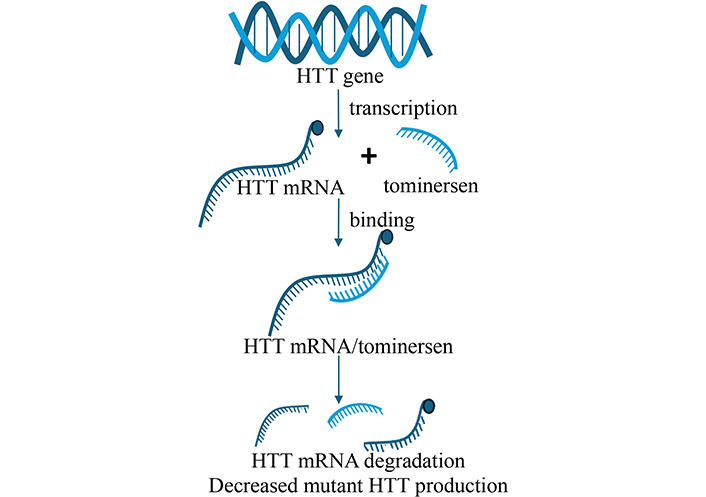
Tominersen as an ASO. Tominersen binds to HTT mRNA and degrades them. As a result, mutant HTT protein production decreases. ASO: antisense oligonucleotide; HTT: huntingtin. Adapted from Van de Roovaart et al. 2023 [77]. © 2023 by the authors. CC BY 4.0
Branaplam initially in phase 2 clinical trial for spinal muscular atrophy (NCT02268552) showed promises against HTT protein and was in phase 2b trial (NCT05111249) until recently [8]. ASO therapy targeting the GGGGGCC repeat expansion in C9ORF72 gene shows promise for ALS/FTD treatment. ASOs effectively reduce GGGGGCC-containing transcripts and poly GP dipeptides in patient-derived cells and C9BAC mice. ASOs with reduced phosphorothioate content improve tolerability without compromising efficacy. Intrathecal delivery of optimal ASO in a patient with mutant C9ORF72 led to significant reductions in cerebrospinal fluid poly GP levels [9]. ASO therapy for SCA3 mice resulted in significant total choline rescue and partial reversals of taurine, glutamine, and total N-acetyl aspartate in the cerebellum and brainstem. ASO treatment fully or partially reversed neurochemical abnormalities in SCA3 mice, indicating the potential for these measures to serve as non-invasive treatment biomarkers in future SCA3 gene silencing trials [10]. As expansion in polyglutamine stretch in ataxin-3 gene is the main reason for SCA3, anti-ATXN3 ASO has been shown effective in several mouse and cell culture models [11–13]. It is also now being investigated in phase I clinical trial (identifier: NCT05160558) [14]. Preclinical studies using ASOs in SBMA mouse models showed improved muscle strength and extended lifespan by reducing toxic proteins [15]. Several ASOs are being evaluated for the treatment of DM1. These ASOs corrected splicing defects, reduced RNA foci, and improved muscle strength in mice [16–19]. Clinical trials: NCT02312011, DYNE-101, IONIS 486178 showed promise in correcting the DM1 phenotype in human. Challenges with ASOs in DM1 include poor blood-brain barrier (BBB) penetration and inadequate tissue uptake in skeletal muscle. Strategies to improve ASO delivery in DM1 include ligand-conjugated ASOs and cell-penetrating peptide conjugates [20]. As ASOs are not selective for the mutant allele, some strategies like mixmer design, nucleotide mismatch, and backbone modification can be used to improve specificity.
RNA interference (RNAi) utilizes small interfering RNA (siRNA) molecules to target and degrade mRNA transcripts encoding toxic proteins. Like ASOs, siRNA-based therapies target mutant gene expression reduction. RNAi has been explored in diseases like DM1, where expanded CUG repeats in the DMPK gene cause toxic RNA foci that sequester proteins, disrupting normal cellular function [21]. A phase 1/2 clinical trial (NCT05027269) has been completed in 2023 for safety check. RNAi approaches targeting these repeats have demonstrated therapeutic potential in preclinical models. In several HD mouse models, RNAi was found beneficial [22–25]. Lipid nanoparticle delivered siRNA has been used to selectively silence polyQ-expanded androgen receptor, causative agent for in SBMA [26]. ASOs using single and double-stranded siRNAs proved promising to reduce gain-of-toxicity features caused by the GGGGCC repeat. Mice with C9ORF72 RNA repeats or inactivated C9orf72 alleles showed reduced RNA foci and dipeptide-repeat proteins using siRNA [27–29].
MicroRNA (miRNA) approach is a promising therapeutic strategy for HD by targeting the HTT protein. This approach involves delivering a therapeutic miRNA precursor using an adeno-associated viral (AAV) vector to reduce overall HTT translation in target cells. AAV5-miHTT intrastriatal injections were developed in 2019 and are in phase I/II clinical trial as of October 2024 (uniQure AMT-130 trial, NCT04120493). Another clinical trial (AMT-130, NCT05243017) using AAV5-miHTT was developed in 2021 and was in phase Ib/II stage as of October 2024. Using AAV-delivered anti-HTT RNAi technique, VY-HTT01 was developed for HD’s clinical trial that was discontinued later [30–32]. AAV-mediated miRNA targets AR gene expression via miRNAs like miR-196a, which enhances androgen receptor mRNA decay, showing promising results in SBMA mouse models [15]. Using antisense technology, antagomiR-23b has been tested against DM1 that upregulated MBLN1 protein, recovered mis-splicing and muscle strength. AntagomiR-23b is now in phase I/II clinical trial [33]. AAV5-miC (artificial anti-C9orf72-targeting miRNAs) in induced pluripotent stem cells (iPSCs) neurons and an ALS mouse model demonstrated successful silencing of C9orf72 and reduction of toxic transcripts and foci [34].
The CRISPR/Cas9 genome-editing tool holds potential for treating REDs by directly targeting and correcting the expanded repeat sequences. In principle, CRISPR/Cas9 can be used to excise the expanded repeats or modify the genes to restore normal function.
CRISPR/Cas9 can be programmed to recognize and cut the expanded repeat sequences in affected genes. This approach has been applied in preclinical studies of SCA, and HD where the expanded CAG repeats excision reduced the toxic protein burden [35, 36]. The system has been effective in iPSCs as creating isogenic control iPSCs could help confirm precise correction of the mutations [37, 38]. In DM1, CRISPR/Cas9 has been implemented to delete large CTG repeats [39]. However, delivering CRISPR components efficiently and specifically to affected tissues remains a significant hurdle [40].
Another approach involves base editing, where specific nucleotides in the expanded repeat sequence are altered without introducing double-stranded breaks. This method may offer a safer alternative to traditional CRISPR/Cas9 editing by minimizing off-target effects and limiting potential genomic instability [41]. A study demonstrated the effectiveness of a CRISPR-based editing approach by skipping exon 13 of the HTT gene. These editors reduced the formation of toxic N-terminal HTT fragments, reduced mutant HTT protein aggregation, and attenuated brain atrophy in HD rodent model. CRISPR base editing showed promising results in the rodent model by producing mutant HTT isoforms that are more resistant to toxic proteolysis, offering potential for long-term treatment [42, 43].
Several small molecules have ability to alleviate the pathological consequences of repeat expansions. These molecules either directly target the expanded RNA repeats or modulate cellular pathways that are disrupted by toxic protein accumulation.
Small molecules that bind selectively to expanded RNA repeats can prevent the formation of toxic RNA foci. For example, compounds targeting the expanded CUG repeats in DM have shown promise in preventing the sequestration of essential RNA-binding proteins, thus restoring normal cellular function. This study explores the interaction of small molecules with trinucleotide repeat expansions in RNA, which are linked to diseases like DM1 and HD [44]. Using nuclear magnetic resonance (NMR) and molecular dynamics, researchers identified how three small molecules (a diguandinobenzoate, an imidazole derivative, and a quinoline) bind to the RNA’s hairpin structure. These findings enhance the understanding of ligand-RNA interactions and offer potential for designing new treatments targeting RNA-related disorders [45]. Another study reported SPI-24 and SPI-77 as Spt5-Pol II small molecule inhibitors for lowering mutant HTT mRNA and protein levels in HD mouse and cell lines [46].
Proteostasis regulators aim at the ubiquitin-proteasome system by enhancing the degradation of toxic proteins. These strategies target various components of the proteolytic machinery to address protein misfolding and aggregation in HD. Overexpression of proteasome activator PA28ɣ improved cell viability in HD neuronal models [47] and salvaged HD phenotypes in YAC128 mice model [48]. Elevated expression of the proteasome’s pbs-5 catalytic subunit improved resistance to proteotoxic stress and alleviated motor dysfunction in nematode HD models [49]. Enhanced expression of E3 ubiquitin ligases (such as CHIP, Ube3, Herp, and UBR5) may reduce mutant HTT aggregation by facilitating its ubiquitination and subsequent degradation via the proteasome [50, 51]. Overexpression of NUB1, which recruits ubiquitin ligases reduced mutant HTT neurotoxicity in fly models [52]. Ubiquilin-2 overexpression with the Hsp70-Hsp110 disaggregation system facilitates the transfer of Hsp70-bound mutant HTT for proteasome degradation [53].
Gene therapy approaches involve the delivery of normal copies of genes or regulatory elements to correct the effects of the expanded repeats. Viral vectors have been employed to deliver therapeutic genes to affected cells such as AAVs.
In cases where loss of function due to expanded repeats is the primary cause of disease, gene replacement therapy can be used to introduce functional copies of the affected gene. This approach has been tested for SBMA in mouse models, where delivery of wild-type androgen receptor genes reduced disease severity [15]. An AAV delivery system AT-466 has been developed for treating DM that inserts a functional version of the MBNL1 gene into cells impacted by DM1 [54–56].
Another gene therapy approach involves modifying regulatory elements to increase the expression of non-expanded alleles or downregulate mutant alleles. This strategy is under investigation for FXS, where CGG repeats expansion in the FMR1 gene causing transcriptional silencing. This study emphasizes that directing transcriptional activators specifically to CGG repeats offers a promising approach for reactivating FMR1 expression in FXS patients. However, challenges remain, such as achieving sufficient FMRP protein production, which is necessary for therapeutic effects [57].
Repeat expansion disorders frequently result in the accumulation of toxic protein aggregates within cells. Various therapeutic approaches focus on preventing or reversing this aggregation.
Heat shock proteins (HSPs) known as molecular chaperones assist in protein folding and help clear misfolded proteins. Small molecules that enhance the expression or function of HSPs are being examined for their ability to reduce protein aggregation in diseases like HD [58].
Chemical chaperones are small molecules that stabilize protein structures and prevent misfolding. Some research shows that chemical chaperones may have the ability to prevent the formation of toxic protein aggregates in various REDs [59–62]. A study showed molecular chaperones named Brichos alleviated the toxicity and aggregation processes by selectively engaging with amyloid fibrils in Alzheimer’s disease [63]. In addition to targeting the root cause of REDs, some therapeutic approaches focus on mitigating downstream cellular pathways that are disrupted by repeat expansions. As neurodegeneration is a hallmark of many REDs, some “neuroprotective agents” are being tested to slow disease progression. These agents aim to reduce oxidative stress, inflammation, and mitochondrial dysfunction, all of which contribute to neuronal death in REDs [64]. Besides, abnormal ion channel function has been implicated in certain repeat expansion disorders, such as SCAs. “Ion channel modulators” like riluzole may help to restore normal neuronal firing patterns and improve motor function in patients [65].
The prevalence of FXS with CGG-repeat mutations is about 1 in 7,000–11,000 in population globally [66]. The global prevalence of HD with CAG repeat mutations is about 1 in 7,000–11,000 in population [67]. For spinal bulbar muscular atrophy with CAG repeat, the prevalence is 1 in 100,000 globally [68]. These findings highlight the significant global burden of REDs, which vary by population and specific disorder. Therapeutic strategies for REDs, while advancing significantly, still face several major challenges. These obstacles affect the development, delivery, and effectiveness of treatments such as gene silencing, gene editing, small molecule therapies, and gene therapy. One of the key challenges is delivery challenges. Many REDs, such as FXS, HD, and ALS primarily affect the central nervous system (CNS). A major challenge is ensuring that therapeutic agents, such as ASOs and viral vectors used in gene therapy, can successfully cross the BBB and reach the target neurons. This obstacle can be bypassed by using ASO intrathecal injection or delivery particles coated ASOs or by recombinant AAV vectors [69]. Gene therapy using AAV might be irreversible and result in high level of target gene increase as the risks [70]. While ASO and AAV do not cross the BBB and require complicated and painful administration methods, small molecules can easily be administered and pass the BBB. In recent years, CRISPR/Cas9 technology has shown promise for correcting mutations in REDs, but concerns remain over off-target cuts, which could potentially cause unintended mutations and lead to negative consequences [71]. Moreover, many treatments, especially those based on ASOs or small molecule therapies, may require repeated administration to maintain efficacy. This escalates the risk of side effects and reduces patient compliance. For example, frequent intrathecal injections of ASOs in HD patients are required, which poses practical and clinical challenges [72]. Furthermore, high doses of gene therapies or ASOs may cause toxicity, particularly if these therapies are administered repeatedly or in high concentrations. The CNS is especially sensitive to such toxicity, and balancing effective doses with safety is crucial [73]. Besides, gene therapy and gene editing technologies, particularly those involving viral vectors or large-scale production of biologics, are complex and costly. This poses issues for scalability, especially in making these therapies widely available to patients across different healthcare systems [74]. Monitoring the effectiveness of therapies is challenging due to the lack of reliable biomarkers that can indicate therapeutic success, especially in slowly progressing diseases such as spinal bulbar muscular atrophy. Without robust biomarkers, it becomes difficult to evaluate treatment effects early in the disease course [75]. Last but not least, ethical and regulatory challenges remain a concern. Gene editing technologies, like CRISPR, present significant ethical concerns, particularly when used for germline editing, as they can result in heritable changes. Regulatory hurdles are high and long-term safety concerns must be addressed before these therapies can be widely adopted [76]. While significant advances have been made in developing therapeutic strategies for REDs, numerous challenges remain, including safe and effective drug delivery to the CNS, minimizing off-target effects in gene editing, sustaining therapeutic efficacy, managing toxicity, and addressing scalability and cost. Overcoming these barriers will be key to translating these therapies from preclinical promise to clinical reality.
AAV: adeno-associated viral
ASOs: antisense oligonucleotides
BBB: blood-brain barrier
CNS: central nervous system
DM1: myotonic dystrophy type 1
FXS: fragile X syndrome
HD: Huntington’s disease
HSPs: heat shock proteins
HTT: huntingtin
iPSCs: induced pluripotent stem cells
miRNA: microRNA
REDs: repeat expansion diseases
RNAi: RNA interference
SBMA: spinal and bulbar muscular atrophy
SCA: spinocerebellar ataxia
SCA3: spinocerebellar ataxia type 3
siRNA: small interfering RNA
AB: Conceptualization, Writing—original draft, Writing—review & editing.
The author declares that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

